

Abstract
ADP-ribosyl cyclases are ubiquitous enzymes responsible for synthesis from NAD+ of the intracellular calcium-releasing signal molecules cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP+). Here, we show that cyclases from lower and higher Metazoa also synthesize three adenylic dinucleotides from cADPR and adenine: diadenosine diphosphate and two isomers thereof. These dinucleotides are present and metabolized in mammalian cells and affect intracellular calcium and cell proliferation. The diadenosine diphosphate isomers are naturally occurring nucleotides containing an N-glycosidic bond different from the usual C1′-N9. The identification of these members of the family of NAD+-derived, calcium-active nucleotides opens new areas of investigation into their functional cooperation with cADPR and NAADP+ and into their involvement in the physiology and pathology of calcium-controlled cell functions.
Keywords: adenylic dinucleotides, NAD+, cyclic ADP-ribose, cytotoxicity
ADP-ribosyl cyclases (ADPRCs) are a family of enzymes present from protists and unicellular algae to higher Metazoa and Metaphyta. They convert NAD+ to cyclic ADP-ribose (cADPR), a universal intracellular Ca2+ mobilizer from intracellular stores (1). Cytosolic Ca2+ movements are perhaps the most ancient and universal signaling system in cell physiology: this fact, and the presence of ADPRCs throughout the living kingdom, suggest involvement of these enzymes in the regulation of pivotal Ca2+-controlled cell functions. Indeed, this proved to be the case as concerns Ca2+-controlled processes of increasing complexity from lower to higher Metazoa and Metaphyta: cell-cycle progression in protists (2), oocyte fertilization in echinoderms (3), filtration in sponges (4), tissue regeneration in hydroids (5), stomatal closure in higher plants (6), stem cell proliferation (7), muscle contraction (8), hormone secretion (9), and other cell responses (10) in mammals. In these various cell functions, cADPR plays a general role as a signal molecule relating cell function to environmental conditions: light exposure in protists and hydroids (2, 5), water potential in plants (6), and hormone and glucose concentration in mammals (9, 11). Significant sequence homologies have indeed been observed between the ADPRCs from lower and higher Metazoa (12), suggesting that present-day cyclases may have evolved from an ancient, environmental stress-activated enzyme, producing signal molecules active on the single most influential intracellular parameter, the cytosolic free calcium concentration ([Ca2+]i).
cADPR is not the only product of ADPRC activity: hydrolysis of cADPR, which is catalyzed to a variable extent by all ADPRCs, produces ADPR, which has been recently shown to activate specific Ca2+ channels on the plasma membrane of mammalian cells (13). Cyclases from lower and higher Metazoa also produce nicotinic acid adenine dinucleotide (NAADP+) by way of a “base exchange reaction” substituting the nicotinamide moiety of NADP+ with nicotinic acid: this reaction has been reported to take place only at acidic pH values (pH 4-5) (14). Microinjected NAADP+ induces Ca2+ release from specific stores in several cell types (15). Moreover, cADPR, ADPR, and NAADP+, together with inositol 1,4,5-trisphosphate (IP3), cooperate in eliciting Ca2+ responses, and the mechanisms affecting the relative concentrations of these Ca2+ agonists inside the cell (cyclase regulation and compartmentation, substrate availability) are the subject of intense research (10).
Here, we add pieces to this intriguing puzzle, extending the family of Ca2+-active nucleotides produced by ADPRCs from lower and higher Metazoa to include three adenylic homodinucleotides, which are synthesized from cADPR in the presence of adenine (Ade): diadenosine diphosphate (Ap2A) and two isomers thereof [hence-forth called peak (P) 18 and P24 from their HPLC retention time]. Ap2A has been described in platelet secretory granules (16) and in cardiac myocytes (17), but the enzyme responsible for its synthesis was unknown. The Ap2A isomers contain an unusual N-glycosidic bond between one of the Ade and the ribose (involving N1 and N3 in P18 and P24, respectively), making them naturally occurring nucleotides featuring an N-glycosidic bond different from the “usual” C1′-N9. P18, P24, and Ap2A are present in ADPRC-positive human HeLa cells and modify the [Ca2+]i of intact HeLa when applied extracellularly: P18 inducing a decrease, P24 and Ap2A inducing an increase of the [Ca2+]i. Because the coculture of human hemopoietic progenitors (HP) over ADPRC-transfected stromal layers has recently been shown to stimulate or to inhibit HP proliferation, depending on the amount of cyclase activity expressed by the stroma (18), the effects of P18, P24, and Ap2A on the “in vitro” proliferation of human HP were investigated. Results indicate that these dinucleotides can stimulate (Ap2A) or inhibit (P18 and P24) colony growth at submicromolar concentrations and may thus be part of the network of growth-stimulatory and -inhibitory signals between ADPRC-positive hemopoietic stroma (19) and HP in the bone marrow.
