

Abstract
Purpose
Imatinib is a small molecule inhibiting the tyrosine kinases bcr-abl, c-kit, PDGFR-α and PDGFR-β. Investigations were performed to screen ovarian cancer cell lines and tumor samples for target receptor expression. Effects of Imatinib on cell proliferation and apoptosis induction were measured with and without additional cytotoxic agents.
Methods
Expression patterns of abl, c-kit, PDGFR-α and PDGFR-β (Imatinib targets) were studied in 5 cell lines and 111 tissue arrays by PCR and immunohistochemistry. Proliferation assays were performed with single agent Imatinib or combined with Paclitaxel and Carboplatin. Apoptosis was measured by DNA fragmentation.
Results
All cell lines expressed abl and PDGFR-β. C-kit was only expressed in 2/5 cell lines and PDGFR-α in 4/5. Imatinib reduced cell growth and lead to pro-apoptotic changes. Combination of Carboplatin, Paclitaxel and Imatinib showed synergistic activity.
Conclusions
Our results suggest that Imatinib may be useful for the specific treatment of ovarian cancer as an add-on to conventional chemotherapy.
Keywords: Ovarian cancer, Targeted therapy, In vitro, Imatinib Mesylate
Introduction
Ovarian cancer is the second most deadly gynecological malignancy with an estimated 22,200 new cases each year and an estimated 16,210 death per year (Jemal et al. 2005). Most patients are diagnosed at a late stage which leads to poor prognosis (Bristow et al. 2002). After cytoreductive surgery, adjuvant chemotherapy with a combination of Carboplatin and Paclitaxel is considered the gold standard therapy (Neijt et al. 2000). In cases of platinum resistance, there are only limited options (Gordon et al. 2001). New drugs targeting cancer-specific molecules are currently widely tested in patients suffering from different solid tumors. In cases of recurrent or platinum-resistant ovarian cancer combinations of new targeted substances with standard chemotherapy could be beneficial.
Imatinib (Gleevec), a small molecule antagonist, has been shown to specifically inhibit the pathognomonic bcr-abl tyrosine kinase in chronic myeloic leukemia (CML). Furthermore, Imatinib was able to inhibit additional receptor tyrosine kinases (RTK) like c-kit and PDGFR-β and intermediates of downstream cascades (e.g. akt). Those are important key receptors in solid cancer types (Frolov et al. 2003). RTK are the initiators of signaling pathways with a wide spectrum of physiological functions like regulation of cell proliferation and differentiation, modulation of cellular metabolism and promotion of cell survival (c-kit and PDGFR). Their ligands are growth factors and hormones like NGF, EGF, PDGF, SCF and insulin. Any mutation of c-kit causing pathological autophosphorylation or other excessive activation of c-kit and PDGFR, respectively, may lead to cancer progression. PDGF is also known as a strong mitogen and a stimulating factor for endothelial cells and smooth muscle cells. Therefore, RTK have gained broad attention. The purpose consists in developing antibodies or substances which are able to inhibit the activation of these kinases. Imatinib is furthermore showing synergistic effects with the established chemotherapeutic substances (Kano et al. 2001) in leukemia and in adenocarcinomas (Faivre et al. 2005). All these observations lead to the consideration, that Imatinib could be useful for the treatment of a wider range of solid cancers (Krystal et al. 2000; Kubler et al. 2005).
Our hypothesis is that ovarian cancer cells express targets (RTK) for a therapy with Imatinib Mesylate and will respond with growth inhibition after incubation with this tyrosine kinase inhibitor. Imatinib targets of standard ovarian cancer cell lines will be analyzed and inhibitory effects of Imatinib with and without additional cytotoxic agents will be investigated in vitro.
Materials and methods
Cell lines
To define potential molecular targets for a drug regimen including Imatinib, five ovarian cancer cell lines were tested for receptor expression and response to Imatinib incubation in vitro. The ovarian cancer cell lines BG-1, HEY, OVCAR 3, SK-OV 3 and OVCAR 8 (American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 with 10% FCS and 30,000 IU penicillin/streptomycin at 37°C and 5% CO2. Cells were tested for mycoplasma infection by PCR Mycoplasma Detection Set (TaKaRa, Japan). Only non-infected cell lines were used for the investigation. Sub-cultivation was performed with non-confluent (70–80% confluency) cells.
