

Abstract
Nonalcoholic fatty liver disease (NAFLD), including its more severe manifestation, nonalcoholic steatohepatitis (NASH), has a global prevalence of 20–25% and is a major public health problem. Its incidence is increasing in parallel to the rise in obesity, diabetes and metabolic syndrome. Progression from NASH to NASH-related hepatocellular carcinoma (HCC) (~2% of cases per year) is influenced by many factors, including the tissue and immune microenvironment, germline mutations in PNPLA3, and the microbiome. NASH-HCC has unique molecular and immune traits compared with other aetiologies of HCC and is equally prevalent in men and women. Comorbidities associated with NASH, such as obesity and diabetes mellitus, can prevent the implementation of potentially curative therapies in certain patients; nonetheless, outcomes are similar in patients who receive treatment. NASH-HCC at the early to intermediate stages is managed with surgery and locoregional therapies, whereas advanced HCC is treated with systemic therapies, including anti-angiogenic therapies and immune-checkpoint inhibitors. In this Review, we present the latest knowledge of the pathogenic mechanisms and clinical management of NASH-HCC. We discuss data highlighting the controversy over varying responses to immune-checkpoint inhibitors according to underlying aetiology and suggest that the future of NASH-HCC management lies in improved surveillance, targeted combination therapies to overcome immune evasion, and identifying biomarkers to recognize treatment responders.
Introduction
Primary liver cancer is the third-leading cause of cancer-related mortality worldwide, with an incidence approaching 1 million cases annually1. Hepatocellular carcinoma (HCC), which accounts for ~90% of primary liver cancers, typically arises in a background of chronic liver disease caused by hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, alcohol-associated liver disease or, increasingly, nonalcoholic fatty liver disease (NAFLD)2,3. NAFLD is defined by the presence of fat comprising at least 5% of the weight of the liver (also known as steatosis), as established either by histology or liver imaging, with no secondary causes to explain the abnormality4. The global prevalence of NAFLD is approximately 20–25%3,5,6. NAFLD is comprised of two main subgroups: those with hepatic fat without evidence of hepatocellular injury (nonalcoholic fatty liver) or those with nonalcoholic steatohepatitis (NASH). Nonalcoholic fatty liver is typically defined by >5% hepatic steatosis without other features, whereas NASH is characterized by the presence of >5% hepatic steatosis plus evidence of hepatocellular injury (‘ballooning’) and inflammation, with or without fibrosis. NASH can progress to cirrhosis or liver failure and confers a rising risk of HCC as the disease progresses5. NASH is observed in 20% of patients with NAFLD and is a chronic liver condition linked to the epidemics of obesity, diabetes and metabolic syndrome7–10. Patients with NASH-related cirrhosis have an annual HCC incidence of approximately 2%5,10,11, and NASH is the leading cause of HCC in patients without cirrhosis12. Overall, NASH-HCC accounts for 20% of HCC in the Western world and is estimated to become the predominant aetiology of HCC globally by 2030 (ref. 11). NASH-HCC development is associated with specific and unique mutational, immunological and microenvironmental features, including increased prevalence of TP53 and ACVR2A mutations in hepatocytes13, subpopulations of dysfunctional immune cells including CD8+PD1+ T cells14, and single-nucleotide polymorphisms (SNPs) in genes such as PNPLA3 (ref. 15). Although the majority of cases of HCC in NASH arise once cirrhosis is present, ~30–40% of tumours develop in patients with advanced fibrosis but without cirrhosis, which has implications for surveillance and early detection16–19. This is in contrast to HBV or HCV infection, in which >80–90% of tumours arise only after cirrhosis is present17. The increased propensity for HCC development without cirrhosis in NASH compared with other aetiologies underscores the unique metabolic milieu and the likely contribution of extrahepatic drivers of cancer associated with metabolic syndrome. NASH-HCC is currently treated using the same approach as other aetiologies of HCC, including transplantation, resection or locoregional therapies for early-stage or intermediate-stage disease20. A NASH aetiology is the fastest growing HCC-related indication for liver transplantation in the USA21; however, systemic therapy is applied in approximately 50% of patients during the course of their disease and includes several combinational therapies (atezolizumab plus bevacizumab or durvalumab plus tremelimumab) and single-agent tyrosine kinase inhibitors (sorafenib, lenvatinib, regorafenib and cabozantinib) or monoclonal anti bodies (ramucirumab)2,22. Whether immune-based agents are equally efficacious in non-viral versus viral-related HCC is unclear14,23,24.
In this Review, we comprehensively discuss the latest knowledge regarding the pathogenic mechanisms and clinical management of NASH-HCC. We describe the pathogenesis of progression from NASH to NASH-HCC, including the contribution of fibrosis, the immune microenvironment, microbiome and genomic features. Next, we focus on surveillance strategies and advances in the clinical management of patients with NASH-HCC, including the tailored use and results of surgical, locoregional and systemic therapies. Finally, we frame future prospects and strategies to overcome the unmet challenges in NASH-HCC.
Epidemiology
Global trends
Globally, ~10% (range 1–38%) of all HCC cases are attributed to NAFLD5, with the higher estimates (>20%) reported in studies from the USA, UK, India, Germany and the Middle East25–29, and lower estimates (1–2%) from China and Japan30 (Fig. 1). Using mathematical models to predict NAFLD-HCC incidence on the basis of historical and projected changes in rates of diabetes and obesity, the incidence of NAFLD-HCC is predicted to increase between 2016 and 2030 by 47% in Japan, 82% in China, 88% in the UK, 117% in France and 130% in the USA7,8. Compared with patients with HCC related to viral hepatitis (HBV or HCV) or alcohol-associated liver disease, those with NAFLD-HCC have a lower male-to-female ratio (1.2:1), are 5–10 years older (mean age 73 years old)5, and are more likely to have metabolic and cardiovascular comorbidities such as type 2 diabetes mellitus (T2DM) and chronic vascular disease8. Although the incidence of NAFLD-related HCC is lower than that of active viral hepatitis-related HCC5,31, the rising prevalence of NAFLD coupled with improvements in treatments for viral hepatitis is highly likely to increase both the proportion and rate of HCC attributed to NAFLD32.
Fig. 1 |. The global incidence of NASH-related HCC.
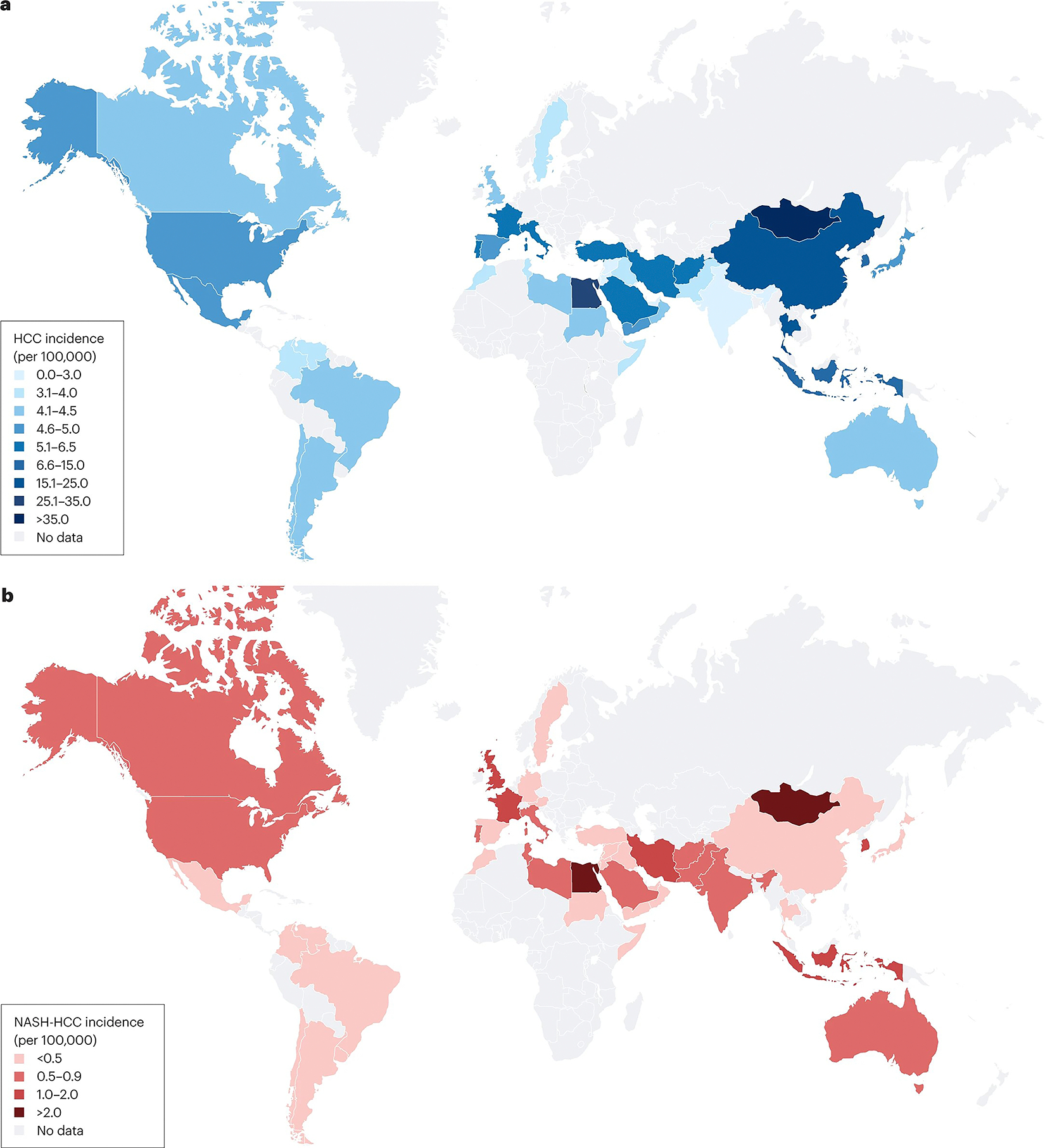
a, The estimated global incidence of hepatocellular carcinoma (HCC) per 100,000 people1. b, The estimated global incidence of nonalcoholic steatohepatitis (NASH)-related HCC per 100,000 people1,5,30,209–211. Please note that Fig. 1 is derived from data from studies with varying methodologies.
Risk factors
A study of ~300,000 patients with NAFLD published in 2018 reported an HCC incidence of 0.21 per 1,000 person-years in persons with NAFLD, which was sevenfold higher than in control individuals without liver disease (absence of viral hepatitis and normal ALT)18. The main risk factor for NAFLD-HCC is cirrhosis. Incidence rates of HCC in NASH cirrhosis cohorts are approximately 2% per year (range 0.3–4.7% per year)5,31,33. The wide variation across studies is explained in part by differing age, metabolic profiles and severity of decompensation. As previously mentioned, HCC also develops in NASH in the absence of cirrhosis in a sizable percentage of patients16,17. However, overall HCC incidence in non-cirrhotic NASH is low (0.01–0.13% per year)5, and even lower in the general NAFLD population16. Therefore, clinical determination of cirrhosis status is the main risk stratifier in NAFLD34. Other risk factors for HCC in NAFLD include older age, Hispanic ethnicity in the USA, presence of features of metabolic dysfunction (particularly T2DM and high BMI), dyslipidaemia, hypertension and genetic variability18,34,35. In European and US cohort studies of individuals with NAFLD, T2DM was the strongest independent metabolic risk factor for the development of HCC18,36. Although overweight and obesity have been associated with a modest (48–83%) increase in the risk of HCC irrespective of aetiology37, NAFLD status was not examined in most of these studies. In a large cohort study (n = 296,707), patients with NAFLD with obesity had a non-significant increase (P = 0.06) in the risk of HCC development, but the risk for developing HCC was significantly elevated 2.6-fold when obesity occurred alongside diabetes, hypertension and hyperlipidaemia18. In addition, smoking has been associated with an increased risk of HCC in general38 but has not been examined specifically in NAFLD. HCC risk calculators, including Veterans Affairs score39 and NAFLD fibrosis score40, have incorporated some of these clinical risk factors, with only modest performance for HCC risk stratification in NAFLD.
Pathogenesis
Pathogenesis from NAFLD to HCC
Role of fibrosis.