Materials and Methods
ADP-Ribosyl Cyclases (ADPRCs). The ADPPRC from the marine sponge Axinella polypoides was purified to electrophoretic homogeneity as described in ref. 4. The purified ADPRCs from the mollusc Aplysia californica was purchased from Sigma. Human recombinant CD38 was kindly provided by Hon Cheung Lee (University of Minnesota, Minneapolis). Human HeLa cells transfected with human CD38, sense (CD38+) and antisense (CD38-), were obtained as described in ref. 20 and were maintained in DMEM supplemented with 10% FCS, penicillin, and streptomycin (complete medium) at 37°C under a 5% CO2 humidified atmosphere.
Production and Purification of P18, P24, and P31. The purified ADPRCs from A. polypoides was used for production of P18 and P24 for structural and functional studies. In a total volume of 0.4 ml of 10 mM Tris·HCl (pH 7.0), purified cyclase (0.05 mg) was incubated with 1 mM cADPR (or NAD+), 5 mM Ade, and 2 mM MgCl2 at 25°C. cADPR (or NAD+) and Ade were resupplemented every 6 h, while every 24 h fresh ADPRCs was added and an aliquot of the incubation was deproteinized with trichloroacetic acid (TCA) (5% final concentration) and analyzed by HPLC (analytical phosphate) to control peak production. After 3-5 days, the incubation was TCA-extracted, and 250-μl aliquots were injected into the preparative phosphate HPLC analysis (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). P18, P24, and P31 were collected, dried in a RotoVapor (Büchi, Uster, Switzerland), redissolved in deionized water, and injected into the preparative formate HPLC (see Supporting Materials and Methods). Eluted peaks were collected, dried, and stored at -20°C; no degradation was detected over several months of storage.
The HPLC-purified peaks produced by the Axinella cyclase were used for “in vitro” experiments on several cell types, for the enzymatic production of the mononucleotides and nucleosides (see below), and as internal or external HPLC standards to identify the dinucleotides in cell extracts, and their absorption spectra were stored in the HPLC spectra library for reference.
Ca2+ Measurements. Adherent (on 20-mm coverslips) CD38+ and CD38- HeLa cells were incubated in the presence of 1 μM P18, P24, or Ap2A (P31) for 18 h. Cells were then washed, incubated with 6 μM Fura 2-acetoxymethyl ester (FURA 2-AM) in standard saline (135 mM NaCl/5.4 mM KCl/1.8 mM CaCl2/1 mM MgCl2/5 mM Hepes/10 mM glucose, pH 7.4) for 45 min at 37°C, and washed several times with standard saline. Ca2+ measurements were performed in a 200-μl recording chamber mounted on an inverted microscope, as described in detail in ref. 20. After determination of the basal [Ca2+]i, 1 μM thapsigargin was added to induce Ca2+ release from thapsigargin-sensitive stores. The parameter values needed for calibration were obtained at the end of each measurement as described in ref. 20. During overnight incubation, limited (≤30%) dinucleotide degradation to adenine and hypoxanthine occurred in CD38+ cell cultures.
Cell-Proliferation Assays. Cord blood-derived mononuclear cells (CB MNC) were isolated by density gradient centrifugation, washed, and resuspended in complete medium (21). Cells (106 per ml) were incubated for 24 h in the presence of the various dinucleotides at concentrations ranging from 0.4 to 20 μM: thereafter, aliquots of cells (1-2 × 104) were seeded in hemopoietic growth factor-supplemented semisolid medium (MethoCult, Stem Cell Technologies, Vancouver). After 2 weeks, colonies were microscopically identified according to standard criteria (CFU-E, CFU-GM, and CFU-GEM) and counted: each colony identifies a colony-forming cell (CFC) (21).
In another set of experiments, CB MNC were cultured in the medium conditioned by CD38+/- HeLa cells. The 3-day medium conditioned by confluent monolayers of CD38+/- HeLa was incubated in the absence (control) or presence of nucleotide pyrophosphatase (NPP) (0.5 units/ml) for 6 h at 37°C. Thereafter, the pH of both samples was brought to 9.0 by addition of 5 N NaOH, and the NPP-treated medium was further digested with calf intestinal phosphatase (CIP) (2 units/ml) for 6 h at 37°C, whereas the control medium was incubated for the same time without additions. Finally, the pH of both media (control and NPP/CIP-digested) was restored to 7.5. CB MNC (106 per ml) were cultured for 10 days in 70% conditioned medium (CM) plus 30% fresh, complete medium, with one medium change after 5 days. Thereafter, aliquots of cells (2-5 × 104) were seeded in semisolid medium to evaluate colony growth.