PCR
Receptor expression of abl, c-kit, PDGFR-α and PDGFR-β was determined in all the five ovarian cancer cell lines by PCR. Therefore, RNA was isolated following the Rneasy mini-kit protocol (Qiagen GmbH, Hilden, Germany). mRNA was selectively transcribed into cDNA using Superscript II RNase H Reverse Transcriptase (both Invitrogen, Madison, WI, USA). PCR was performed using Hot Star Taq (Qiagen GmbH, Hilden, Germany) and the DNA Engine PTC-200 Peltier Thermal Cycler (MJ Research, Waltham, MA, USA). A temperature of 95°C was used for 15 min to ensure denaturation of short fragments of DNA before the first cycle. The following cycles started with denaturation at 94°C for 1 min. Then a primer-specific temperature was chosen to induce primer annealing for 1 min, followed by elongation for 1 min at 72°C. After the last cycle a temperature of 72°C was applied for 10 min to allow final elongation. G3PDH (forward primer 5′-ACC ACA GTC CAT GCC ATC ACT GCC-3′, reverse primer 5′-CAT GTG GGC CAT GAG GTC CAC CAC-3′) (Hartman et al. 2001) a common example for a housekeeping gene, that is involved in the glycolysis pathway, was used as a positive control for successful transcription. PCR results were obtained by electrophoresis using 2% agarose gels with ethidium bromide development. All results were documented by Image Master VDS and Fujifilm Thermal Imaging System FTI-500.
To detect the expression of the Abl-tyrosine kinase, we used the primer pair with the forward sequence 5′-GAG GGC GTG TGG AAG AAA TA-3′ and the reverse part 5′-GCT GTG TAG GTG TCC CCT GT-3′ (annealing temperature of 57°C for 35 cycles) to get a 412 bp fragment (Roche-Lestienne et al. 2002). C-kit expression pattern was screened by using the forward primer 5′-CTG AAC ACG CAC CTG CTG AA-3′ and as reverse primer 5′-AAG CTA CGT TGC TAT TGG GAA-3′ (Palumbo et al. 2002) at again 57°C for 35 cycles to gain a 512 bp sized fragment. The primer pair for the detection of PDGFR-α used the forward sequence 5′-GAG TGA GGA TTC TTT GGA ACC-3′ and the reverse sequence 5′-CTT CTT CCT GAC GTA TTC CAC C-3′ (Westphal et al. 2000) with an annealing temperature of 60°C for 35 cycles and showed a product with a size of 322 bp. Finally, the forward primer 5′-TGA CCA CCC AGC CAT CCT TTC-3′ and the reverse primer 5′-GAG GAG GTG TTG ACT TCA TTC-3′ (Satomura et al. 1998) were used to detect the PDGFR-β. An annealing temperature of 56°C for 38 cycles was used to gain a product of 228 bp. Primers used in this study were specific for the detection of each transcript.
Immunohistochemistry assay
To confirm PCR results (RNA level), immunohistochemistry (IHC) was performed to evaluate receptor expression on a post-translational protein level. Staining was performed according to the standard protocol (DAKO EnVisionT System, DakoCytomation, Glostrup, Denmark). Attached cells were washed twice with PBS and then fixed in cold acetone for 10 min. After washing, slides (LabTec® Chamber Slides, Nalge Nunc International, Rochester, NY, USA) were covered with methanol (Merck, Darmstadt, Germany) for 1 min and then washed with methanol plus 3% H2O2 (Merck) for 20 min and rinsed with purified water. DAKO Peroxidase Blocking Reagent was then applied for 10 min, followed by another washing step with aqua. Now the first antibody was applied. Primary antibodies included rabbit anti-c-kit (C-19: sc-168), rabbit anti-PDGFR-α (C-20: sc-338) and rabbit anti-PDGFR-β (P20 Sc-339) (all Santa Cruz Biotechnology, Santa Cruz, CA, USA) and were used in a 1:200 dilution of PBS containing 1% BSA. In total, 100 μl of antibody solution were applied to each slide and incubated for 30 min at room temperature. Negative controls were incubated without primary antibody. Slides were washed with Tris buffer solution (Merck) and second antibodies, goat anti-rabbit and anti-mouse (Dako Kit) were applied. After 30-min incubation at room temperature, slides were washed with Tris buffer solution. For visualization the developing solution (DAB) was added, counterstaining was performed using Mayer’s Hamalaun solution (Merck).
In vitro proliferation assays
A tetrazolium (MTT) assay (Promega Medicine, WI, USA) was performed as described before (Dunigan et al. 1995). For single and combination treatment Carboplatin, Paclitaxel (both Sigma Aldrich, Munich, Germany) and Imatinib (Novartis Pharmaceutical, Basel, Switzerland) were used. Carboplatin was dissolved in PBS, Paclitaxel and one part of Imatinib in DMSO; all three were stored at −20°C. The remaining Imatinib was dissolved in H2O and stored at 4°C.