NASH is a multifactorial disease with many dysregulated pathways that contribute to injury and fibrosis. To date, we lack a temporal hierarchy of causality, which has confounded efforts to identify key therapeutic vulnerabilities. As ~80% of patients with NAFLD do not have NASH, efforts have been focused on defining those elements that distinguish patients with inflammation, cell injury and fibrosis (NASH) from those who have steatosis alone. The concept of ‘lipotoxicity’, or the presence of hepatocellular injury owing to dysregulation of fat metabolism, remains a critical concept in uncovering drivers that precipitate NASH41. Lipotoxicity is the result of multiple inputs, including increased delivery of fatty acids and exosomes from peripheral fat to the liver, enhanced insulin resistance, and inflammatory signals derived from abnormal adipose42,43. The cellular consequences of enhanced lipotoxicity-related injury in hepatocytes are endoplasmic reticulum stress, oxidative stress, activation of the inflammasome and cell death44,45. From the perspective of HCC, these hepatocellular damage responses contribute to pre-malignant events, including oxidant-induced DNA damage and accumulation of mutations in master metabolism genes such as FOXO1, CIDEB and GPAM46,47 (Fig. 2). Interestingly, these mutations can result from selective pressure to protect hepatocytes from the effects of lipotoxicity, yet they also enhance the risk of malignancy47.
Fig. 2 |. NASH-HCC pathogenesis and progression.
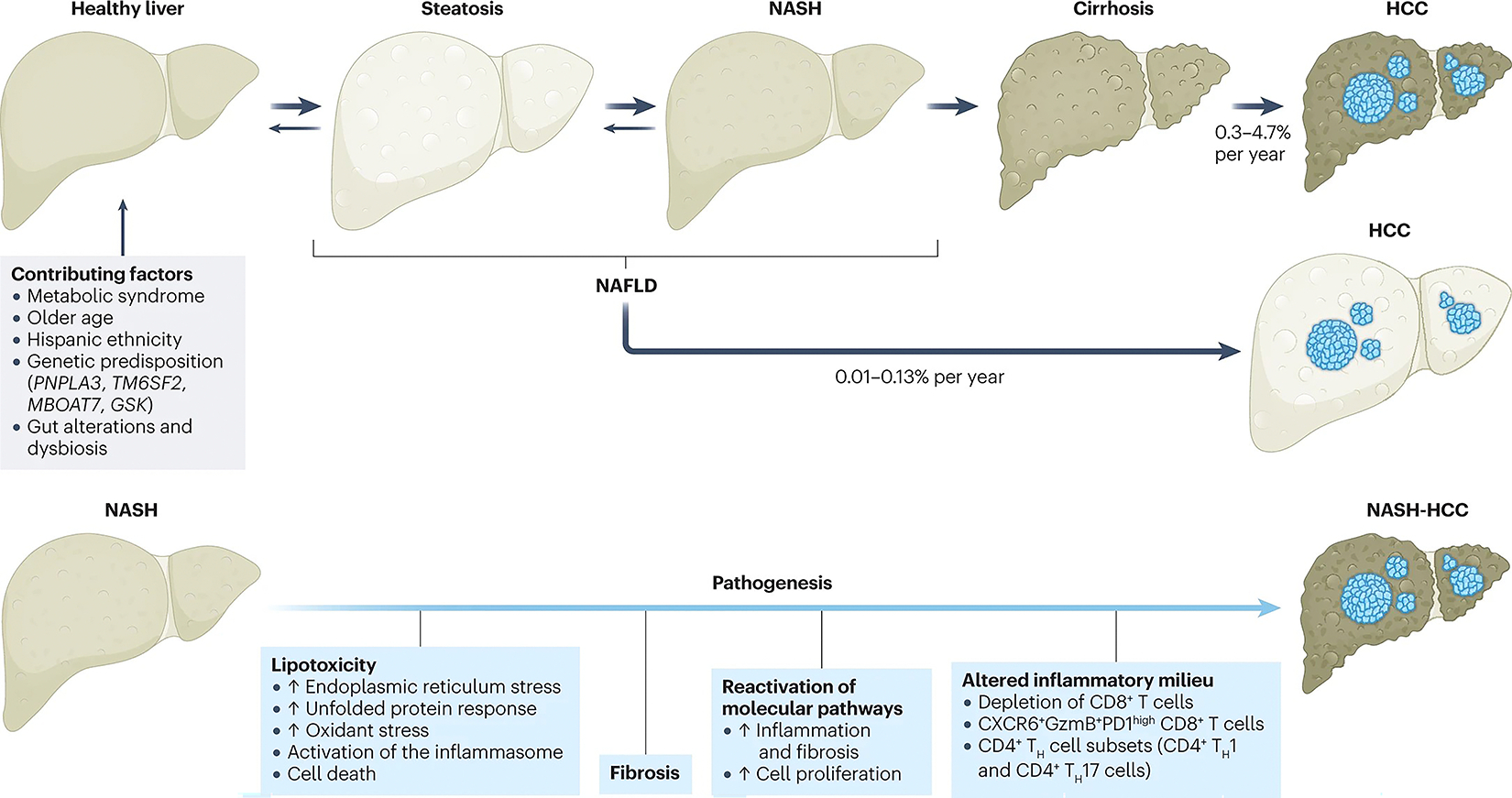
Contributing factors and mechanisms behind the pathogenesis from a healthy liver to nonalcoholic fatty liver disease (NAFLD) and subsequent hepatocellular carcinoma (HCC)5,14,31,33,44,75,91. NASH, nonalcoholic steatohepatitis; TH, T helper.
In an effort to restore hepatocellular mass in the setting of NASH, developmental pathways, such as YAP–TAZ, Notch and Hedgehog signalling48,49, are reactivated in hepatocytes, the consequences of which can include cell proliferation, inflammation and cancer50. Interestingly, progressive loss of hepatocyte proliferation and reduced regenerative capacity are hallmarks of advanced NASH, and these dysregulated hepatocytes can also directly provoke further inflammation and fibrosis51. Hepatocellular dysregulation and damage contribute to an altered inflammatory milieu that promotes chronic inflammation. All immune cell types are affected (see section ‘Role of the immune cells’), including T cells and B cells, dendritic cells, macrophages, plasma cells, natural killer (NK) cells, and NK T cells52,53.
The stage of hepatic fibrosis in NASH is the key determinant of clinical outcomes because it may lead to cirrhosis and liver failure54, and fibrosis also contributes to a milieu that promotes cancer. The activation, or transdifferentiation, of resident hepatic stellate cells (HSCs; liver-specific pericytes) into fibrogenic, proliferating myofibroblasts results in progressive accumulation of extracellular matrix (or scar formation). Powerful single-cell sequencing technologies have uncovered substantial heterogeneity of HSCs in NASH, although the functional implications of this cellular diversity are largely unknown55,56. Indeed, targeting subsets of activated HSCs, for example, those that are senescent, might have therapeutic benefit in improving fibrosis based on in vivo studies in mouse models; however, the effect on HCC is not known57.
Angiogenesis also has a role in NASH and possibly in NASH-HCC. Earlier studies identified increased CD34 expression in new blood vessels of both humans and rodents, which reflects vascularization58. Indeed, vascular endothelial growth factor (VEGF), which is the best-understood angiogenic signal, is increased in experimental models of NASH, and its antagonism reduces vascularization, inflammation and steatosis59. More recently, signalling by the TIE1 and TIE2 angiopoietin receptors has been implicated in NASH60. Dysregulation of liver sinusoidal endothelial cells also occurs in NASH as a result of altered angiogenic signalling as demonstrated in mouse models61.
Fibrosis is a risk factor for cancer in the liver and other organs (for example, pancreas and lung)62, although it is unclear which of these fibrotic mechanisms are unique to NASH compared with other aetiologies of chronic liver disease. The precise mechanisms by which NASH-HCC develops in the absence of cirrhosis are unknown, but they might relate to fibrosis status. Stiffness of the liver conferred by the accumulation of extracellular matrix can promote tumour cell emergence or enhance tumour cell growth (reviewed previously63). The accumulating scar matrix is also a rich repository of bound growth factors, some of which, such as insulin-like growth factors, might enhance survival of pre-neoplastic hepatocytes to promote tumour initiation or perpetuation. At the same time, HSCs are immunoregulatory and are thought to contribute to the unique immune tolerance of the liver64,65, which could influence its resistance to checkpoint blockade.
As therapies that reduce hepatic fibrosis emerge, their effect on NASH-HCC will be anxiously awaited. It is not known whether a liver with advanced fibrosis is ‘imprinted’ with a risk of cancer even if fibrosis later regresses. On the basis of studies of patients who have been successfully treated for HBV or HCV infection, one might anticipate a reduction but not complete attenuation of HCC risk66–68, especially in patients with cirrhosis or advanced fibrosis.
Currently, there are no approved therapies for NASH; therefore, the effect of medical therapy on NASH-HCC cannot be assessed. Nonetheless, a reduced risk of HCC has been reported in patients with NASH after bariatric surgery69, which is a hopeful indication that medical therapy for NASH might also reduce HCC incidence. A detailed review of investigational drugs for NASH is beyond the scope of this article but is the subject of several excellent reviews70–73.
Role of the immune system.
The immune system has a critical role in the context of both NAFLD and HCC, for which different immunogenomic classes are described74. Lobular inflammatory responses characterize NASH and are considered the driving force in disease progression to fibrosis, cirrhosis or HCC75. Studies have demonstrated an important role for innate and adaptive immune mechanisms promoting hepatic inflammation in NASH. Resident Kupffer cells as well as the recruitment of leukocytes, such as neutrophils, monocytes, NK cells and NK T cells, promote inflammatory immune responses to NASH by releasing cytokines, chemokines, eicosanoids, nitric oxide and reactive oxygen species44,75. Similarly, CD4+ T helper cell subsets (specifically, CD4+ T helper 1 (TH1) and CD4+ TH17 cells) were increased in NASH livers in mice76. Nonetheless, T cells also have an anti-tumorigenic effect. Depletion of CD8+ T cells greatly enhanced tumour growth in a murine model of NASH-driven HCC77, whereas depletion of CD4+ T cells promoted tumour growth78,79. This depletion of CD4+ cells greatly limited the efficacy of immune-based therapeutic approaches in murine HCC models80, indicating that underlying liver disease might affect response to immunotherapy outcomes. Several preclinical models have been instrumental in elucidating the role of fibrosis and the immune response in the development of NASH-HCC (Box 1).
Box 1. Murine models of NASH-related HCC.
Experimental models are essential to elucidate the pathophysiological mechanisms of nonalcoholic steatohepatitis (NASH)-related hepatocellular carcinoma (HCC) and identify new therapeutic targets. Briefly, murine models of NASH-HCC can be categorized into four types:
Diet-induced models: these models closely resemble the natural progression of HCC development on a background of NASH aetiology. They include choline deficiency with a high-fat diet (25% HCC incidence at 48 weeks)76, choline-deficient and amino acid-defined diet alone (~20% HCC incidence at 84 weeks) or in combination with a high-fat diet (~27% HCC incidence at 60 weeks)212, Western diet combined with sugar water (89% HCC incidence at 32 weeks)213 and the American lifestyle-induced obesity syndrome diet (~85% HCC incidence at 48 weeks)214. These models are able to closely recapitulate the histological, physiological and metabolic features of human NASH. However, tumour penetrance is low, with long latency periods.
Chemical plus diet-induced models: these models have the advantage of rapid disease progression and high rates of HCC formation. Most commonly, streptozotocin (100% HCC incidence at 20 weeks)215, diethylnitrosamine (100% HCC incidence at 32–36 weeks)216,217 and carbon tetrachloride (100% HCC incidence at 24 weeks)218 are used in combination with modified dietary regimes (such as those detailed above). However, some models fail to recapitulate key characteristics of metabolic syndrome, including obesity and impaired glucose tolerance.
Genetic plus diet-induced models: these models involve inducing genetic alterations that lead to substantial hepatic lipid accumulation and, in combination with a modified diet, lead to inflammation and fibrosis and the development of NASH-HCC. For example, MUP-uPA mice fed a high-fat diet (50% HCC incidence at 40 weeks)219 or URI and IL17 transgenic mice fed a high-fat diet (40% HCC incidence at 65 weeks)220 develop HCC on a NASH background. Compared to this diet alone, these combined models exhibit a more severe disease phenotype.
Implantation models: these models involve orthotopically implanting hepatoma cells into a liver in which NASH has been induced by a dietary regime85. They stand out due to their reliable and rapid (100% HCC incidence at ~14 weeks) time frame for NASH-HCC formation but are limited by the fact that tumour development is not autochthonous.
A detailed review of nonalcoholic fatty liver disease/NASH and HCC animal models is beyond the scope of this Review and can be found elsewhere3,221.