The effect of P18 and of P24 on the proliferation of several cell lines was determined with the MTT-reduction assay (22). Briefly, cells were seeded at 3 × 104 per well in 96-well, flat-bottom tissue-culture plates and cultured in complete medium in the presence of various concentrations (ranging from 0.4 to 20 μM) of the purified dinucleotides. After 24 h, 10 μl of a 5 mg/ml solution of MTT was added to each well, the plates were further incubated for 2 h, the supernatant was removed from each well, and the blue crystals formed by reduction of the dye were dissolved in 200 μl per well DMSO. The optical absorbance at 570 nm was determined with a Bio-Rad plate reader (22).
Results and Discussion
Production and Degradation of P18, P24, and P31 by ADPRCs from Lower and Higher Metazoa. ADPRCs from Axinella polypoides (porifera, demospongiae) and from Aplysia californica (molluscs) and the human cyclase CD38 all produced three unidentified peaks from cADPR and Ade. Fig. 5, which is published as supporting information on the PNAS web site, shows representative chromatograms obtained from incubations with the purified cyclases from A. polypoides, A. californica, and CD38 (Fig. 5 A-C, respectively) and with intact human HeLa cells transfected with human CD38 (Fig. 5D, blue chromatogram): the new products were identified by their HPLC retention times as peak (P) 18, P24, and P31. Similar results were obtained when the cyclases were incubated with NAD+ and Ade (data not shown), whereas no peak formation was observed in the absence of cyclase activity [i.e., without addition of the purified enzymes or with HeLa transfected with antisense CD38 (Fig. 5D, red chromatogram)]. Optimal conditions for peak production were similar for all cyclases, i.e., neutral pH and an Ade/cADPR (or an Ade/NAD+) ratio of 10 or higher. Zn2+ significantly inhibited peak formation at 2 mM, whereas other cations (Ca2+,Mg2+) were without effect. Quantitative differences were observed between the various ADPRCs, regarding both the synthesis and the degradation of P18, P24, and P31. In Table 3, which is published as supporting information on the PNAS web site, the absolute values of the peak-forming and -degrading activities are compared with the cADPR-forming and -hydrolyzing activities for each enzyme. The invertebrate enzymes had much higher ADPRC and peak-forming specific activities, compared with recombinant human CD38. None of the purified ADPRC was able to hydrolyze P31, whereas the dinucleotide was degraded by intact CD38+ HeLa, but not by CD38- HeLa, with production of Ade and hypoxanthine. The invertebrate ADPRCs did not hydrolyze P24 (a minor activity was detectable only at pH 4), which was a poor substrate also to CD38. P18 was hydrolyzed to Ade and ADPR by all enzymes, with the exception of the Axinella cyclase, which converted P18 to cADPR and Ade, a feature that was exploited to develop an assay for the detection of P18 in cell lysates (see below).
Because the specific activities of the enzymes varied over a very wide range [almost 4 logs (Table 3)], the peak-forming activities were also expressed as a percentage of the cyclase or of the hydrolase activity displayed by each ADPRC (Table 1). Peak production apparently correlated better with the hydrolase activity than with the cyclase, as the relevant percentage values for the various enzymes varied within a narrower range (0.3-50% of the hydrolase activity as compared with 0.004-36% of the cyclase activity). This consideration, together with the fact that dinucleotide synthesis does occur from cADPR, i.e., independently of the cyclization reaction, suggests that the catalytic mechanism of peak formation is similar to the hydrolysis of cADPR, with Ade substituting for water. In the presence of Ade, peak production appears to be a significant percentage of the cyclase activity of CD38, both the recombinant, soluble enzyme (36%) and the native transmembrane protein expressed on transfected cells (31%). Because of the very high specific activity of the ADPRCs purified from A. polypoides (Table 3), this enzyme was utilized to produce the new dinucleotides for structure determination.