Ovarian cancer cells were diluted to 10,000 cells per 1 ml. In total, 100 μl of cell dilution was applied per well on a 96-well plate (Sarstedt, Nümbrecht, Germany). Drugs were diluted in standard medium as described earlier and added after overnight cell attachment. Cells were incubated for 72 h at 37°C. The number of viable cells was determined by the MTT assay utilizing an ELISA reader at a wavelength of 590 nm (Dynatech MR 5000 Microplate Reader, Dynatech Corp., Burlington, MA, USA).
The inhibiting concentrations (IC values) for the drugs used were evaluated for each cell line separately. IC 50 represents the concentration of a drug that is required for 50% inhibition in vitro. The detected IC values were used as basic information for the experimental design of the drug combination proliferation assays. These assays were performed to quantify growth inhibition and to characterize subtractive, additive and possible synergistic effects of drug combinations in vitro. Negative controls for proliferation statistics were carried out by adding the dissolving solution (PBS, DMSO or H2O) in the referring drug concentration to standard medium. Each experiment was set up in triplicates. For Imatinib incubation, cells were applied at 1,000 cells/well on a 96-well plate and fresh Imatinib solution was added daily for 7 days.
Statistical analysis was carried out using the Students’ t test between controls and combination therapy including Imatinib. P values <0.05 were considered statistical significant. By definition, additive effects were observed when cell proliferation was inhibited according to the sum of the individual IC of the agents combined. Synergism was defined as inhibition of cell proliferation that exceeds the additive effect at least of further 5% points.
Apoptosis asssay
Apoptosis induction by Imatinib in ovarian cancer cell lines was examined. Also, it had to be distinguished between specific apoptosis and unspecific toxicity of higher drug concentrations. Therefore, testing for early and late signs of apoptosis was performed according to the ApopLadder Ex(tm) protocol (Takara Bio Inc., Otsu, Shiga, Japan). Cells were incubated with Imatinib for 72 h and genomic DNA was isolated according to the manufacturer’s protocol. DNA fragmentation was then visualized by electrophoresis in 2% agarose gels with ethidium bromide and documented as described above.
Tissue microarrays
Microarrays of 111 formalin embedded ovarian cancer cases were produced. For detection of PDGFR-β and c-kit antigen retrieval was performed using 1 mM EDTA or 10 mM citrate buffer. After incubation with peroxidase blocking reagent (DAKO), slides were washed and incubated with goat serum for further reduction of background signals. Primary antibodies used were Dako CD 117 (1:100), sc-338 rabbit anti-PDGFR-α IgG (1:100) (Santa Cruz) and rabbit anti-PDGFR-β mAB 3169(1:50) (Cell signaling, Beverly, MA, USA). Negative controls were carried out by incubation omitting primary antibodies. Dako Envision kit was used for secondary labeling. Receptor expression was evaluated according to the IRS (immunoreactive score combining intensity of receptor staining and percentage of receptor positive cells) (Remmele and Stegner 1987). The cutoff value for positive staining was defined as IRS ≥ 3.
Results
Receptor status
Expression of the tyrosine kinases abl and PDGFR-β could be detected in mRNA transcripts of all the five examined ovarian cancer cell lines (shown in Fig. 1). Expression of PDGFR-α was clearly positive in four of the five cell lines. PCR analysis of SK-OV 3 cells revealed a hardly detectable signal. C-kit was detectable with PCR only in OVCAR 3 and SK-OV 3 cells. These results for receptor status were confirmed by IHC and correlate with findings from other research groups (Matei et al. 2006).
Fig. 1.

PCR receptor results for the five ovarian cancer cell lines HEY, BG-1, OVCAR 8, OVCAR 3 and SK-OV 3. Product size for the screened receptor is mentioned in the brackets
Cell proliferation
Table 1 shows the results for the proliferation assays performed with Imatinib as mono therapy in vitro. These experiments show significant differences in Imatinib activity, depending on the solution agent used (DMSO or aqua). For Imatinib dissolved in aqua, a much lower concentration between 10 and 15 μM was needed to reach the IC50 in all the five examined ovarian cancer cell lines. For Imatinib, dissolved in DMSO, concentrations between 13 and 28 μM were necessary to reach the IC50. A further dramatic reduction of Imatinib’s IC 50 has been seen when Imatinib and fresh medium were applied daily for 7 days. This experimental design describes the lowest active dose for Imatinib as single therapy in all the examined ovarian cancer cell lines. Concentrations from 3 to 5 μM reduced cell proliferation to 50% in comparison to controls without Imatinib treatment (see Table 1). There is only a weak correlation between response to Imatinib monotherapy and the expression of c-kit. C-kit is the only tyrosine kinase receptor, which is only expressed in two cell lines. The c-kit negative cell line HEY needs the lowest concentration (IC 50:3 μM) of Imatinib to inhibit cell proliferation. The inhibitory activity is similar in the two c-kit-positive cell lines OVCAR 3 and SK-OV 3 (IC 50:3.5 and 4.0 μM).