Disruption of the immune system in NASH, and ultimately in NASH-HCC, has been associated with response to immunotherapies. In the context of NASH, both adaptive and innate immune cells have been reported to shape the liver microenvironment towards NASH-HCC transition, including CD4+ T cells78, metabolically activated CD8+ T cells76, platelets81 or dendritic cells (for example, XCR1+cDC1 cells)82. Moreover, neutrophils populate the liver in several stages of NASH and might critically affect the transition from fatty liver to steatohepatitis as demonstrated in mouse studies83. For example, the production of neutrophil extracellular traps during tissue injury promotes an immunosuppressive liver microenvironment through PDL1 signalling and is responsible for CD8+ T cell exhaustion in NASH-induced HCC, which also possibly affects the response to immunotherapy84,85. It has become apparent that different NASH-inducing diets, similar to high-fat diets linked to NASH in humans, might alter the function of these cells and drive the NASH-HCC transition3,86. Antigen-specific T cells can be present in mice with NALFD but their function is impaired by macrophages87. Tumour infiltration of CCR2+ and CX3CR1+ macrophages has been linked to non-response to immune-checkpoint inhibition in patients with advanced HCC, whereas pro-inflammatory PDL1-expressing CXCL10+ macrophages have been reported to drive response on the basis of single-cell RNA sequencing data from pretreatment biopsy samples88. A study published in 2022 described an impaired ‘fitness’ of T cells in NAFLD (that is, loss of their effector functions), potentially contributing to the lack of response to immune-checkpoint inhibitor (ICI) therapy89. Systemic metformin treatment rescued the effect of ICI therapy in mice with NAFLD. Neutrophil reprogramming using a CXCR2 antagonist is an alternative approach to overcome NAFLD-dependent impairment of ICI therapy in HCC by increasing intratumoural dendritic cell activation and CD8+ T cell numbers in mouse models of NASH-HCC85. Together, these studies indicate that various mechanisms to overcome NASH-dependent impairment of ICI therapy might exist. Future studies on other pivotal immune cells in the tumour microenvironment, such as dendritic cells and myeloid-derived suppressor cells90, as well as on their metabolic state are needed to better understand the complex metabolic and cellular interactions in the NAFLD-specific tumour microenvironment.
In two intriguing studies in mice and humans, hepatic abundance of CD8+PD1+ T cells increased with the progression of NASH. These cells recapitulate an auto-aggressive state, in which liver-resident CD8+PD1+CD103+ T cells, although exhausted (TOX+; TIGIT+), display an activated phenotype expressing high levels of cytokines such as tumour necrosis factor (TNF), CCL2, IL-10 or granzyme B14,91. In the context of NASH-HCC immunotherapy in mouse models, CD8+PD1+ cells remained virtually unaltered in their transcriptomes and proteomes and were augmented in size over time, which further contributed to increased hepatic inflammation, hepatocyte cell death and oncogenic signalling14. Thus, instead of eradicating HCC, these cells became dysfunctional in tumour surveillance and even pro-tumorigenic. In NASH-HCC mouse models, this led to non-responsiveness towards ICI in a therapeutic setting as well as to a worsening of HCC development in a prophylactic setting14. Similar CD8+ T cell phenotypes have been described in the context of human NASH-HCC, including in the setting of ICI14. Thus, it is possible that, in human NASH-HCC, peritumoural and intratumoural CD8+PD1+ T cells might be a positive predictor for treatment success or resistance to ICIs. Linking the immune microenvironment composition to outcomes could help determine the most effective therapies in future studies.
Role of the microbiome.
The gut microbiome is an important determinant of altered hepatic responses in NASH, in part via its effects on hepatic bile acid metabolism92 as well as by the translocation of gut-derived signals through a progressively leaky gut epithelium93. In all disease stages of NASH, the gut–liver axis is ‘activated’ and the interplay between liver damage, liver regeneration and enhanced gut permeability can amplify inflammatory or pro-fibrogenic and pro-carcinogenic pathways44. As a result of this permeability defect, direct (for example, bacteria per se) as well as indirect (for example, bacterial metabolites) interactions between the mucosal microbiome and liver are initiated, affecting liver metabolism and promoting NASH and HCC.
Several studies have implicated the gut microbiome as a major trigger of NAFLD (previously reviewed94). The mucosal microbiome drives liver steatosis by increasing energy harvest, monosaccharide absorption and aberrant acetate production in the intestine94. Moreover, the dysbiotic leaky gut enables transit of intestinal mucosa-derived pathogen-associated molecular patterns and danger-associated molecular patterns into the liver, activating immune cells and hepatic Toll-like receptors (TLRs) to induce pro-inflammatory and fibrotic pathways43. This defect in gut permeability can also be initiated by bacteria themselves: a microbiota-driven disruption of the gut vascular barrier has been described as a prerequisite of NASH based on a mouse model of NASH95. Notably, it is conceivable that innate and adaptive inflammatory cells in the gut might contribute to NASH by transmigrating from the gut mucosa to the liver, thereby contributing to the translocation of bacteria5. Owing to the common co-existence of NASH, metabolic syndrome and altered dietary habits, the effects of diet-induced metabolic changes can only be scarcely dissected from effects caused by an altered microbiome under the same conditions. However, several bacterial species in humans were found to be associated with NAFLD (for example, Proteobacteria, Enterobacteriaceae and Escherichia species)96. Bacteroides have also been found to be elevated in patients with NASH compared with matched healthy individuals97. Treating mice with NAFLD or NASH with non-absorbable antibiotics, such as rifaximin98, reinforces a potential role for the gut microbiome in disease pathogenesis as treatment with antibiotics might improve liver function as shown in patients with NASH99,100.
Molecular alterations in NASH and NASH-HCC
Genetic risk of NAFLD, NASH and NASH-HCC development. Several SNPs associated with abnormal lipid handling in hepatocytes have been linked to NASH risk and progression to HCC15,101–107 (Table 1). The most widely reported SNP is in the gene PNPLA3 (encoding patatin-like phospholipase domain-containing protein 3); the SNP rs738409 impairs the degradation of lipid droplets in hepatocytes108. As a result, this PNPLA3 variant is associated with reduced triglyceride lipolysis, thereby promoting hepatic steatosis109 and leading to a more than 2-fold increased risk of NASH (compared with healthy control individuals)102 and a 2.2-fold increased risk of NAFLD-related HCC (compared with patients with NAFLD without the SNP variant)15. Second, an SNP in TM6SF2 (encoding transmembrane 6 superfamily member 2), which regulates liver fat metabolism, increases hepatic triglyceride content110,111. The TM6SF2 SNP rs58542926 has been linked to a 1.6-fold increased risk of NASH (compared with healthy control individuals)102 and a 1.9-fold increased risk of NAFLD-related HCC (compared with patients with NAFLD without the SNP variant)103. Third, an SNP near MBOAT7 is associated with increased hepatic triglyceride content112 and is twice as common in patients with NAFLD-HCC than in those with NAFLD alone104. Additionally, a loss-of-function variant in GCKR (encoding glucokinase regulator) results in increased de novo lipogenesis by inducing glycolysis, insulin resistance and inflammation in people with metabolic syndrome113. This GCKR SNP has been linked to a 1.5-fold increased risk of NASH (compared with healthy control individuals)106 and a 1.8-fold increased risk of NASH-HCC (compared with the general population)107. A polygenic risk score combining these 4 SNPs has been proposed for risk stratification of HCC development in patients of European ancestry with NASH cirrhosis and was shown to predict HCC more robustly than the individual SNPs (P < 10−13)114.
Table 1.
SNPs associated with NAFLD/NASH-related HCC
| Gene | Variant | Normal protein function and variant mechanism | NAFLD/NASH OR (95% CI) | NAFLD/NASH-related HCC OR (95% CI) |
|---|---|---|---|---|
| PNPLA3 (refs. 15,102,108,109) | rs738409 c.444C>G p.I148M |
WT: regulates lipid droplets in hepatocytes Variant: protein ubiquitination is impaired, resulting in the accumulation of hepatic lipid droplets that promote hepatic steatosis |
OR for the G allele in patients with NASH 2.05 (1.86–2.27) versus healthy controls | OR for the G allele in patients with NAFLD-related HCC 2.26 (1.23–4.14) versus patients with NAFLD alone |
| TM6SF2 (refs. 102,103,110,111) | rs58542926 c.449C>T p.E167K |
WT: regulates liver fat metabolism Variant: associated with upregulation of cholesterol and fatty acid biosynthesis pathways |
OR for the T allele in patients with NASH 1.61 (1.34–1.92) versus healthy controls | OR for the T allele in patients with NAFLD-related HCC 1.92 (1.31–2.81) versus patients with NAFLD alone |
| MBOAT7 (refs. 104,105,112) | rs641738 C>T p.G17E |
WT: participates in the regulation of triglyceride metabolism Variant: associated with increased hepatic triglyceride content and steatosis development |
OR for the T allele 1.18 (1.00–1.40) versus C allele in patients with NASH | OR for the T allele in NAFLD-related HCC 2.10 (1.33–3.31) versus patients with NAFLD alone |
| GCKR 106,107,113 | rs1260326 c.1337C>T p.P446L |
WT: regulates glucokinase activity Variant: modulates insulin resistance and inflammatory markers on interaction with plasma n-3 PUFAs |
OR for the T allele in patients with NASH 1.55 (1.10–2.17) versus healthy controls | OR for the T allele in patients with NASH-related HCC 1.84 (1.23–2.75) versus general population |
Odds ratios (ORs) depict the increased presence of each single-nucleotide polymorphism (SNP) associated with developing nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH) and NAFLD/NASH-related hepatocellular carcinoma (HCC). Data included for ORs are from different cohorts and therefore are not directly comparable across diseases and variants. PUFAs, polyunsaturated fatty acids; WT, wild type.
Molecular traits of NASH-HCC.
Several genetic features are specifically linked to NASH-HCC (Table 2). NASH-HCC is characterized by an enrichment of ACVR2A and TP53 mutations13 and by the proliferative class S1-WNT/TGFβ115,116. A de novo mutational signature (called MutSig-NASH-HCC), present in 25% of patients with NASH-HCC (compared with 2% in other aetiologies), has been reported to be characterized by a higher frequency of C>T and C>A transitions13. Patients with NASH-HCC also have higher levels of hepatic oxidative DNA damage compared with patients with other aetiologies117,118, which has been associated with a lower DNA damage response in experimental models46. Finally, epigen etic events repressing the transcription of genes related to bile and fatty acid metabolism and upregulating proliferative pathways have also been implicated in patients with NASH-HCC and epigenetic reprogramming has been shown to revert hepatocarcinogenesis in experimental models119.
Table 2.
Molecular differences between NASH-HCC and other aetiologies
| NASH-related HCC | HCC not related to NASH |
|---|---|
| Aetiology | |
| Obesity, diabetes, metabolic syndrome | HBV infection, HCV infection, alcohol |
| ~10% of HCCs globally; ~20% in Western countries | ~90% of HCCs globally ~80% in Western countries |
| Male-to-female ratio 1.2:1 | Male-to-female ratio 2–3:1 |
| Molecular alterations | |
| ↑ ACVR2A and TP53 mutations | |
| ↑ MutSig-NASH-HCC | ↑ MutSig24 |
| ↑ WNT/TGFβ proliferation | ↓ WNT/TGFβ proliferation |
| SNPs in PNPLA3, TM6SF2, MBOAT7 and GCKR | SNPs in GTSM1 and GSTT1 |
| Immune | |
| Activation of dysfunctional immune cells, including CD8+PD1+ cells, IgA+ plasma cells, NK cells and TH17 cells, that disrupt tumour immune surveillance | HBV: exhaustion of effector CD8+ T cells and infiltration of immunosuppressive T and B cells HCV: CD8+ T cell exhaustion and immune evasion by interference with MHC-I-dependent antigen presentation ALD: increased M2 macrophages and gMDSC infiltration |
| Microenvironment | |
| ↓ Underlying cirrhosis (60–70%) | ↑ Underlying cirrhosis (>80%) |
| ↑ Oxidative DNA damage, microbial signals generated by gut bacterial metabolism | Necroinflammation from chronic hepatitis viral exposure |
ALD, alcohol-associated liver disease; gMDSC, granulocytic myeloid-derived suppressor cells; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; NASH, nonalcoholic steatohepatitis; NK, natural killer; SNPs, single-nucleotide polymorphisms; TH, T helper. Data retrieved from refs. 3,5,13,14,22,101.