Table 1. Peak production relative to cyclase or to hydrolase activity of various ADPRC.
| Percentage cyclase or hydrolase
|
|||
|---|---|---|---|
| P18 | P24 | P31 | |
| CD38 (recombinant) | 36 | 11 | 6.0 |
| 24 | 0.7 | 0.4 | |
| HeLa CD38+* intact cells | 31 | 26 | 2.4 |
| 4.5 | 3.7 | 0.3 | |
| A. polypoides | 0.02 | 0.05 | 0.004 |
| 18 | 50 | 3.6 | |
| A. californica | 0.08 | 0.06 | 0.11 |
| 6.9 | 5.8 | 10.4 | |
Production of P18, P24, and P31 from cADPR and adenine by the indicated ADPRC is expressed as a percentage of their cyclase or of their cADPR-hydrolase (bold numbers) activity. Mean results from at least three experiments are shown; SD ≤ 15% of the mean value.
No synthesis or degradation detectable in CD38– 3T3
Structure Determination of P18, P24, and P31. UV spectral analysis and MS of the nucleotide fragments obtained by enzymatic digestion. P31 coeluted in HPLC with standard Ap2A and showed an identical absorbance spectrum; thus, it was tentatively identified as Ap2A (Ap2A*). P18 and P24 had been observed to be sensitive to acid hydrolysis, yielding Ade and ADPR, both identified by their HPLC coelution and spectral identity with standard compounds: this fact suggested the presence of an “unusual,” acid-sensitive N-glycosidic bond in the structure of P18 and P24. P18, P24, and P31, produced by incubation of Axinella cyclase with cADPR and Ade and HPLC-purified, were digested with NPP: the products were separated by HPLC (Fig. 6, which is published as supporting information on the PNAS web site). NPP digestion (see Supporting Materials and Methods) of P18 yielded a peak at 8 min (P8) and a peak coeluting with, and having the same absorbance spectrum as, standard AMP and thus tentatively identified as AMP (AMP*) (Fig. 6A); digestion of P24 yielded P11 and AMP* (Fig. 6B); hydrolysis of P31 produced AMP* only (Fig. 6C), in line with the tentative identification of P31 with Ap2A. These results demonstrated the dinucleotide structure of P18, P24, and P31. The mononucleotides obtained by NPP digestion of P18, P24, and P31 were subjected to UV spectral analysis and MS.
Comparison of the UV spectra (at pH 5.0) of the mononucleotides derived by NPP digestion of P18, P24, and P31 showed λmax values of 260 nm for AMP*, 264 nm for P8, and 274 nm for P11 (Fig. 7B, which is published as supporting information on the PNAS web site), similar to those reported for N9-methyl-Ade and its isomers, N1-methyl-Ade and N3-methyl-Ade, respectively (23). The pH is known to differently affect the absorbance spectrum of the various methyl-adenine isomers: whereas the λmax value of N1-methyl adenine changes considerably over a pH range from 5 to 11, λmax values of N9- and N3-methyl-adenine remain unchanged (23). Thus, we compared the effect of pH on λmax of P8, P11, and AMP*. Whereas the λmax of P8 shifted from 264 nm at pH 5 to 272 nm at pH 11 (Fig. 8A, which is published as supporting information on the PNAS web site), the λmax of P11 remained unchanged (at 274 nm) at all pH values (Fig. 8B), as did the λmax of AMP* and of standard AMP [at 260 nm (data not shown)]. These results were consistent with P8 and P11 being N1-isoAMP and N3-isoAMP, respectively.
MS analysis of the products of NPP digestion of P18, P24, and P31 yielded an identical mass (m/z) of 346.6 for each mononucleotide (P8, P11, and AMP*), identical to that of standard AMP (Fig. 9, which is published as supporting information on the PNAS web site). Moreover, the dinucleotides themselves showed an m/z value of 675.3, identical to that of standard Ap2A (data not shown).
Based on these results, P31 can be identified as Ap2A, because of the identity of mass, elution time, and absorbance spectrum of the dinucleotide, and of the mononucleotide resulting from NPP digestion, with the relative standard (Ap2A and AMP). P18 and P24 are isomers of Ap2A, where one of the N-glycosidic bonds between Ade and ribose is different from the usual C1′-N9, probably involving N1 in P18 and N3 in P24.
From the area of known amounts of standard AMP, the conversion factor between peak area and nmol was calculated for P18 and P24 and for the iso-AMP moieties derived therefrom by NPP digestion.