Table 1.
Imatinib doses needed to reach cell inhibition in monotherapy for 72 h and 7 days. The inhibiting concentration (IC) was acquired by mean value of triplicates of three experiments
| Cell line | IC | Imatinib (DMSO) for 72 h (µM) | Imatinib (H2O) for 72 h (µM) | Imatinib for 7 days (daily change of medium) (µM) |
|---|---|---|---|---|
| HEY | IC0 | 5 | 2 | 0.25 |
| IC5 | 11 | 4 | 0.5 | |
| IC25 | 12 | 6 | 1.5 | |
| IC50 | 13 | 10 | 3 | |
| IC75 | 18 | 25 | 4.5 | |
| BG-1 | IC0 | 5 | 2 | 1 |
| IC5 | 11 | 4 | 2 | |
| IC25 | 12 | 6 | 3.5 | |
| IC50 | 13 | 10 | 5 | |
| IC75 | 18 | 25 | Not reached | |
| OVCAR8 | IC0 | 5 | 2 | 1 |
| IC5 | 11 | 4 | 2.5 | |
| IC25 | 16 | 8 | 3.5 | |
| IC50 | 20 | 10 | 4.5 | |
| IC75 | 30 | Not reached | 7 | |
| OVCAR3 | IC0 | 5 | 2 | 0.5 |
| IC5 | 13 | 4 | 1.5 | |
| IC25 | 18 | 11 | 2 | |
| IC50 | 25 | 18 | 3.5 | |
| IC75 | 35 | 25 | 4.5 | |
| SK-OV3 | IC0 | 5 | 1 | 1 |
| IC5 | 18 | 2 | 2 | |
| IC25 | 23 | 5 | 3.5 | |
| IC50 | 28 | 15 | 4 | |
| IC75 | 40 | Not reached | 4.5 |
The inhibiting concentration (IC) data derived from the 72-h assays were used in the combination experiment. Figure 2 shows the combination of Carboplatin in IC50 dose and Imatinib in increasing concentrations for 72 h of incubation. A significant decrease in cell proliferation (P < 0.05) could be observed for the ovarian cancer cell lines HEY, OVCAR 8, OVCAR 3 and SK-OV 3 when Imatinib was used in doses of 1–2 μM. At these low Imatinib doses no inhibitory effects in the monotherapy setting could be seen. The strongest reduction in cell proliferation was observed in OVCAR 3 cells. The growth inhibition in BG-1 cells was not significant. Likewise, results could be observed when the Imatinib concentration was increased to 2 and 4 μM (IC5). The c-kit positive cell lines OVCAR 3 and SK-OV 3 also showed a significant further inhibition of cell proliferation when treated with a combination of Carboplatin (IC50) and Imatinib (IC25) dose concentration (i.e. 5, 6, 8 and 11 μM), compared to the IC5 data. Further increase of Imatinib concentration did not show additional effects. When Paclitaxel was combined in IC50 concentration with Imatinib consistent data resulted. A combination of Paclitaxel with Imatinib in a concentration of 1or 2 μM, respectively, again showed a significant decrease of cell proliferation in comparison to the control group treated with Paclitaxel only for four of the examined cell lines. HEY did not show a significant decrease in cell growth. If Imatinib concentration was increased to the IC5 (2 and 4 μM) in the combination therapy, a significant inhibition of cell proliferation in all the five examined cell lines in comparison to control groups could be observed. If Imatinib concentration was further increased, inhibition of cell proliferation could not be enhanced. Finally, the triple therapy of Carboplatin and Paclitaxel (common IC50) with Imatinib led to identical observations, as cell growth could significantly be lowered for all ovarian cancer cell lines except BG-1 by introducing Imatinib in the combination in concentrations of 1–2 μM. Increase of Imatinib to 2–4 μM could inhibit cell growth for these four cell ovarian cell lines further significantly. As shown in Fig. 2, the c-kit positive cell lines OVCAR 3 and SK-OV 3 showed significant decrease in cell proliferation when Imatinib was increased to levels of 5, 6, 8 or 11 μM, respectively. In OVCAR 3 Imatinib showed synergistic cell growth inhibition with the cytotoxic drugs. In SK-OV 3 cells, Imatinib showed additive effects with cytotoxic drugs.