Clinical management
Prevention
Success in preventing HCC development in patients with chronic liver disease has been achieved for viral-related HCC through global vaccination programmes120 as well as through antiviral treatments121 for patients with HBV and direct-acting antiviral therapy for patients with HCV122. Numerous observational, retrospective, population-based studies have suggested a role for metformin123 and, to a lesser extent, statins124, coffee125 and aspirin126 in HCC prevention, regardless of the aetiology of liver disease. Metformin inhibits the mammalian target of rapamycin (mTOR) pathway, which has a role in proliferation via activation of AMP-activated protein kinase (AMPK)127, inhibits angiogenesis, blocks the cell cycle and induces p53-independent apoptosis128. In addition, metformin induces moderate weight loss, reducing the effects of hyperinsulinaemia on the cell cycle and inflammation, and confers an improvement in liver biochemistry and histology in patients with NAFLD129–132. Statins have pleiotropic anticancer properties: they inhibit the oncogenic drivers MYC, AKT, Rho-dependent kinase, and extracellular signal-regulated kinase 1 and 2 (refs. 133–135); they activate hepato-protective AMPK and p38-mitogen-activated protein kinase (MAPK) pathways136,137; and they induce p53-dependent apoptosis138. Coffee contains antioxidants, mainly phenolic compounds (for example, chlorogenic, caffeic, ferulic and coumaric acids), melanoidins and diterpenes (such as cafestol and kahweol), some of which have an inhibitory effect on liver carcinogenesis139–142. Part of the favourable effect of coffee on HCC prevention might be mediated by its prevention of T2DM, a known risk factor for HCC143–145. With a generally favourable benefit-to-harm profile, coffee drinking is encouraged for patients with chronic liver disease by the European Association for the Study of the Liver (EASL)146, but none of the other agents has shown sufficient efficacy to be recommended for HCC prevention2, and most of the studies were not conducted in well-defined NAFLD or NASH populations. A retrospective cohort study (n = 85,963) of patients with NAFLD reported a 22% reduction of HCC risk in patients treated with metformin compared with no treatment147. In patients with NAFLD and T2DM, sustained glycaemic control (HbA1C <7) and a lack of diabetic complications are associated with a reduced risk of HCC147.
For NASH-HCC specifically, prevention strategies recommended by the American Association for the Study of Liver Diseases (AASLD), EASL and the Asia–Pacific Working Party on NAFLD practice guidelines involve combining a hypocaloric or Mediterranean diet with moderate-intensity exercise to sustain weight loss4,148,149. Likewise, physical activity has been shown to correlate with a decreased risk of HCC in a large multinational cohort study150. Finally, although there is currently no direct evidence indicating that weight loss leads to a reduction in NAFLD-related HCC risk151, observational studies indicate a reversal of steatosis and possibly fibrosis in patients with NASH who lose weight152, thereby lending credence to the likelihood of weight loss-related benefit in reducing HCC risk.
Surveillance
Clinical practice guidelines from AASLD and EASL recommend semi-annual HCC surveillance using abdominal ultrasound, with or without α-fetoprotein (AFP) assessment, in patients with cirrhosis of any aetiology146,153. These recommendations are supported by several cohort studies that have demonstrated a consistent association between surveillance and improved early detection, receipt of curative treatment and overall survival154. However, only two studies have specifically examined potential surveillance benefits in patients with NAFLD cirrhosis. Although HCC surveillance was associated with increased detection of early-stage HCC (69.6% versus 30.0%; P = 0.001)155, no association with the applicability of curative treatment (45.5% versus 51.5%; P = 0.72) was found in a second study156.
Data for patients with NAFLD specifically are critical given that this patient population presents several unique challenges to the traditional surveillance paradigm. First, approximately one-third of NAFLD-HCC cases occur in patients in the absence of cirrhosis; these patients are not included in the defined at-risk population warranting surveillance; hence, this represents a clinical unmet need16. Patients with NAFLD are also less likely to have recognized liver disease and cirrhosis at the time of HCC diagnosis, further contributing to lower surveillance utilization in this population157,158. Second, patients with NASH are more prone to inadequate ultrasound visualization and surveillance failure, resulting in more late-stage diagnoses even when surveillance is conducted159,160. These data suggest that the sensitivity of ultrasound-based surveillance in patients with NASH is likely lower than the 63% observed in patients with HCC of other aetiologies161. These data highlight the need for alternative imaging modalities, such as CT or MRI scan10, and blood-based biomarker strategies in this population. Finally, patients with NAFLD often have comorbidities, and therefore higher competing risks of mortality, highlighting the importance of careful patient selection to maintain the benefits of surveillance and early tumour detection162.
Overview of treatment
Management of HCC has improved dramatically since the first Barcelona Clinic Liver Cancer (BCLC) classification in 1999 (ref. 163), when there was a lack of solid evidence for any effective treatment for patients at intermediate and advanced stages. However, we can now count on several therapies for early and intermediate HCC with different levels of evidence and recommendation, and 10 systemic therapies for patients with HCC at intermediate or advanced stages2,146,153,164. Consequently, there has been a noticeable improvement in median survival rates for early HCC (beyond 60 months), intermediate HCC (25–30 months) and advanced HCC (first line ~19–20 months; second-line 10–14 months)2,22,146,153. NAFLD-related HCC is associated with unique features that jeopardize early detection, either because ~30–40% of patients do not have cirrhosis and are therefore not included in surveillance programmes16, or because the performance of ultrasound is suboptimal in the setting of a fatty liver160. As previously mentioned, patients with NASH-HCC have a high prevalence of comorbidities (such as cardiovascular disease) that might preclude the implementation of curative therapies, particularly surgical procedures156,165–167. However, a systematic review found that, despite the increased prevalence of comorbidities and larger tumours at the time of diagnosis, there were no significant differences in curative treatment allocation between patients with NAFLD and those without168. Furthermore, when severe comorbidities are excluded, outcomes after curative and locoregional therapies in NASH-HCC are similar or even better than in patients without NASH (Table 3). Finally, immunotherapies might not be as effective in non-viral compared with viral aetiologies owing to the previously discussed impairments in the immune system14,23,80.
Table 3.
Summary of treatment outcomes reported for patients with early-stage or intermediate-stage HCC, stratified by NASH aetiology
| Treatment | n | Outcomes in patients with NASH versus other aetiologies |
|---|---|---|
| Surgical resection172 | 7,226 | Meta-analysis: improved disease-free survival (HR 0.81, 95% CI 0.70–0.94) and overall survival (HR 0.78, 95% CI 0.67–0.90) |
| Liver transplantation174,175 | 41,289 68,950 |
UNOS database: higher post-transplant survival (HR 0.69, 95% CI 0.63–0.77) and lower graft failure (HR 0.76, 95% CI 0.69–0.83) European Liver Transplant Registry: no significant difference in post-transplant survival (HR 1.10, 95% CI 0.97–1.24) or graft survival (HR 1.02, 95% CI 0.90–1.15) |
| Local ablation167 | 17,664 | SEER-Medicare database: similar overall survival (median 1.3 years (range 0–6.9) for NAFLD-HCC versus HBV-HCC (2.0 years, 0–5.9), HCV-HCC (1.6 years, 0–6.0) and ALD-HCC (1.2 years, 0–6.3)) |
| Transarterial chemoembolization182 | 220 | Propensity score-matched analysis: similar time-to-progression (13.0 versus 8.5 months; P = 0.25) and overall survival (23.2 versus 28.0 months; P = 0.48) |
| Transarterial radioembolization183 | 149 | Retrospective cohort study: no significant difference in overall survival or toxicity |
ALD, alcohol-associated liver disease; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; SEER, The Surveillance, Epidemiology, and End Results; UNOS, United Network for Organ Sharing.
Treatment of early or intermediate stages
Surgical therapies.
Curative surgical options for patients with BCLC stage A HCC include surgical resection and liver transplantation, with both affording a 5-year survival of >70%2. Resection is the recommended therapy for patients without cirrhosis and for those with compensated cirrhosis with no portal hypertension, whereas liver transplantation is the optimal therapy for patients with liver dysfunction severe enough to preclude resection2,146,153. Patients with NAFLD can be at risk of increased intra-operative complications and worse post-surgical outcomes given their increased risk of metabolic syndrome comorbidities. Obesity169 and T2DM170 are both associated with poorer survival in patients with cancer, including those undergoing surgical therapies.
Even after proper selection for surgery according to clinical practice guidelines, most studies that included patients with NASH-HCC reported a higher prevalence of hypertension, hyperlipidaemia and ischaemic heart disease compared with patients with other aetiologies; these features of metabolic dysfunction are associated with higher post-surgical morbidity and complications165. Previous studies have also suggested that the degree of steatosis can be associated with worse surgical outcomes171. However, once properly selected patients are treated with resection, results are similar to or even better than those with viral-related HCC. A systematic review and meta-analysis of 14 studies including 7,226 patients with HCC (~20% with NAFLD-HCC) reported improved disease-free survival (HR 0.81, 95% CI 0.70–0.94) and overall survival (HR 0.78, 95% CI 0.67–0.90) after resection in those with NAFLD compared with other aetiologies172. These results were confirmed by another meta-analysis173, and might be a reflection of a higher proportion of patients with NASH without cirrhosis and that patients with concurrent comorbidities have been excluded from these complex treatments. No neoadjuvant or adjuvant therapies before or after resection for HCC, including for NASH-HCC specifically, have been shown to improve recurrence-free survival in phase III trials23.
Liver transplantation.
An analysis of the United Network for Organ Sharing (UNOS) registry between 2002 and 2012 reported that patients with NAFLD had significantly higher post-transplantation survival (HR 0.69, 95% CI 0.63–0.77) and lower risk of graft failure (HR 0.76, 95% CI 0.69–0.83) than patients with other aetiologies of HCC despite a higher prevalence of diabetes and cardiovascular disease174. By contrast, data from the European Liver Transplant Registry showed no statistically significant difference in post-transplantation survival or graft survival between patients with HCC with and without NAFLD175, although there were differences in causes of mortality (Table 3). Although some single-centre studies suggest that patients with NAFLD might have a higher risk of post-transplantation complications176, current data demonstrate a similar survival between patients with NAFLD and those with other aetiologies.
Locoregional therapies.
Locoregional therapies, defined as imaging-guided liver tumour-directed procedures, are utilized in half of patients with HCC177. Tumour destruction of HCC can be achieved with chemicals, such as percutaneous ethanol injection, or thermal ablation, which includes radiofrequency ablation, microwave ablation, cryoablation and laser interstitial thermotherapy177. Local ablative therapy, primarily with radiofrequency ablation, is an alternative curative option for patients who are not eligible for surgical therapies, particularly for those with BCLC stage 0–A with HCC nodules <3–4 cm in diameter146,153. Transarterial radioembolization (TARE) also produces objective responses in 88.3% of patients with single tumours with a median diameter of 2.7 cm (ref. 178). Personalized dosimetry, with the aim of delivering at least 205 Gy to tumours, has been shown to increase response rates with TARE compared with standardized dosimetry179. Observational studies testing stereotactic body radiation therapy have shown high local responses but there are fewer data rigorously evaluating progression-free survival or overall survival180. Data on the efficacy of these therapies specifically in patients with NASH-HCC are lacking and few studies have compared outcomes according to liver disease aetiology, although one retrospective study using The Surveillance, Epidemiology, and End Results (SEER)-Medicare database found similar overall survival between patients with NAFLD-HCC and those with HCC of other aetiologies167 (Table 3).
Transarterial chemoembolization (TACE) is the primary therapeutic option for patients with BCLC stage B disease177. A meta-analysis of several studies has demonstrated that TACE significantly improves overall survival compared with best supportive care23. Currently, estimates of the clinical benefits from TACE are a median overall survival of 30 months and an objective response rate (ORR) of ~50%177. TARE is an alternative to TACE according to phase II data177, including a small phase II randomized controlled trial (RCT; n = 179) demonstrating that TARE prolonged time-to-progression compared with TACE (HR 0.12, 95% CI 0.03–0.56), with similar overall survival181 (18.6 versus 17.7 months, respectively; P = 0.99). Few studies have compared outcomes after TACE or TARE between patients with NAFLD-HCC and those with HCC from other aetiologies. One small propensity score-matched cohort study (n = 220) found no difference in the time-to-progression (13.0 versus 8.5 months; P = 0.25) and overall survival (23.2 versus 28.0 months; P = 0.48) between patients with HCC with NAFLD and without NAFLD, respectively182. Similarly, a small cohort study of 87 patients with NAFLD-HCC and 62 patients with HBV-related HCC reported no significant difference in treatment-related adverse events (38.7% versus 39.2%; P = 0.91) and overall survival (11.1 versus 9.3 months; P = 0.38)183. Overall, these data suggest that TACE and TARE can likely be safely used in patients with NAFLD, with the expectation of similar outcomes across cirrhosis aetiologies (Table 3).
Systemic therapies in advanced NASH-HCC
The options for systemic management of advanced HCC have grown substantially over the past 10–15 years2,22,164. In 2007, the approval of sorafenib as the first systemic therapy for HCC marked the start of a new era in HCC treatment, which was then followed in the next decade by the subsequent approval of several single-agent systemic therapies in the first line (lenvatinib) and second line (regorafenib, cabozantinib and ramucirumab). Next, the approval of atezolizumab and bevacizumab in 2020 marked the dawn of a third era dominated by combination regimens involving immunotherapies164. As the therapeutic options have expanded, the question of how to best individualize treatment decisions has evolved. While all phase III studies in advanced HCC have largely recruited patients with well-compensated liver disease (Child–Pugh A cirrhosis), treatment decisions and clinical trial design did not historically consider the aetiology of liver disease. For data reporting, most studies report efficacy data by the typical stratification factors (Eastern Cooperative Oncology Group-performance status, region, presence or absence of macrovascular invasion, and elevation of AFP) as well as other clinical factors of interest, including aetiology, which is often reported as HBV, HCV and ‘non-viral’, the latter encompassing all remaining patients, including those with alcohol-related disease, NASH and other aetiologies. Meta-analyses of individual data of randomized trials comparing sorafenib versus placebo identified a greater benefit in patients with HCV-related liver cancer versus other aetiologies when exposed to the active agent184,185.