NMR spectral analyses. To identify the nitrogen atom involved in the N-glycosidic bond in the iso-AMP moieties obtained by NPP digestion of P18 and P24, the mononucleotides P8 and P11 (see Fig. 6 A and B) were dephosphorylated to obtain the corresponding nucleosides, which were then analyzed by NMR spectroscopy. Both P8 and P11 proved to be resistant to 5′-nucleotidase (which conversely readily dephosphorylated standard AMP), but they were substrate of alkaline phosphatase (CIP). Thus, the sequential digestion of P24 and of P18 with NPP and CIP yielded two nucleosides for each dinucleotide, as summarized:
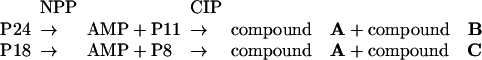 |
Compound A was easily identified as adenosine (Ado) because its 1H and 13C NMR spectra were identical to those of an authentic sample of Ado. On the other hand, 1H (Fig. 10A, which is published as supporting information on the PNAS web site) and 13C NMR spectra of compound B were different from the corresponding spectra of A, more closely resembling those reported for 3-isoadenosine (3-β-d-ribofuranosyladenine). Unfortunately, NMR data reported in the literature (24-26) for 3-isoadenosine are rather incomplete and, in some cases, conflicting, most likely because of the effect of pH and concentration changes on NMR resonances. Thus, to unambiguously deduce the chemical structure of compound B, a detailed 2D NMR analysis appeared to be necessary. Thus, the 2D 1H-1H COSY spectrum, whose cross-peaks correlate protons showing scalar coupling, was instrumental to assign the proton resonances of the sugar spin systems, while the gradient-heteronuclear single quantum correlation (g-HSQC) spectrum allowed the association of the proton resonances with those of the directly attached carbon atoms. Finally, the heteronuclear multiple bond correlation experiment, showing correlation peaks in correspondence of proton and carbon atoms separated by two or three bonds, provided essential information to clarify the chemical structure of compound B. In particular, the key 3JH-C heteronuclear multiple bond correlations H-8/C-5 and H-2/C-6 and those of the anomeric proton H-1′ with both C-2 and C-4 appeared to be unambiguously indicative of the linkage of the sugar unit at N-3, thus establishing the 3-isoadenosine nature of compound B. The complete 1H and 13C NMR assignment for compound B is reported in Table 4, which is published as supporting information on the PNAS web site.
A similar approach was used to deduce the structure of compound C, obtained through NPP and CIP digestion of P18 followed by HPLC separation of the products. 1H (Fig. 10B) and 13C NMR spectra of compound C appeared to be different from those of both compounds A and B, and thus, also in this case, structural analysis was guided by combined inspection of 2D NMR spectra. In particular, the g-HSQC spectrum (Fig. 10C) allowed the association of all of the proton resonances (the sugar signals and the two singlets at δH 7.66 and 7.21) with those of the relevant carbon atoms. On the other hand, the 3JH-C heteronuclear multiple bond correlation (Fig. 10D) correlations H-2/C-6, H-8/C-4, H-8/C-5, and those of the anomeric proton H-1′ with both C-2 and C-6 indicated the linkage of the sugar moiety at N-1. Because the sugar unit can be confidently assigned as a β-ribofuranose on the basis of comparison of its NMR data with those reported in the literature, compound C must be 1-isoadenosine (1-β-d-ribofuranosyladenine). Isolation of 1-isoadenosine is a particularly remarkable result, because a literature survey revealed that this molecule has never been obtained before from either a biological material or a synthetic procedure. The complete 1H and 13C NMR assignment for 1-isoadenosine (C) is reported in Table 4.
The deduced structures of the isoadenosine moieties obtained by enzymatic digestion of P18 and of P24 are shown in Fig. 1.
Fig. 1.

Structures of the isoadenosine moieties derived by NPP and CIP digestion of P18 and of P24. N3-isoadenosine, from P24N; N1-isoadenosine, from P18.