Fig. 2.

Cell proliferation after exposure to different combinations of Imatinib, Carboplatin and Paclitaxel for 72 h. Drug concentration of each drug used in the particular combination is given below each cell line
Apoptosis
HEY, OVCAR 3 and SK-OV 3 were exposed to the preliminary determined IC50 concentrations of Imatinib for 72 h. A positive DNA ladder pattern for the cell lines OVCAR 3 and HEY (Fig. 3) suggests pro-apoptotic effects caused by Imatinib.
Fig. 3.

DNA fragmentation can be shown for the cell lines OVCAR 3 and HEY after exposure to Imatinib in IC50 concentration for 72 h, negative control is shown on the right (atifact for OVCAR 3)
Immunohistochemistry
Among the 111 ovarian cancer cases, 70 were papillary serous, 12 mucinous, 15 endometrial and 14 solid ovarian cancers by histology. We observed that of the papillary serous tumors, 62 showed an expression of PDGFR-α, 60 of PDGFR-β and 56 of c-kit (Fig. 4). In the group of mucinous tumors nearly all samples (Roche-Lestienne et al. 2002) expressed PDGFR-α and PDGFR-β. C-kit was detected by eight samples (Table 2).
Fig. 4.
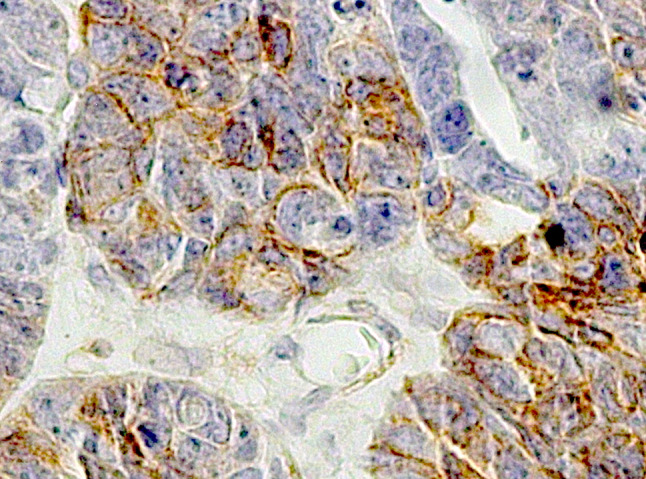
Immunohistochemical expression of PDGFR-β in serous papillary ovarian cancer. Tissue arrays were stained with primary antibody against PDGFR-β
Table 2.
Expression of PDGFR-α, PDGFR-β and c-kit among 111 samples of ovarian cancer
| PDGFR-β | PDGFR-α | c-kit | ||
|---|---|---|---|---|
| Histology | Total | Positive (%) | Positive (%) | Positive (%) |
| Papillary serous | 70 | 85.71 | 88.57 | 80.00 |
| Mucinous | 12 | 91.67 | 91.67 | 66.67 |
| Endometrial | 15 | 80.00 | 86.67 | 73.33 |
| Solid | 14 | 92.86 | 85.71 | 78.57 |
The receptor expression patterns of endometrial and solid ovarian cancers were similar: 12 solid and 13 endometrial type ovarian cancers were positive for PDGFR-α. PDGFR-β expression could be found in 13 solid and 12 endometrial type samples. 11 samples of each type were positive for c-kit expression. The majority of receptor positive ovarian cancers had also a tumor stroma which showed positive receptor expression.
Discussion
This study was designed to examine the effects of the selective tyrosine kinase inhibitor Imatinib in ovarian cancer cell lines. Previously we had hypothesized that Imatinib could be considered for targeted therapy in ovarian cancer.
We could show that the tyrosine kinases abl and PDGFR-β are expressed in all the five examined ovarian cancer cell lines. PDGFR-α was detected in four cell lines. The expression in SK-OV 3 was too weak for photo documentation. C-kit expression could only be detected in OVCAR 3 and SK-OV 3 cells on the transcriptional (RNA) level and as well on the post-translational (protein) level. The three other cell lines neither expressed c-kit on the transcriptional nor on the post-translational level. This broad expression of Imatinib target receptors has also been confirmed by immunohistochemistry in tissue arrays of 111 ovarian cancer cases. High expression of PDGFR-α and PDGFR-β in ovarian tumors and ovarian cancer cell lines has also been described by other groups (Apte et al. 2004), but the percentage of receptor positive tumors variegates among the studies. Moreover, an over-expression of c-kit and PDGFR-α among serous ovarian carcinoma was described which was associated with aggressive tumor characteristics, but no mutations were found (Lassus et al. 2004). Although investigation of larger series of ovarian cancer subtypes would be helpful, the examined number of mucinous, endometrial and solid tumors is already among the larger cohorts published (Matei et al. 2006).