While there are currently several agents approved in the first-line and second-line setting, they are broadly classified into two categories: multi-kinase VEGFR-targeting small molecules and VEGFR2 monoclonal antibody approaches, and immunotherapy-based approaches. The former group includes the small molecules sorafenib184, lenvatinib186, regorafenib187, cabozantinib188 and the monoclonal antibody ramucirumab189. Both sorafenib and lenvatinib have been studied in the first-line setting, whereas the others have been approved as options for patients who are intolerant to first-line therapy or who progress after it. When evaluating overall survival, which is the primary end point for these studies, the efficacy of these drugs does not differ significantly based on aetiology when comparing the hazard ratios for overall survival of the study arm versus the control arm (Table 4); this is generally true also for the secondary end points of progression-free survival and ORRs (Table 4).
Table 4.
Systemic therapies for HCC: summary of subgroup analyses of HBV versus HCV versus non-viral HCC in key phase III studies
| Trial | Treatment | Trial aetiology (n, %) | Overall survival HR (95% CI) | Progression-free survival HR (95% CI) |
|---|---|---|---|---|
| Immunotherapies — first line | ||||
| IMbrave150 (ref. 190) | Atezolizumab plus bevacizumab versus sorafenib | HBV (240, 48%) HCV (108, 22%) Non-viral (153, 31%) |
0.51 (0.32–0.81) 0.43 (0.22–0.87) 0.91 (0.52–1.60) |
0.47 (0.33–0.67) 0.69 (0.39–1.20) 0.71 (0.47–1.08) |
| HIMALAYA191 | Tremelimumab plus durvalumab versus sorafenib | HBV (241, 31%) HCV (214, 27%) Non-viral (327, 42%) |
0.64 (0.48–0.86) 1.06 (0.76–1.49) 0.74 (0.57–0.95) |
NA |
| COSMIC-312 (ref. 204) | Atezolizumab plus cabozantinib versus sorafenib | HBV (190, 29%) HCV (202, 31%) Non-viral (257, 40%) |
0.53 (0.33–0.87) 1.10 (0.72–1.68) 1.18 (0.78–1.79) |
0.46 (0.29–0.73) 0.64 (0.38–1.09) 0.92 (0.60–1.41) |
| ORIENT-32 (ref. 205) | Sintilimab plus IBI305 versus sorafenib | HBV+ (538, 94%) HBV− (33, 6%) |
0.58 (0.43–0.76) 0.80 (0.22–2.87) |
0.56 (0.40–0.76) 0.38 (0.14–1.06) |
| CheckMate 459 (ref. 193) | Nivolumab versus sorafenib | HBV (233, 31%) HCV (173, 23%) Non-viral (336, 45%) |
0.77 (0.56–1.05) 0.71 (0.49–1.01) 0.95 (0.74–1.22) |
NA |
| Immunotherapies — second line | ||||
| KEYNOTE-240 (ref. 194) | Pembrolizumab versus placebo | HBV (101, 24%) HCV (64, 15%) Non-viral (248, 60%) |
0.57 (0.35–0.94) 0.96 (0.48–1.92) 0.88 (0.64–1.20) |
0.70 (0.44–1.13) 0.46 (0.24–0.90) 0.75 (0.56–1.01) |
| Tyrosine kinase inhibitors — first line | ||||
| SHARP184,185 | Sorafenib versus placebo | HBV (111, 18%) HCV (169, 28%) Alcohol (159, 26%) |
0.76 (0.38–1.50) 0.50 (0.32–0.77) 0.76 (0.50–1.16) |
NA |
| Asia–Pacific206,207 | Sorafenib versus placebo | HBV+ (165, 73%) HBV− (61, 27%) |
0.74 (0.51–1.06) 0.57 (0.29–1.13) |
NA |
| REFLECT186 | Lenvatinib versus sorafenib | HBV (479, 50%) HCV (217, 23%) Alcohol (57, 6%) |
0.83 (0.68–1.02) 0.91 (0.66–1.26) 1.03 (0.47–2.28) |
0.62 (0.50–0.75) 0.78 (0.56–1.09) 0.27 (0.11–0.66) |
| Tyrosine kinase inhibitors – second line | ||||
| CELESTIAL188 | Cabozantinib versus placebo | HBV (267, 38%) HCV (168, 24%) Non-viral (272, 38%) |
0.69 (0.51–0.94) 1.11 (0.72–1.71) 0.72 (0.54–0.96) |
0.31 (0.23–0.42) 0.61 (0.42–0.88) 0.48 (0.36–0.63) |
| RESORCE187 | Regorafenib versus placebo | HBV (216, 38%) HCV (119, 21%) Alcohol (145, 25%) |
0.58 (0.41–0.82) 0.79 (0.49–1.26) 0.92 (0.61–1.38 |
0.39 (0.29–0.54) 0.59 (0.39–0.90) 0.53 (0.37–0.77) |
| REACH-2 (ref. 208) | Ramucirumab versus placebo | HBV (107, 37%) HCV (76, 26%) Other (109, 37%) |
0.84 (0.52–1.35) 0.76 (0.44–1.33) 0.63 (0.38–1.06) |
0.43 (0.28–0.68) 0.33 (0.19–0.60) 0.57 (0.35–0.95) |
HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; NA, not available.
Several phase III studies have reported on the use of monoclonal antibodies that disrupt critical immune checkpoints in HCC, although no specific study targeting patients with NASH-HCC has been conducted22. These drugs include the monoclonal antibodies nivolumab and pembrolizumab, which target the PD1 receptor on T cells, and durvalumab and atezolizumab, which target the PD1 ligand (PDL1) on tumour cells. These antibodies essentially block the negative signalling induced by the interaction of PD1 and PDL1 and restore anticancer immunity22. Unlike the drugs discussed in the previous paragraph, ICIs have not only demonstrated a survival benefit but have also induced significant response rates associated with a long duration of response beyond 20 months. Atezolizumab, in combination with the anti-VEGF antibody bevacizumab, has been approved globally on the basis of improving overall survival in the first-line setting versus sorafenib in the phase III IMbrave150 study (median overall survival 19.2 versus 13.2 months)190. Data from the phase III HIMALAYA study evaluating the combination of durvalumab and the anti-CTLA4 antibody tremelimumab have also demonstrated an improvement in overall survival compared with sorafenib in this setting (median overall survival 16.4 versus 13.8 months)191. Nivolumab, while demonstrating meaningful single-agent activity in earlier studies192, did not meet its primary end point of improving overall survival versus sorafenib in the CheckMate 459 study, but this study did confirm the safety of nivolumab in the first-line setting and demonstrated an ORR of 15%193. Pembrolizumab was evaluated against placebo in two phase III studies in the second-line setting. KEYNOTE-240 was a global study that did not demonstrate statistical significance for overall survival, despite an ORR of 13.8% (95% CI 7.7–19.5)194, whereas KEYNOTE-394 (ref. 195) — a study conducted in China with a similar study design — did meet its primary end point of improving overall survival (P = 0.0180) (Fig. 3 and Supplementary Figure 1).
Fig. 3 |. ICIs for advanced HCC: summary of subgroup analyses of HBV versus HCV versus non-viral HCC in key phase III studies.

Meta-analyses of trials expanding on data previously reported about three randomized controlled trials (RCTs) in ref. 23. Two meta-analyses are reported, one involving single-agent immune-checkpoint inhibitor (ICI) therapy with or without targeted therapies (IMbrave150 (ref. 190), CheckMate 459 (ref. 193), KEYNOTE-240 (ref. 194) and COSMIC-312 (ref. 204) trials; four RCTs) and the other including five studies (previous four plus HIMALAYA191), testing combinations of ICIs. P*, indicates P of heterogeneity. For single RCT subgroup analysis according to aetiology, see Supplementary Figure 1.
The results of these studies highlight the need to identify predictive markers of response to best select patients who are most likely to benefit from these regimens, and there have been many efforts to identify biomarkers in the blood or tissue of patients. Only ramucirumab is approved based on a prospective study associated with a specific biomarker, namely AFP ≥400 ng/ml (ref. 189). An effort to identify patients who respond best to ICIs is not unique to the HCC field but is of great interest in oncology owing to the wide approval of these agents. Biomarkers that have been used in other tumour types, such as expression of PD1 or PDL1, elevated tumour mutational burden, and microsatellite instability, either have not been correlated with survival benefit in HCC22 or occur at a very low frequency (microsatellite instability <1%)196. Four studies have identified signatures predicting response to single-agent ICI therapy197–199 or to an atezolizumab–bevacizumab combination regime200. In all circumstances, the gene sets include features of inflamed tumours such as genes related to interferon signalling and MHC-II-mediated antigen presentation.
Evaluating clinical characteristics for markers of benefit, particularly those directed at distinct pathogenic pathways and immune profiles associated with specific aetiologies, has been of interest. Data from two studies14,23 have raised the question of whether immunotherapies are as effective in NASH-related HCC compared with viral-related HCC (Table 4, Fig. 3 and Supplementary Figure 1). Unfortunately, the percentage of patients with NASH-related HCC aetiology has not been disclosed in any of the ~35 phase III RCTs reported so far in advanced HCC23. Thus, indirect measurement to capture any survival effect associated with aetiology has involved exploration of the results of ICIs in non-viral aetiologies. A meta-analysis (three RCTs: IMbrave150, CheckMate 459 and Keynote-240) assessing the effect of immunotherapies on overall survival according to aetiology led to the conclusion that viral-related HCC responds better (HR 0.64, 95% CI 0.50–0.83) than non-viral-related HCC (HR 0.92, 95% CI 0.77–1.11; P = 0.2)23. After the publication of a subgroup analysis of the COSMIC-312 trial assessing the efficacy of cabozantinib plus atezolizumab201, a meta-analysis of four RCTs confirmed the difference in efficacy (P = 0.01) (Fig. 3 and Supplementary Figure 1). This difference is still significant but less so (P = 0.046) when including the HIMALAYA trial (meta-analysis of five RCTs) combining two ICIs (Fig. 3 and Supplementary Figure 1). Thus, the results suggest that immunotherapies might work better in viral-related HCC than in other aetiologies of HCC and are aligned with the observation that NASH-HCC tumours have dysfunctional T cells that limit the anti-tumoural effect of ICIs in this aetiology14,23. Nonetheless, it is important to note that these subgroup observations are not powered for statistical conclusions and are not controlled for other relevant prognostic factors. Additionally, the classification of ‘non-viral aetiologies’ is not specific to NASH-related liver disease but includes alcohol-related HCC as well as idiopathic and other metabolic causes. Furthermore, overall survival might be subject to other clinical features such as post-progression therapy and other comorbidities, which again are not accounted for. Compounding these findings is that ORR varied according to aetiology; for instance, ORRs of 19%, 17% and 12% were reported for HBV, HCV and non-viral HCC, respectively, in the CheckMate 459 trial comparing nivolumab with sorafenib in the first-line setting193. Taken together, these data support the concept of stratification according to aetiology in future studies based on the observed distinct outcomes, although the ultimate role of aetiology in outcome will require dedicated prospective studies. While biologically distinct, the clinical approach to NASH-HCC is not recommended to be different than for other non-viral aetiologies, including alcohol-related HCC. In clinical trials conducted in the future, there is a need to specify which patients have NASH-related HCC aetiology as this is the sole approach that can clarify the effect of immunotherapies on survival in patients with NASH-HCC.
Toxicity of systemic therapies for NASH-HCC
The adverse effect profiles of the agents used in the treatment of NASH-HCC are well defined. All VEGF-targeting or VEGFR-targeting agents induce hypertension to various degrees and carry a risk of bleeding, highlighting the importance of managing hypertension and comorbidities before treatment initiation. In addition to non-specific adverse effects, such as fatigue, the small molecules have a broader adverse effect profile that includes gastrointestinal toxicities such as anorexia, weight loss and diarrhoea as well as hand–foot skin reaction, which can be a dose-limiting toxicity202. ICIs are well tolerated and their adverse events are related to their mechanism of action and, as such, are immune mediated203, including immune-mediated rash, colitis and hypothyroidism. These adverse effects can generally be managed with supportive care but, at times, they might be severe enough to require immunosuppression with high-dose steroids or other agents. Therefore, patients with NASH-HCC after liver transplantation or with severe underlying autoimmune disease might not be eligible for treatment with this drug class although those with mild autoimmune disease can typically be treated safely. How to identify those who will develop adverse effects and their severity is not well established and is not linked to specific clinical factors such as aetiology. Therefore, presently, the clinical management of adverse effects in all patients is approached in the same way: close follow-up and management of any adverse effects as proactively as possible.