Metabolism and Presence of P18, P24, and Ap2A in ADPRC-Positive Cells. Indirect evidence that the dinucleotides produced by cyclase activity may play a physiological role comes from the observation that peak degradation takes place in ADPRC+ but not ADPRC- HeLa cells (Table 3). In CD38- cells, uptake of exogenously added (15 μM) P18, P24, and Ap2A could be observed: undetectable at zero time, their intracellular concentration increased over 5 h at a similar rate of ≈2.4 pmol/min/mg of protein for all dinucleotides. Intact CD38+ cells hydrolyzed P18, P24, and Ap2A with extracellular production of Ade and hypoxanthine: ADPR was not detectable, probably because of its uptake and rapid recycling into the adenylic nucleotide pool. In lysates from CD38+ cells, peak degradation occurred at a 20-fold higher rate than in intact cells, but yielded the same products, i.e., Ade and hypoxanthine, together with N3-iso-AMP, in the presence of P24. These results indicate that P18, P24, and Ap2A can cross the plasma membrane of intact cells and that the “uncommon” N-glycosidic bonds present in P18 and in P24 are hydrolyzed, both extracellularly and intracellularly. The fact that recombinant human CD38 does not hydrolyze Ap2A (Table 3) may be due either to structural differences between the purified enzyme and the one expressed on transduced cells (the former is unglycosylated and lacks the transmembrane and short intracellular domain) or to the presence, in CD38+ cells, of other enzymes, induced by CD38 expression and active on the dinucleotides. The latter hypothesis seems to be supported by the presence of N3-iso-AMP in lysates from CD38+ but not CD38- cells, incubated with P24, indicating the presence of a pyrophosphatase activity, unknown of CD38. Interestingly, N3-iso-AMP proved to be resistant to 5′-nucleotidase but to be a substrate of myokinase, which readily phosphorylated it in the presence of ATP to the corresponding nucleotide diphosphate, raising the possibility of an intracellular biosynthetic metabolism of N3-iso-ADP in CD38+ cells. Lysates from A. polypoides cells also showed a pyrophosphatase activity on P24 and a hydrolyzing activity on Ap2A, yielding Ade and ADPR: instead, the purified cyclase did not show any hydrolyzing activity on either substrate (Table 3), suggesting the presence in sponge cells of other enzymes active on these dinucleotides. Synthesis and degradation (i.e., metabolism) of P18, P24, and Ap2A in ADPRC+ cells from sponges and mammals prompted us to investigate the presence of the dinucleotides in these cells.
In acid extracts from A. polypoides cells, P18, P24, and Ap2A were indeed detected by HPLC at 0.32 ± 0.06, 1.79 ± 0.2, and 0.15 ± 0.02 nmol/mg, respectively (mean ± SD from three determinations). The isolated peaks were identified by comparison of retention time and absorbance spectrum with standards (obtained with the purified Axinella cyclase) and by HPLC analysis of the products of their digestion with NPP (see Supporting Materials and Methods). These values are in the range of the cADPR concentration in A. polypoides tissue (0.24 ± 0.06 nmol/mg) (4).
P24 was also detected by HPLC analysis (see Supporting Materials and Methods) in acid extracts of CD38+ but not CD38- HeLa cells, at a concentration of 9.8 ± 0.9 pmol/mg, i.e., in the same order of magnitude as that of cADPR (50 ± 0.2 pmol/mg). P24 identification was simplified by its peculiar UV spectrum (Fig. 7A) and by that of the N3-iso-AMP derived from it by NPP digestion (Fig. 7B).
Positive identification of P18 in CD38+ HeLa cells by HPLC proved more difficult, probably because of its lower intracellular concentration compared to P24. Thus, a modification of the cycling assay used to measure the cADPR concentration in cell extracts (27) was developed, taking advantage of (i) the resistance of P18 to NAD-ase treatment (a step required during the cycling assay) and (ii) the ability of the Axinella cyclase to convert P18 into cADPR and Ade (Table 3) and cADPR into NAD+ in the presence of excess nicotinamide (similarly to the Aplysia cyclase).
P18 was detected in acid extracts from CD38+ but not CD38- HeLa cells (Fig. 11C, which is published as supporting information on the PNAS web site), at 0.14 pmol/mg, a concentration 40 times lower than that of P24. The reason for this may lie in the higher pyrophosphatase activity displayed by CD38+ cells on P18 compared with P24 (Table 3). Ap2A has already been described in platelet secretory granules (16) and in rat cardiac myocytes (17). Notably, both cell types are cyclase-positive (28, 29). The extremely low synthesizing activity displayed by CD38+ HeLa (Table 3) may account for our failure to determine its intracellular concentration by HPLC.
Effects of P18, P24, and Ap2A on the [Ca2+]i of CD38+ and CD38- HeLa Cells. Thus, in ADPRC+ cells from sponges and mammals, production of the Ca2+ mobilizer cADPR is accompanied by the synthesis of three adenine homodinucleotides: this prompted us to investigate their effects on the [Ca2+]i in the presence or absence of cADPR. Again, we took advantage of the experimental cell system provided by CD38+/- HeLa cells: in these cells, the basal [Ca2+]i and the content of the thapsigargin-sensitive Ca2+ stores [i.e., the endoplasmic reticulum (ER)] (30) were measured after 18 h of incubation with each of the three membrane-permeant (see above) adenine dinucleotides. The results obtained are shown in Table 2. In CD38+ cells, the [Ca2+]i was 60% higher, and ER stores contained 50% less calcium, compared with CD38- cells, owing to the Ca2+-releasing effect of cADPR, as reported in ref. 20. Incubation with 1 μM P18 induced a decrease of the basal [Ca2+]i and of the ER Ca2+ stores, which was more pronounced in CD38+ HeLa cells (43% and 48% of control values, for basal and ER Ca2+, respectively) compared with CD38- HeLa cells (52% and 78% of control values, respectively; see Fig. 12, which is published as supporting information on the PNAS web site). Because mitochondrial calcium does not appear to be increased by P18 in CD38+/- 3T3 (S.B., unpublished data), these results suggest an overall depletion of cell calcium, whose mechanism remains to be elucidated. CD38+ cells are more sensitive than CD38- cells to the ER-depleting effect of P18 (Table 2), possibly because of the concomitant Ca2+-releasing action of cADPR.