In our studies, Imatinib was used in a therapeutic range from 1.45 μM (through) to 4.6 μM (peak). These are dose levels which are comparable to plasma levels of patients treated with a regimen of 200 mg Imatinib twice daily (Buchdunger et al. 2002).
In vitro Imatinib as a single agent (from 3 to 5 μM) showed a dramatic reduction of cell proliferation by 50% (IC50) for all the five examined cell lines in comparison to controls without Imatinib treatment. Furthermore we could observe apoptosis induction in OVCAR 3 and HEY cells by Imatinib. Therefore, we suggest that the combination of direct antiproliferative activities and apoptosis induction might lead to the observed cytoreductive effects among the afore mentioned cell lines. In the clinical setting (recurrent ovarian cancer in heavily pretreated patients) Imatinib has so far failed to show relevant clinical activity as a single agent (Alberts et al. 2007; Coleman et al. 2006).
Combination therapy with the established cytotoxic drugs like Carboplatin and Paclitaxel for 72 h (within the therapeutic range) (Alberts et al. 2001) led to synergistic effects in four of the five ovarian cancer cell lines when Imatinib was added in concentrations of 1–2 μM. In the BG-1 cell line only limited reduction of cell proliferation was seen when combined with Carboplatin. Cell growth of the two c-kit positive cancer cell lines OVCAR 3 and SK-OV 3 seems to be slightly stronger inhibited by Imatinib compared to proliferation rates reached by combination therapy in c-kit negative cell lines. When concentrations of 5 and 8 μM Imatinib are introduced in the combination treatments containing Carboplatin, cell proliferation can even further be decreased. This effect did not occur in c-kit-negative cell lines.
Synergistic effects of Imatinib and Paclitaxel were also described in a mouse ovarian cancer model. The authors of this study concluded that Imatinib lead to PDGF-receptor-mediated inhibition of endothelial cell proliferation in the interstitium and was responsible for anti-angiogenic effects and the following reduction of tumor progression (Apte et al. 2004). Our experimental design did not include the examination of an extracellular effect induced by Imatinib, which could be mediated through PDGF-β receptor.
Additionally to the above mentioned effects, recent publications have discussed the impact of Imatinib on the Akt pathway. Akt is a pro-apoptotic protein, which has a central function in signaling pathways induced by abl, c-kit as well as PDGF-α and PDGF-β receptors (Thompson and Thompson 2004). As shown previously by other groups, Imatinib down-regulates the signaling through the Akt-dependent pathway in concentrations of 1 μM and completely inhibits the Akt-dependent pathway in concentration of 5 μM in ovarian cancer cells (Matei et al. 2004; Shaw and Vanderhyden 2007). Although not further examined, this observation could explain our findings of synergistic effects of Imatinib in combinations with Paclitaxel and Carboplatin. Mechanisms leading to Paclitaxel and Carboplatin resistance in ovarian cancer are often associated with high expression of Akt (Hu et al. 2002; Page et al. 2000; Yuan et al. 2003). Re-sensitizing of ovarian cancer cells to these drugs was observed, when the Akt pathway was inhibited upstream by a phosphatidyl- inositol 3-kinase inhibitor (Shaw and Vanderhyden 2007; Arimoto-Ishida et al. 2004; Mabuchi et al. 2002).
As a further way of action Imatinib can reduce the increased hydrostatic pressure in the tumor-associated matrix. Mediated through a PDGF-β-receptor inhibition, Imatinib facilitates the transport of substances and especially drugs from the capillary vessels into tumor tissue (Pietras et al. 2001).
We conclude that Imatinib as a single agent has direct inhibitory effects on ovarian cancer cells in low, clinically relevant concentrations (<5 μM). Single drug use of Imatinib in heavily pre-treated ovarian cancer patients did not result in clinical responses (Alberts et al. 2007; Coleman et al. 2006).
Until today, Imatinib was considered to reduce tumor growth mainly by anti-angiogenic effects and a PDGFR-β-mediated effect on the extracellular matrix. Therefore, it is of great importance, that Imatinib (doses low as 1–2 μM) was able to show significant and synergistic reduction in ovarian cancer cell proliferation if added to Carboplatin and or Paclitaxel.
Imatinib may therefore offer a helpful addition to the established chemotherapy regimens in ovarian cancer therapy. Imatinib could help enhancing the efficacy of standard chemotherapy and reduce unwanted side effects of these cytotoxic therapies. These in vitro results have to be confirmed in clinical trials.