Future prospects and unmet needs
There has been substantial progress in elucidating the pathophysiology of NASH-HCC over the past 10–20 years, and key features distinguishing NASH-HCC from HCC of other aetiologies have now been identified. Looking to the future, progress is already under way to address the remaining unmet needs in NASH-HCC, summarized in Box 2. It is of the utmost importance to identify the key molecular drivers that lead to the development of HCC in patients with non-cirrhotic NAFLD and to develop effective therapies for NASH to prevent the onset of HCC. Thus far, there are no approved therapies for NASH; therefore, the effect of attenuating the underlying disease on HCC is uncertain. In HCV-associated HCC, cure of the underlying disease with antivirals reduces but does not eliminate the risk of cancer204. There is a need to increase the implementation of surveillance in patients with NASH with cirrhosis and to explore alternatives to ultrasound in patients with obesity. With respect to therapies, a proper refinement of patient selection for surgery will mitigate post-operative complications. For local and systemic therapies, it is vital to report NASH-HCC as a standalone entity in clinical trials in HCC, thereby enabling specific analyses of outcomes. Immunotherapies remain the backbone treatment for advanced tumours but prospective studies should ultimately clarify whether NASH-HCC-related T cell dysfunction has an effect on the decreased efficacy of ICIs. In addition, there is a need to define biomarkers of response and to search for combinations that improve the response to molecular treatments. The number of important ongoing phase III investigations and emerging strategies against HCC are encouraging and will likely yield further improvements in the outcomes of patients with NASH-HCC.
Box 2. Unmet needs in NASH-related HCC.
Pathogenesis: identify the key molecular drivers and specific mutational signatures leading to the development of hepatocellular carcinoma (HCC) in patients with non-cirrhotic nonalcoholic fatty liver disease (NAFLD).
Prevention: develop effective therapies for nonalcoholic steatohepatitis (NASH) to prevent disease progression and the development of HCC, and explore the effect of statins and weight loss.
Surveillance: increase implementation of surveillance in patients with NASH with cirrhosis, refine populations at-risk in non-cirrhotic NAFLD, and explore alternative surveillance strategies to ultrasound, such as fastMRI, in patients with obesity.
Surgery: refine the selection of patients to mitigate post-operative complications.
Locoregional therapies: clarify and improve the efficacy of locoregional therapies in patients with NASH-HCC: transarterial chemoembolization versus radioembolization.
Systemic therapies: rigorously compare the efficacy of immunotherapies in NASH-HCC to other aetiologies, clarify and exploit mechanisms to increase immune surveillance in NASH-HCC to improve response to immune-checkpoint inhibitors, and explore the use of cell-based therapies (CAR T cells) for NASH-HCC.
Identify predictive biomarkers to systemic treatment for NASH-HCC based on recent discoveries in single-cell studies.
Identify aetiology-specific adverse effects of treatments for patients with NASH-HCC.
Establish evidence-based guidelines to optimize the sequence of systemic agents in patients with NASH-HCC.
Report NASH-HCC as a standalone entity in clinical trials in HCC.
Conclusions
NASH is a major global health problem and is predicted to become the leading cause of HCC by 2030 (ref. 11). Progression from NASH to HCC is influenced by a multitude of factors, including molecular alterations and germline variants, fibrosis status, immune microenvironment and microbiome. Data suggest that lifestyle measures are critical to preventing the progression of NAFLD and that surveillance in patients with NASH cirrhosis is associated with earlier detection and improved survival. Currently, NASH-HCC is treated according to the same guidelines for other aetiologies; however, comorbidities associated with NASH, such as obesity and T2DM, can complicate the implementation of surgical and locoregional therapies. Furthermore, both preclinical and clinical data indicate that distinct immune microenvironmental factors could be influencing response rates to immunotherapies, which are now the standard of care for advanced HCC. It is recommended that NASH-HCC aetiology be specifically described in clinical trials so that more effective, personalized treatment approaches can be implemented in the near future.
Supplementary Material
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41575-023-00754-7.
Key points.
Nonalcoholic steatohepatitis (NASH)-related hepatocellular carcinoma (HCC) is a major public health problem, the incidence of which is increasing in parallel to the rise in obesity, diabetes and metabolic syndrome.
Progression from NASH to NASH-HCC occurs at an approximate rate of 2% per year and is influenced by many factors, including the tissue and immune microenvironment, germline mutations in PNPLA3, and the microbiome.
HCC surveillance in these at-risk patients with NASH-related cirrhosis is associated with earlier detection and improved survival.
NASH-HCC in the early to intermediate stages is managed with surgery and locoregional therapies; however, comorbidities associated with NASH, such as obesity and type 2 diabetes mellitus, can complicate the implementation of potentially curative therapies.
Advanced NASH-HCC is treated with systemic therapies, including anti-angiogenic therapies and immune-checkpoint inhibitors. Distinct microenvironmental factors could be limiting the response of NASH-HCC to immunotherapy.
Acknowledgements
The authors acknowledge U. Balaseviciute and J. Huguet Pradell (Translational Research in Hepatic Oncology, Liver Unit, Institut d’Investigacions Biomèdiques August Pi i Sunyer, Barcelona), and F. Castet (Gastrointestinal and Endocrine Tumour Unit, Vall d’Hebron Institute of Oncology, Vall d’Hebron University Hospital, Barcelona) for their help in preparing the original versions of the figures for this manuscript.
Footnotes
Competing interests
J.M.L. received research support from Bayer HealthCare Pharmaceuticals, Eisai Inc., Bristol Myers Squibb, Boehringer-Ingelheim and Ipsen and consulting fees from Merck, Eli Lilly, Eisai Inc., Bayer HealthCare Pharmaceuticals, Bristol Myers Squibb, Exelixis, Ipsen, Genentech, Roche, Glycotest, Nucleix, Mina Alpha Ltd. and AstraZeneca. A.G.S. has served as a consultant or on advisory boards for Bayer, Eisai, Genentech, BMS, Exelixis, AstraZeneca, Wako Diagnostics, Exact Sciences, Roche, Glycotest, GRAIL and TARGET PharmaSolutions. H.B.E.-S. received research support from Glycotest, Gilead Sciences, Merck Sharp & Dohme BV and AbbVie. R.S.F. reports consulting fees from AstraZeneca, Bayer, CStone, BMS, Eisai, Exilixis, Eli Lilly, Pfizer, Merck, Roche/Genentech and Hengrui. S.L.F. is a consultant to 89 Bio, Amgen, Axcella Health, Blade Therapeutics, Bristol Myers Squibb, Can-Fite Biopharma, Casma Therapeutics, ChemomAb, Escient Pharmaceuticals, Forbion, Galmed, Gordian Biotechnology, Glycotest, Glympse Bio, Insitro, Morphic Therapeutics, North Sea Therapeutics, Novartis, Ono Pharmaceuticals, Pfizer Pharmaceuticals, Scholar Rock and Surrozen and has stock options (all less than 1% of company value) in Blade Therapeutics, Escient, Galectin, Galmed, Genfit, Glympse, Hepgene, Lifemax, Metacrine, Morphic Therapeutics, Nimbus, North Sea Therapeutics, Scholar Rock and Surrozen. All other authors declare no competing interests.
Peer review information Nature Reviews Gastroenterology & Hepatology thanks Jean-Francois Dufour, James Harding and Frank Tacke for their contribution to the peer review of this work.
Data availability
For the data underlying the meta-analyses reported in Fig. 3, see Supplementary Figure 1.
References
- 1.Sung H et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 7, 6 (2021). This review provides a comprehensive up-to-date overview of the epidemiology, pathogenesis and management of HCC.
- 3.Anstee QM, Reeves HL, Kotsiliti E, Govaere O & Heikenwalder M From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16, 411–428 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Chalasani N et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Huang DQ, El-Serag HB & Loomba R Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 18, 223–238 (2021). This manuscript thoroughly analyses the global epidemiology, projections and risk factors for NAFLD-related HCC.
- 6.Younossi ZM et al. Global epidemiology of nonalcoholic fatty liver disease — meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Estes C et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 69, 896–904 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Estes C, Razavi H, Loomba R, Younossi Z & Sanyal AJ Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazarus JV et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat. Rev. Gastroenterol. Hepatol. 19, 60–78 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Loomba R, Lim JK, Patton H & El-Serag HB AGA clinical practice update on screening and surveillance for hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: expert review. Gastroenterology 158, 1822–1830 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gawrieh S et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: a United States multicentre study. Aliment. Pharmacol. Ther. 50, 809–821 (2019). [DOI] [PubMed] [Google Scholar]
- 12. Ioannou GN Epidemiology and risk-stratification of NAFLD-associated HCC. J. Hepatol. 75, 1476–1484 (2021). This review covers the risk factors associated with NAFLD-HCC, and how the identification of at-risk patients is needed for effective surveillance strategies.
- 13.Pinyol R et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol. 75, 865–878 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pfister D et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 592, 450–456 (2021). This study demonstrates the existence of a CD8+PD1+ subset of pro-tumorigenic cells in NASH that favour the development of HCC and hamper response to ICIs.
- 15. Liu YL et al. Carriage of the PNPLA3 rs738409 C>g polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 61, 75–81 (2014). This study demonstrates that the PNPLA3 rs738409 C>G polymorphism is associated with greater risk of progressive steatohepatitis, fibrosis and HCC development.
- 16.Mittal S et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 14, 124–131.e1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stine JG et al. Systematic review with meta-analysis: risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 48, 696–703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanwal F et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 155, 1828–1837.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piscaglia F et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 63, 827–838 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Vogel A, Meyer T, Sapisochin G, Salem R & Saborowski A Hepatocellular carcinoma. Lancet 400, 1345–1362 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Puigvehí M et al. Liver transplant for hepatocellular carcinoma in the United States: evolving trends over the last three decades. Am. J. Transplant. 20, 220–230 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Llovet JM et al. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 19, 151–172 (2022). Comprehensive overview of the immune microenvironment role according to aetiologies, mechanisms of response and role of immunotherapies for the treatment of HCC.
- 23. Haber PK et al. Evidence-based management of hepatocellular carcinoma: systematic review and meta-analysis of randomized controlled trials (2002–2020). Gastroenterology 161, 879–898 (2021). Meta-analysis of RCTs of therapies in HCC from 2002 to 2020, including all relevant studies leading to a paradigm change in the management of HCC.
- 24.Kelley RK & Greten TF Hepatocellular carcinoma — origins and outcomes. N. Engl. J. Med. 385, 280–282 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Dyson J et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 60, 110–117 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Patkar S, Parray A, Mahendra B, Kurunkar S & Goel M Performance of Hong Kong liver cancer staging system in patients of hepatocellular carcinoma treated with surgical resection: an Indian validation study. J. Surg. Oncol. 120, 1119–1125 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Mittal S et al. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin. Gastroenterol. Hepatol. 13, 594–601.e1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aljumah AA et al. Clinical presentation, risk factors, and treatment modalities of hepatocellular carcinoma: a single tertiary care center experience. Gastroenterol. Res. Pract. 2016, 1989045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganslmayer M et al. A large cohort of patients with hepatocellular carcinoma in a single European centre: aetiology and prognosis now and in a historical cohort. Swiss Med. Wkly 144, w13900 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Park JW et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE study. Liver Int. 35, 2155–2166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White DL, Kanwal F & El-Serag HB Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 10, 1342–1359.e2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balakrishnan M & El-Serag HB Editorial: NAFLD-related hepatocellular carcinoma — increasing or not? With or without cirrhosis? Aliment. Pharmacol. Ther. 47, 437–438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orci LA et al. Incidence of hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: a systematic review, meta-analysis, and meta-regression. Clin. Gastroenterol. Hepatol. 20, 283–292.e10 (2022). [DOI] [PubMed] [Google Scholar]
- 34.Natarajan Y et al. Risk of cirrhosis and hepatocellular cancer in patients with NAFLD and normal liver enzymes. Hepatology 72, 1242–1252 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JD et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 71, 907–916 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexander M et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 17, 95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Wang X, Wang J, Yan Z & Luo J Excess body weight and the risk of primary liver cancer: an updated meta-analysis of prospective studies. Eur. J. Cancer 48, 2137–2145 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Abdel-Rahman O et al. Cigarette smoking as a risk factor for the development of and mortality from hepatocellular carcinoma: an updated systematic review of 81 epidemiological studies. J. Evid. Based Med. 10, 245–254 (2017). [DOI] [PubMed] [Google Scholar]
- 39. Ioannou GN, Green P, Kerr KF & Berry K Models estimating risk of hepatocellular carcinoma in patients with alcohol or NAFLD-related cirrhosis for risk stratification. J. Hepatol. 71, 523–533 (2019). Study developing models available as web-based tools for estimating HCC risk in patients with NAFLD-cirrhosis or alcohol-associated liver disease with cirrhosis.