Table 2. Effects of P18, P24, and Ap2A on the [Ca2+]i and on the content of thapsigargin-sensitive Ca2+ stores in CD38+/- HeLa cells.
| [Cai2+], nM
|
||||
|---|---|---|---|---|
| Cells | Control | P18 | P24 | Ap2A |
| HeLa CD38+ | 37 | 16 | 57 | 79 |
| 254 | 123 | 203 | 184 | |
| HeLa CD38– | 23 | 12 | 44 | 35 |
| 470 | 366 | 290 | 398 | |
Basal [Ca2+]i and thapsigargin-induced Ca2+ release (bold numbers) were measured on FURA 2-loaded CD38+ and CD38– HeLa incubated in the absence (control) or in the presence of 1 M dinucleotides for 18 hr. During this time, partial (≤30%) dinucleotide degradation occurred in CD38+ cell cultures, with production of Ade and hypoxanthine (see Table 3). Mean results from at least eight different determinations are shown; SD ≤ 15% of the mean value.
Conversely, P24 induced an increase of the basal [Ca2+]i, which was higher in CD38- than in CD38+ cells (191% vs. 154%, respectively; see Fig. 12) and was accompanied by a depletion of the ER stores, more pronounced in CD38- cells (60% vs. 80%). This rules out a synergistic effect of cADPR and P24 and suggests instead a different mechanism of action for the two agonists (see below). The effects of P24 are slightly more pronounced on CD38- cells, possibly because of the absence of intracellular degradation (Table 3).
Finally, Ap2A induced an increase of the [Ca2+]i and a depletion of the ER stores; both effects were more evident in CD38+ cells (213% vs. 152% and 72% vs. 85%, respectively; see Fig. 12). Ap2A is known to act synergistically with cADPR in rat brain, skeletal muscle cells, and cardiac muscle cells by increasing the sensitivity of the ryanodine receptor to the cyclic nucleotide (31); these results are in line with this observation.
The short-term effects of the three dinucleotides on HeLa cell calcium were also explored (Fig. 13, which is published as supporting information on the PNAS web site). The addition of 1 μM P18 induced a slow and progressive [Ca2+]i decrease, starting from addition of the dinucleotide. P24 at 1 μM conversely induced a steady [Ca2+]i increase, which was completely prevented by extracellular EGTA, indicating that the dinucleotide induces extracellular Ca2+ influx. This, in turn, could trigger intracellular Ca2+ release, justifying the partial depletion of the ER stores observed after long-term incubation of the cells (see above). Qualitatively similar results were observed with P18 and P24 on CD38+/- cells. Finally, 1 μM Ap2A induced a slow and steady [Ca2+]i increase, which was unaffected by extracellular EGTA. The short-term effect of Ap2A was detectable only in CD38+ HeLa cells, in agreement with the described synergism of Ap2A with cADPR (31) and with the more marked [Ca2+]i increase observed in CD38+ compared to CD38- HeLa cells after overnight incubation (Fig. 12).
Effects of P18, P24, and Ap2A on Hemopoietic Cell Proliferation. cADPR, both exogenously added and paracrinally produced by ADPRC+ stroma, has been demonstrated to behave as a hemopoietic growth factor, stimulating (via Ca2+-mediated mechanisms) the proliferation of committed (CFCs) as well as of uncommitted [long-term culture-initiating cells (LTC-IC) and stem cells] HP (7, 18, 21, 32). The facts that the dinucleotides described here are produced together with cADPR by ADPRC+ cells and that they affect cell Ca2+ (see above) prompted us to investigate their functional effects on the proliferation of human HP “in vitro.” P18 and P24 strongly inhibited CFC growth: IC50 values were 1.0 and 0.18 μM, respectively (Fig. 2). The iso-AMP moieties of P18 and of P24 showed similar toxicity as the parent dinucleotide, whereas the corresponding iso-adenosines were significantly less toxic (Fig. 2): IC50 values were 37 and 5 μM for the iso-adenosine from P18 and from P24, respectively. Prolonging the incubation time increased toxicity; e.g., IC50 for P18 was 30 μM with 2 h of incubation and 1 μM with 24 h of incubation. This fact suggests a slow influx of the dinucleotides into sensitive cells. Excess adenosine (200-fold) had no protective effect on CFCs, indicating that toxicity of the dinucleotides is not due to an antimetabolic effect of the iso-adenosine or iso-AMP moieties of the dinucleotides.