Acknowledgment
We are thankful for an unrestricted research grant from Novartis Pharmaceutical, Germany supporting this study.
Footnotes
C. Mundhenke and M. T. Weigel contributed equally to this work.
References
- Alberts DS, Hallum AV III, Stratton-Custis M, Garcia DJ, Gleason-Guzman M, Salmon SE, Santabarbara P, Niesor EJ, Floret S, Bentzen CL (2001) Phase I pharmacokinetic trial and correlative in vitro phase II tumor kinetic study of Apomine (SR-45023A), a novel oral biphosphonate anticancer drug. Clin Cancer Res 7:1246–1250 [PubMed] [Google Scholar]
- Alberts DS, Liu PY, Wilczynski SP, Jang A, Moon J, Ward JH, Beck JT, Clouser M, Markman M (2007) Phase II trial of imatinib mesylate in recurrent, biomarker positive, ovarian cancer (Southwest Oncology Group Protocol S0211). Int J Gynecol Cancer 17:784–788 [DOI] [PubMed] [Google Scholar]
- Apte SM, Bucana CD, Killion JJ, Gershenson DM, Fidler IJ (2004a) Expression of platelet-derived growth factor and activated receptor in clinical specimens of epithelial ovarian cancer and ovarian carcinoma cell lines. Gynecol Oncol 93:78–86 [DOI] [PubMed] [Google Scholar]
- Apte SM, Fan D, Killion JJ, Fidler IJ (2004b) Targeting the platelet-derived growth factor receptor in antivascular therapy for human ovarian carcinoma. Clin Cancer Res 10:897–908 [DOI] [PubMed] [Google Scholar]
- Arimoto-Ishida E, Ohmichi M, Mabuchi S, Takahashi T, Ohshima C, Hayakawa J, Kimura A, Takahashi K, Nishio Y, Sakata M, Kurachi H, Tasaka K, Murata Y (2004) Inhibition of phosphorylation of a forkhead transcription factor sensitizes human ovarian cancer cells to cisplatin. Endocrinology 145:2014–2022 [DOI] [PubMed] [Google Scholar]
- Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ (2002) Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol 20:1248–1259 [DOI] [PubMed] [Google Scholar]
- Buchdunger E, O’Reilly T, Wood J (2002) Pharmacology of imatinib (STI571). Eur J Cancer 38(Suppl 5):S28–S36 [DOI] [PubMed] [Google Scholar]
- Coleman RL, Broaddus RR, Bodurka DC, Wolf JK, Burke TW, Kavanagh JJ, Levenback CF, Gershenson DM (2006) Phase II trial of imatinib mesylate in patients with recurrent platinum- and taxane-resistant epithelial ovarian and primary peritoneal cancers. Gynecol Oncol 101:126–131 [DOI] [PubMed] [Google Scholar]
- Dunigan DD, Waters SB, Owen TC (1995) Aqueous soluble tetrazolium/formazan MTS as an indicator of NADH- and NADPH-dependent dehydrogenase activity. Biotechniques 19:640–649 [PubMed] [Google Scholar]
- Faivre S, Raymond E, Casiraghi O, Temam S, Berthaud P (2005) Imatinib mesylate can induce objective response in progressing, highly expressing KIT adenoid cystic carcinoma of the salivary glands. J Clin Oncol 23:6271–6273; author reply 6273–6274 [DOI] [PubMed] [Google Scholar]
- Frolov A, Chahwan S, Ochs M, Arnoletti JP, Pan ZZ, Favorova O, Fletcher J, von Mehren M, Eisenberg B, Godwin AK (2003) Response markers and the molecular mechanisms of action of Gleevec in gastrointestinal stromal tumors. Mol Cancer Ther 2:699–709 [PubMed] [Google Scholar]
- Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ (2001) Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 19:3312–3322 [DOI] [PubMed] [Google Scholar]
- Hartman M, Piliponsky AM, Temkin V, Levi-Schaffer F (2001) Human peripheral blood eosinophils express stem cell factor. Blood 97:1086–1091 [DOI] [PubMed] [Google Scholar]
- Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB (2002) Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 62:1087–1092 [PubMed] [Google Scholar]
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ (2005) Cancer statistics. CA Cancer J Clin 55:10–30 [DOI] [PubMed] [Google Scholar]
- Kano Y, Akutsu M, Tsunoda S, Mano H, Sato Y, Honma Y, Furukawa Y (2001) In vitro cytotoxic effects of a tyrosine kinase inhibitor STI571 in combination with commonly used antileukemic agents. Blood 97:1999–2007 [DOI] [PubMed] [Google Scholar]