- 40.Younes R et al. Long-term outcomes and predictive ability of non-invasive scoring systems in patients with non-alcoholic fatty liver disease. J. Hepatol. 75, 786–794 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Marra F & Svegliati-Baroni G Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 68, 280–295 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Fuchs A et al. Associations among adipose tissue immunology, inflammation, exosomes and insulin sensitivity in people with obesity and nonalcoholic fatty liver disease. Gastroenterology 161, 968–981.e12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parthasarathy G, Revelo X & Malhi H Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol. Commun. 4, 478–492 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Friedman SL, Neuschwander-Tetri BA, Rinella M & Sanyal AJ Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 24, 908–922 (2018). Overview of the pathogenic and clinical features of NAFLD, preclinical models of NAFLD and emerging targets for drug development.
- 45.Hotamisligil GS Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Daugherity EK et al. The DNA damage checkpoint protein ATM promotes hepatocellular apoptosis and fibrosis in a mouse model of non-alcoholic fatty liver disease. Cell Cycle 11, 1918–1928 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng SWK et al. Convergent somatic mutations in metabolism genes in chronic liver disease. Nature 598, 473–478 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Zhu C et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl Med. 10, eaat0344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michelotti GA et al. Smoothened is a master regulator of adult liver repair. J. Clin. Invest. 123, 2380–2394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu C, Tabas I, Schwabe RF & Pajvani UB Maladaptive regeneration — the reawakening of developmental pathways in NASH and fibrosis. Nat. Rev. Gastroenterol. Hepatol. 18, 131–142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dewhurst MR et al. Loss of hepatocyte cell division leads to liver inflammation and fibrosis. PLoS Genet. 16, e1009084 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huby T & Gautier EL Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 22, 429–443 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kazankov K et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 16, 145–159 (2019). [DOI] [PubMed] [Google Scholar]
- 54. Sanyal AJ et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 385, 1559–1569 (2021). Prospective multicentre study of patients with NAFLD demonstrating that bridging fibrosis and cirrhosis is associated with increased risks of liver-related complications and death.
- 55.Xiong X et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramachandran P et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amor C et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kitade M et al. Neovascularization and oxidative stress in the progression of non-alcoholic steatohepatitis. Mol. Med. Rep. 1, 543–548 (2008). [PubMed] [Google Scholar]
- 59.Coulon S et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology 57, 1793–1805 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Lefere S et al. Angiopoietin-2 promotes pathological angiogenesis and is a therapeutic target in murine nonalcoholic fatty liver disease. Hepatology 69, 1087–1104 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Ibrahim SH Sinusoidal endotheliopathy in nonalcoholic steatohepatitis: therapeutic implications. Am. J. Physiol. Gastrointest. Liver Physiol. 321, G67–G74 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piersma B, Hayward MK & Weaver VM Fibrosis and cancer: a strained relationship. Biochim. Biophys. Acta Rev. Cancer 1873, 188356 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang DY & Friedman SL Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 56, 769–775 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dunham RM et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 190, 2009–2016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lei H, Reinke P, Volk HD, Lv Y & Wu R Mechanisms of immune tolerance in liver transplantation-crosstalk between alloreactive T cells and liver cells with therapeutic prospects. Front. Immunol. 10, 2667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Der Meer AJ et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. J. Am. Med. Assoc. 308, 2584–2593 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Kamp WM, Sellers CM, Stein S, Lim JK & Kim HS Impact of direct acting antivirals on survival in patients with chronic hepatitis C and hepatocellular carcinoma. Sci. Rep. 9, 17081 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colombo M & Lleo A The impact of antiviral therapy on hepatocellular carcinoma epidemiology. Hepatic Oncol. 5, HEP03 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kwak M et al. Bariatric surgery is associated with reduction in non-alcoholic steatohepatitis and hepatocellular carcinoma: a propensity matched analysis. Am. J. Surg. 219, 504–507 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Vuppalanchi R, Noureddin M, Alkhouri N & Sanyal AJ Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 18, 373–392 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Konerman MA, Jones JC & Harrison SA Pharmacotherapy for NASH: current and emerging. J. Hepatol. 68, 362–375 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Aggarwal P, Noureddin M, Harrison S, Jeannin S & Alkhouri N Nonalcoholic steatohepatitis (NASH) cirrhosis: a snapshot of therapeutic agents in clinical development and the optimal design for clinical trials. Expert Opin. Investig. Drugs 31, 163–172 (2022). [DOI] [PubMed] [Google Scholar]
- 73.Rowe IA, Wong VWS & Loomba R Treatment candidacy for pharmacologic therapies for NASH. Clin. Gastroenterol. Hepatol. 20, 1209–1217 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montironi C et al. Inflamed and non-inflamed classes of HCC: a revised immunogenomic classification. Gut 72, 129–140 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sutti S & Albano E Adaptive immunity: an emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 17, 81–92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolf MJ et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26, 549–564 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Shalapour S et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma C et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brown ZJ et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis. 9, 620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Heinrich B et al. Steatohepatitis impairs T-cell-directed immunotherapies against liver tumors in mice. Gastroenterology 160, 331–345.e6 (2021). This study reveals how steatohepatitis reduces CD4+ T cell and effector memory cell infiltration into liver tumours, which reduces the anti-tumour activity of immunotherapeutic agents.
- 81.Malehmir M et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 25, 641–655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deczkowska A et al. XCR1+ type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 27, 1043–1054 (2021). [DOI] [PubMed] [Google Scholar]
- 83.Ou R et al. Neutrophil depletion improves diet-induced non-alcoholic fatty liver disease in mice. Endocrine 57, 72–82 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Zhang H, van der Windt DJ, Ren J, Tsung A & Huang H The role of neutrophil extracellular traps in nonalcoholic steatohepatitis-associated hepatocellular carcinoma. J. Immunol. 202 (Suppl. 1), 135.2 (2019). [Google Scholar]
- 85.Leslie J et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut 71, 2093–2106 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramadori P, Kam S & Heikenwalder M T cells: friends and foes in NASH pathogenesis and hepatocarcinogenesis. Hepatology 75, 1038–1049 (2022). [DOI] [PubMed] [Google Scholar]
- 87.McVey JC et al. NAFLD indirectly impairs antigen-specific CD8+ T cell immunity against liver cancer in mice. iScience 25, 103847 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cappuyns S et al. Pretreatment immune cell composition and checkpoint ligand expression define the response to immunotherapy in advanced HCC: a study using single-cell sequencing. Clin. Cancer Res. 28 (Suppl. 17), PR03 (2022). [Google Scholar]
- 89.Wabitsch S et al. Metformin treatment rescues CD8+ T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J. Hepatol. 77, 748–760 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoechst B et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology 135, 234–243 (2008). [DOI] [PubMed] [Google Scholar]
- 91.Dudek M et al. Auto-aggressive CXCR6+CD8 T cells cause liver immune pathology in NASH. Nature 592, 444–449 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Ma C et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aron-Wisnewsky J et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 17, 279–297 (2020). [DOI] [PubMed] [Google Scholar]
- 94.Jadhav K & Cohen TS Can you trust your gut? Implicating a disrupted intestinal microbiome in the progression of NAFLD/NASH. Front. Endocrinol. 11, 592157 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mouries J et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 71, 1216–1228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu L et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57, 601–609 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Boursier J et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jian J et al. Rifaximin ameliorates non-alcoholic steatohepatitis in mice through regulating gut microbiome-related bile acids. Front. Pharmacol. 13, 841132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gangarapu V et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 27, 840–845 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Madrid AM, Hurtado C, Venegas M, Cumsille F & Defilippi C Long-term treatment with cisapride and antibiotics in liver cirrhosis: effect on small intestinal motility, bacterial overgrowth, and liver function. Am. J. Gastroenterol. 96, 1251–1255 (2001). [DOI] [PubMed] [Google Scholar]
- 101. Zucman-Rossi J, Villanueva A, Nault JC & Llovet JM Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149, 1226–1239.e4 (2015). Review describing the landscape of genetic alterations and mutations in HCC and their potential as biomarkers as well as the molecular classification of HCC.
- 102.Anstee QM et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 73, 505–515 (2020). [DOI] [PubMed] [Google Scholar]
- 103.Liu YL et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 5, 4309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Donati B et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 7, 4492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Teo K et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: a meta-analysis. J. Hepatol. 74, 20–30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tan HL et al. Association of glucokinase regulatory gene polymorphisms with risk and severity of non-alcoholic fatty liver disease: an interaction study with adiponutrin gene. J. Gastroenterol. 49, 1056–1064 (2014). [DOI] [PubMed] [Google Scholar]
- 107.Kawaguchi T et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE 13, e0185490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.BasuRay S, Smagris E, Cohen JC & Hobbs HH The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 66, 1111–1124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.BasuRay S, Wang Y, Smagris E, Cohen JC & Hobbs HH Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl Acad. Sci. USA 116, 9521–9526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kozlitina J et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46, 352–356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Prill S et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 9, 11585 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mancina RM et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150, 1219–1230. e6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Perez-Martinez P et al. Glucokinase regulatory protein genetic variant interacts with omega-3 PUFA to influence insulin resistance and inflammation in metabolic syndrome. PLoS ONE 6, e20555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bianco C et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 74, 775–782 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hoshida Y et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 69, 7385–7392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiang DY et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 68, 6779–6788 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Seki S et al. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 37, 56–62 (2002). [DOI] [PubMed] [Google Scholar]
- 118.Tanaka S et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 48, 1249–1258 (2013). [DOI] [PubMed] [Google Scholar]
- 119.Jühling F et al. Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma. Gut 70, 157–169 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chang M-H et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. N. Engl. J. Med. 336, 1855–1859 (1997). [DOI] [PubMed] [Google Scholar]
- 121.Papatheodoridis GV, Chan HLY, Hansen BE, Janssen HLA & Lampertico P Risk of hepatocellular carcinoma in chronic hepatitis B: assessment and modification with current antiviral therapy. J. Hepatol. 62, 956–967 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Kanwal F et al. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology 153, 996–1005.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 123.Zhou YY et al. Systematic review with network meta-analysis: antidiabetic medication and risk of hepatocellular carcinoma. Sci. Rep. 6, 33743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Singh S, Singh PP, Singh AG, Murad MH & Sanchez W Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology 144, 323–332 (2013). [DOI] [PubMed] [Google Scholar]
- 125.Kennedy OJ et al. Coffee, including caffeinated and decaffeinated coffee, and the risk of hepatocellular carcinoma: a systematic review and dose-response meta-analysis. BMJ Open 7, e013739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Simon TG et al. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N. Engl. J. Med. 382, 1018–1028 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zheng L et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 19, 5372–5380 (2013). [DOI] [PubMed] [Google Scholar]
- 128.Buzzai M et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 67, 6745–6752 (2007). [DOI] [PubMed] [Google Scholar]
- 129.Forslund K et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bugianesi E et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 100, 1082–1090 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Lee H & Ko G Effect of metformin on metabolic improvement and gut microbiota. Appl. Environ. Microbiol. 80, 5935–5943 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang X et al. Continuation of metformin use after a diagnosis of cirrhosis significantly improves survival of patients with diabetes. Hepatology 60, 2008–2016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cao Z et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 71, 2286–2297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roudier E, Mistafa O & Stenius U Statins induce mammalian target of rapamycin (mTOR)-mediated inhibition of Akt signaling and sensitize p53-deficient cells to cytostatic drugs. Mol. Cancer Ther. 5, 2706–2715 (2006). [DOI] [PubMed] [Google Scholar]
- 135.Relja B et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int. J. Oncol. 38, 879–885 (2011). [DOI] [PubMed] [Google Scholar]
- 136.Yang PM et al. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 70, 7699–7709 (2010). [DOI] [PubMed] [Google Scholar]
- 137.Sutter AP et al. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J. Hepatol. 43, 808–816 (2005). [DOI] [PubMed] [Google Scholar]
- 138.Kah J et al. Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol. Rep. 28, 1077–1083 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Tanaka T et al. Inhibitory effects of chlorogenic acid, reserpine, polyprenoic acid (E-5166), or coffee on hepatocarcinogenesis in rats and hamsters. Basic Life Sci. 52, 429–440 (1990). [DOI] [PubMed] [Google Scholar]
- 140.Cavin C, Holzhäuser D, Constable A, Huggett AC & Schilter B The coffee-specific diterpenes cafestol and kahweol protect against aflatoxin B1-induced genotoxicity through a dual mechanism. Carcinogenesis 19, 1369–1375 (1998). [DOI] [PubMed] [Google Scholar]
- 141.Cavin C et al. Cafestol and kahweol, two coffee specific diterpenes with anticarcinogenic activity. Food Chem. Toxicol. 40, 1155–1163 (2002). [DOI] [PubMed] [Google Scholar]
- 142.Majer BJ et al. Coffee diterpenes prevent the genotoxic effects of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and N-nitrosodimethylamine in a human derived liver cell line (HepG2). Food Chem. Toxicol. 43, 433–441 (2005). [DOI] [PubMed] [Google Scholar]
- 143.Huxley R et al. Coffee, decaffeinated coffee, and tea consumption in relation to incident type 2 diabetes mellitus: a systematic review with meta-analysis. Arch. Intern. Med. 169, 2053–2063 (2009). [DOI] [PubMed] [Google Scholar]
- 144.El-Serag HB, Hampel H & Javadi F The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 4, 369–380 (2006). [DOI] [PubMed] [Google Scholar]
- 145.Polesel J et al. The impact of obesity and diabetes mellitus on the risk of hepatocellular carcinoma. Ann. Oncol. 20, 353–357 (2009). [DOI] [PubMed] [Google Scholar]
- 146.Galle PR et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J. Hepatol. 69, 182–236 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Kramer JR et al. Effect of diabetes medications and glycemic control on risk of hepatocellular cancer in patients with nonalcoholic fatty liver disease. Hepatology 75, 1420–1428 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marchesini G et al. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 64, 1388–1402 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Chitturi S et al. The Asia–Pacific working party on non-alcoholic fatty liver disease guidelines 2017 — part 2: management and special groups. J. Gastroenterol. Hepatol. 33, 86–98 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Baumeister SE et al. Association between physical activity and risk of hepatobiliary cancers: a multinational cohort study. J. Hepatol. 70, 885–892 (2019). [DOI] [PubMed] [Google Scholar]
- 151.Promrat K et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 51, 121–129 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lange NF, Radu P & Dufour JF Prevention of NAFLD-associated HCC: role of lifestyle and chemoprevention. J. Hepatol. 75, 1217–1227 (2021). [DOI] [PubMed] [Google Scholar]
- 153.Marrero JA et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 68, 723–750 (2018). [DOI] [PubMed] [Google Scholar]
- 154. Singal AG et al. HCC surveillance improves early detection, curative treatment receipt, and survival in patients with cirrhosis: a meta-analysis. J. Hepatol. 77, 128–139 (2022). Meta-analysis demonstrating that HCC surveillance is associated with improved early detection and survival in patients with cirrhosis.
- 155.Lo ST, Gane E, Bartlett A & Orr D Clinical features and survival of non-alcoholic fatty liver disease-related hepatocellular carcinoma. Hepatol. Int. 10, S437 (2016). [Google Scholar]
- 156.Aby E, Phan J, Truong E, Grotts J & Saab S Inadequate hepatocellular carcinoma screening in patients with nonalcoholic steatohepatitis cirrhosis. J. Clin. Gastroenterol. 53, 142–146 (2019). [DOI] [PubMed] [Google Scholar]
- 157.Singal AG et al. Failure rates in the hepatocellular carcinoma surveillance process. Cancer Prev. Res. 5, 1124–1130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wolf E, Rich NE, Marrero JA, Parikh ND & Singal AG Use of hepatocellular carcinoma surveillance in patients with cirrhosis: a systematic review and meta-analysis. Hepatology 73, 713–725 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chong N et al. Association between ultrasound quality and test performance for HCC surveillance in patients with cirrhosis: a retrospective cohort study. Aliment. Pharmacol. Ther. 55, 683–690 (2022). [DOI] [PubMed] [Google Scholar]
- 160.Schoenberger H et al. Dynamic changes in ultrasound quality for hepatocellular carcinoma screening in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 20, 1561–1569.e4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tzartzeva K et al. Surveillance imaging and α-fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: a meta-analysis. Gastroenterology 154, 1706–1718.e1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Singal AG, Lok AS, Feng Z, Kanwal F & Parikh ND Conceptual model for the hepatocellular carcinoma screening continuum: current status and research agenda. Clin. Gastroenterol. Hepatol. 20, 9–18 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Llovet JM, Brú C & Bruix J Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin. Liver Dis. 19, 329–337 (1999). This paper describes the BCLC classification widely used in American and European guidelines for the management of HCC.
- 164.Llovet JM et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat. Cancer 3, 386–401 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Koh YX et al. Liver resection for nonalcoholic fatty liver disease-associated hepatocellular carcinoma. J. Am. Coll. Surg. 229, 467–478.e1 (2019). [DOI] [PubMed] [Google Scholar]
- 166.Foerster F, Gairing SJ, Müller L & Galle PR NAFLD-driven HCC: safety and efficacy of current and emerging treatment options. J. Hepatol. 76, 446–457 (2022). [DOI] [PubMed] [Google Scholar]
- 167.Wong CR, Njei B, Nguyen MH, Nguyen A & Lim JK Survival after treatment with curative intent for hepatocellular carcinoma among patients with vs without non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 46, 1061–1069 (2017). [DOI] [PubMed] [Google Scholar]
- 168. Tan DJH et al. Clinical characteristics, surveillance, treatment allocation, and outcomes of non-alcoholic fatty liver disease-related hepatocellular carcinoma: a systematic review and meta-analysis. Lancet Oncol. 23, 521–530 (2022). Meta-analysis revealing that NAFLD-HCC is associated with lower rates of surveillance and cirrhosis compared with HCC due to other causes.
- 169.Petrelli F et al. Association of obesity with survival outcomes in patients with cancer: a systematic review and meta-analysis. JAMA Netw. Open 4, e213520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang YG et al. Diabetes mellitus and poorer prognosis in hepatocellular carcinoma: a systematic review and meta-analysis. PLoS ONE 9, e95485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Su CW et al. Impact of steatosis on prognosis of patients with early-stage hepatocellular carcinoma after hepatic resection. Ann. Surg. Oncol. 22, 2253–2261 (2015). [DOI] [PubMed] [Google Scholar]
- 172.Molinari M et al. Hepatic resection for hepatocellular carcinoma in nonalcoholic fatty liver disease: a systematic review and meta-analysis of 7226 patients. Ann. Surg. 2, e065 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chin KM et al. Outcomes after curative therapy for hepatocellular carcinoma in patients with non-alcoholic fatty liver disease: a meta-analysis and review of current literature. HPB 23, 1164–1174 (2021). [DOI] [PubMed] [Google Scholar]
- 174.Wong RJ et al. Improved survival outcomes in patients with non-alcoholic steatohepatitis and alcoholic liver disease following liver transplantation: an analysis of 2002–2012 United Network for Organ Sharing data. Clin. Transplant. 28, 713–721 (2014). [DOI] [PubMed] [Google Scholar]
- 175.Haldar D et al. Outcomes of liver transplantation for non-alcoholic steatohepatitis: a European Liver Transplant Registry study. J. Hepatol. 71, 313–322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kern B et al. High incidence of hepatocellular carcinoma and postoperative complications in patients with nonalcoholic steatohepatitis as a primary indication for deceased liver transplantation. Eur. J. Gastroenterol. Hepatol. 31, 205–210 (2019). [DOI] [PubMed] [Google Scholar]
- 177. Llovet JM et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 18, 293–313 (2021). Comprehensive analysis of the use of locoregional therapies for early-stage and intermediate-stage HCC.
- 178.Salem R et al. Yttrium-90 radioembolization for the treatment of solitary, unresectable HCC: the LEGACY study. Hepatology 74, 2342–2352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Garin E et al. Personalised versus standard dosimetry approach of selective internal radiation therapy in patients with locally advanced hepatocellular carcinoma (DOSISPHERE-01): a randomised, multicentre, open-label phase 2 trial. Lancet Gastroenterol. Hepatol. 6, 17–29 (2021). [DOI] [PubMed] [Google Scholar]
- 180.Wahl DR et al. Outcomes after stereotactic body radiotherapy or radiofrequency ablation for hepatocellular carcinoma. J. Clin. Oncol. 34, 452–459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Salem R et al. Y90 radioembolization significantly prolongs time to progression compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology 151, 1155–1163.e2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Young S et al. Transarterial chemoembolization of hepatocellular carcinoma: propensity score matching study comparing survival and complications in patients with nonalcoholic steatohepatitis versus other causes cirrhosis. Cardiovasc. Interv. Radiol. 43, 65–75 (2020). [DOI] [PubMed] [Google Scholar]
- 183.Schotten C et al. NAFLD-associated comorbidities in advanced stage HCC do not alter the safety and efficacy of Yttrium-90 radioembolization. Liver Cancer 8, 491–504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Llovet JM et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008). This study demonstrates survival benefit for systemic therapy (sorafenib) in advanced HCC compared with placebo.
- 185.Bruix J et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J. Hepatol. 57, 821–829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kudo M et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018). [DOI] [PubMed] [Google Scholar]
- 187.Bruix J et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 389, 56–66 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Abou-Alfa GK et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 379, 54–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhu AX et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 16, 859–870 (2015). [DOI] [PubMed] [Google Scholar]
- 190. Finn RS et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020). Phase III trial demonstrating that combination therapy (atezolizumab plus bevacizumab) is superior in terms of survival benefit compared with sorafenib in advanced-stage HCC.
- 191.Abou-Alfa GK et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evidence 1, EVIDoa2100070 (2022). [DOI] [PubMed] [Google Scholar]
- 192.El-Khoueiry AB et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yau T et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 23, 77–90 (2022). [DOI] [PubMed] [Google Scholar]
- 194.Finn RS et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J. Clin. Oncol. 38, 193–202 (2020). [DOI] [PubMed] [Google Scholar]
- 195.Qin S et al. Pembrolizumab plus best supportive care versus placebo plus best supportive care as second-line therapy in patients in Asia with advanced hepatocellular carcinoma (HCC): phase 3 KEYNOTE-394 study. J. Clin. Oncol. 40, 383 (2022). [Google Scholar]
- 196.Hause RJ, Pritchard CC, Shendure J & Salipante SJ Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 22, 1342–1350 (2016). [DOI] [PubMed] [Google Scholar]
- 197.Sangro B et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 73, 1460–1469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Haber PK et al. Molecular markers of response to anti-PD1 therapy in advanced hepatocellular carcinoma. J. Clin. Oncol. 39, 4100 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Neely J et al. Genomic and transcriptomic analyses related to the clinical efficacy of first-line nivolumab in advanced hepatocellular carcinoma from the phase 3 CheckMate 459 trial [abstract]. Cancer Res. 82 (Suppl. 12), 2145 (2022). [Google Scholar]
- 200.Zhu AX et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 28, 1599–1611 (2022). [DOI] [PubMed] [Google Scholar]
- 201.Kelley RK et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 23, 995–1008 (2022). [DOI] [PubMed] [Google Scholar]
- 202.Llovet JM, Montal R, Sia D & Finn RS Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 15, 599–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Greten TF et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immunotherapy for the treatment of hepatocellular carcinoma. J. Immunother. Cancer 9, e002794 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Beste LA, Green P, Berry K, Belperio P & Ioannou GN Hepatitis C-related hepatocellular carcinoma incidence in the Veterans Health Administration after introduction of direct-acting antivirals. JAMA 324, 1003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ren Z et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2–3 study. Lancet Oncol. 22, 977–990 (2021). [DOI] [PubMed] [Google Scholar]
- 206.Cheng AL et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 10, 25–34 (2009). [DOI] [PubMed] [Google Scholar]
- 207.Cheng AL et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: subset analyses of the phase III sorafenib Asia-Pacific trial. Eur. J. Cancer 48, 1452–1465 (2012). [DOI] [PubMed] [Google Scholar]
- 208.Zhu AX et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 282–296 (2019). [DOI] [PubMed] [Google Scholar]
- 209.Fassio E et al. Etiology of hepatocellular carcinoma in Latin America: a prospective, multicenter, international study. Ann. Hepatol. 9, 66–69 (2010). [PubMed] [Google Scholar]
- 210.Yapali S & Tozun N Epidemiology and viral risk factors for hepatocellular carcinoma in the Eastern Mediterranean countries. Hepatoma Res. 4, 24 (2018). [Google Scholar]
- 211.Candia J et al. The genomic landscape of Mongolian hepatocellular carcinoma. Nat. Commun. 11, 4383 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ikawa-Yoshida A et al. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int. J. Exp. Pathol. 98, 221–233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Asgharpour A et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 65, 579–588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zaki MYW et al. Key features of the environment promoting liver cancer in the absence of cirrhosis. Sci. Rep. 11, 16727 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fujii M et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 46, 141–152 (2013). [DOI] [PubMed] [Google Scholar]
- 216.Liang JQ et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 9, 4490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Park EJ et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tsuchida T et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 69, 385–395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Nakagawa H et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Gomes AL et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30, 161–175 (2016). [DOI] [PubMed] [Google Scholar]
- 221.Febbraio MA et al. Preclinical models for studying NASH-driven HCC: how useful are they? Cell Metab. 29, 18–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For the data underlying the meta-analyses reported in Fig. 3, see Supplementary Figure 1.