- Krystal GW, Honsawek S, Litz J, Buchdunger E (2000) The selective tyrosine kinase inhibitor STI571 inhibits small cell lung cancer growth. Clin Cancer Res 6:3319–3326 [PubMed] [Google Scholar]
- Kubler HR, van Randenborgh H, Treiber U, Wutzler S, Battistel C, Lehmer A, Wagenpfeil S, Hartung R, Paul R (2005) In vitro cytotoxic effects of imatinib in combination with anticancer drugs in human prostate cancer cell lines. Prostate 63:385–394 [DOI] [PubMed] [Google Scholar]
- Lassus H, Sihto H, Leminen A, Nordling S, Joensuu H, Nupponen NN, Butzow R (2004) Genetic alterations and protein expression of KIT and PDGFRA in serous ovarian carcinoma. Br J Cancer 91:2048–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishio Y, Adachi K, Takahashi K, Arimoto-Ishida E, Nakatsuji Y, Tasaka K, Murata Y (2002) Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem 277:33490–33500 [DOI] [PubMed] [Google Scholar]
- Matei D, Chang DD, Jeng MH (2004) Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor alpha and Akt inactivation. Clin Cancer Res 10:681–690 [DOI] [PubMed] [Google Scholar]
- Matei D, Emerson RE, Lai YC, Baldridge LA, Rao J, Yiannoutsos C, Donner DD (2006) Autocrine activation of PDGFRalpha promotes the progression of ovarian cancer. Oncogene 25:2060–2069 [DOI] [PubMed] [Google Scholar]
- Neijt JP, Engelholm SA, Tuxen MK, Sorensen PG, Hansen M, Sessa C, de Swart CA, Hirsch FR, Lund B, van Houwelingen HC (2000) Exploratory phase III study of paclitaxel and cisplatin versus paclitaxel and carboplatin in advanced ovarian cancer. J Clin Oncol 18:3084–3092 [DOI] [PubMed] [Google Scholar]
- Page C, Lin HJ, Jin Y, Castle VP, Nunez G, Huang M, Lin J (2000) Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res 20:407–416 [PubMed] [Google Scholar]
- Palumbo C, van Roozendaal K, Gillis AJ, van Gurp RH, de Munnik H, Oosterhuis JW, van Zoelen EJ, Looijenga LH (2002) Expression of the PDGF alpha-receptor 1.5 kb transcript, OCT-4, and c-KIT in human normal and malignant tissues. Implications for the early diagnosis of testicular germ cell tumours and for our understanding of regulatory mechanisms. J Pathol 196:467–477 [DOI] [PubMed] [Google Scholar]
- Pietras K, Ostman A, Sjoquist M, Buchdunger E, Reed RK, Heldin CH, Rubin K (2001) Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res 61:2929–2934 [PubMed] [Google Scholar]
- Remmele W, Stegner HE (1987) Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 8:138–140 [PubMed] [Google Scholar]
- Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, Fenaux P, Preudhomme C (2002) Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood 100:1014–1018 [DOI] [PubMed] [Google Scholar]
- Satomura K, Derubeis AR, Fedarko NS, Ibaraki-O’Connor K, Kuznetsov SA, Rowe DW, Young MF, Gehron Robey P (1998) Receptor tyrosine kinase expression in human bone marrow stromal cells. J Cell Physiol 177:426–438 [DOI] [PubMed] [Google Scholar]
- Shaw TJ, Vanderhyden BC (2007) AKT mediates the pro-survival effects of KIT in ovarian cancer cells and is a determinant of sensitivity to imatinib mesylate. Gynecol Oncol 105:122–131 [DOI] [PubMed] [Google Scholar]
- Thompson JE, Thompson CB (2004) Putting the rap on Akt. J Clin Oncol 22:4217–4226 [DOI] [PubMed] [Google Scholar]
- Westphal JR, Van’t Hullenaar R, Peek R, Willems RW, Crickard K, Crickard U, Askaa J, Clemmensen I, Ruiter DJ, De Waal RM (2000) Angiogenic balance in human melanoma: expression of VEGF, bFGF, IL-8, PDGF and angiostatin in relation to vascular density of xenografts in vivo. Int J Cancer 86:768–776 [DOI] [PubMed] [Google Scholar]
- Yuan ZQ, Feldman RI, Sussman GE, Coppola D, Nicosia SV, Cheng JQ (2003) AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: implication of AKT2 in chemoresistance. J Biol Chem 278:23432–23440 [DOI] [PubMed] [Google Scholar]