Fig. 2.

Effects of P18 and P24 and of their respective iso-adenosines on CFC growth. CB MNC were incubated in the presence of various concentrations of P18 (filled triangles), P24 (filled squares), and the isoadenosine moiety derived from P18 (open triangles) and from P24 (open squares) by enzymatic (NPP plus CIP) digestion (see Supporting Materials and Methods). After 24 h, aliquots of cells (1-2 × 104) were seeded in semisolid medium to allow for colony growth. Results are expressed as percentage of inhibition of colony growth compared with controls, which were incubated with the same dilution of chromatographic buffer as present in the HPLC-purified compounds. The IC50 values were determined from the logarithmic regression curves (R ≥ 0.97). Results are the mean from at least three different experiments.
Toxicity of P18 and of P24 was also determined on a number of different human cell lines. The hemopoietic cell lines HL60 and K252 proved to be somewhat less sensitive than CFCs to P18 and P24 (IC50 values were 5-40 times higher) but still significantly more sensitive compared to the nonhemopoietic cell lines, where IC50 values were 50-200 times higher (Table 5, which is published as supporting information on the PNAS web site). No significant differences were observed regarding toxicity of P18 and P24 between CD38+ and CD38- HeLa cells (Table 5), indicating that cADPR, which is present in the ADPRC+ cells but not in the ADPRC- cells, does not protect from the toxic effects of the dinucleotides. These results suggest a peculiar sensitivity of hemopoietic cells, particularly the CFCs, to the toxic effect of these dinucleotides.
Ap2A, on the contrary, increased CFC output similarly to cADPR and showed a synergistic effect with the cyclic nucleotide at suboptimal concentrations of both compounds (Fig. 3). Ap2A also stimulated growth of the more immature HP, the long-term culture-initiating cells (LTC-IC): priming for 24 h with 1 μM Ap2A doubled LTC-IC output compared with untreated controls, similarly to 1 μM cADPR (2.0 ± 0.3 vs. 2.0 ± 0.2, respectively; n = 4).
Fig. 3.

Synergistic effect of Ap2A and cADPR on CFC growth. CB MNC were incubated in the absence (control) or presence of Ap2A (open bars), cADPR (gray bars), or of both nucleotides together (black bars) at the indicated concentrations for 24 h. Aliquots of cells were then seeded in semisolid medium, and colonies (A) and the total cell number (B) were scored after 2 weeks of culture. Results are the mean from three separate experiments (SD ≤ 16% of the mean values).
Production of growth-inhibiting P18 and P24 by CD38+ 3T3, along with growth-stimulating cADPR, may be in part responsible for the inhibition of CFC and LTC-IC output observed after long-term coculture (2-5 weeks) of human HP over this feeder (21). To test this hypothesis, we took advantage of the fact that NPP and CIP digestion substantially reduce P18 and P24 toxicity on HP (40 and 30 times, respectively; Fig. 2), whereas cADPR is resistant to these enzymes (27). Thus, human CB MNC were cultured for 2 weeks in the medium conditioned by CD38+ or CD38- HeLa cells, previously incubated with NPP and CIP: colony output from HP cultured in the enzyme-treated CD38+ CM increased 10- and 5-fold, respectively, compared with that from HP cultured in the untreated CD38+ CM and in the enzyme-digested CD38- CM (Fig. 4). Thus, enzyme treatment of the CD38+ CM enabled its stimulatory effect on CFC growth, afforded by cADPR, to become apparent: this observation suggests that toxic dinucleotides are involved in the growth inhibitory effect of the CD38+ CM on CFCs. This observation may bear some relevance in the clinical setting, where infiltration of the bone marrow by activated (CD38+) lymphocytes has been advocated in the pathogenesis of bone-marrow-failure syndromes (aplasia and graft failure) (33).
Fig. 4.

NPP and CIP digestion of the medium conditioned by CD38+/- HeLa cells removes toxic effect on CFCs. The 3-day CM from confluent monolayers of CD38+/- HeLa cells was incubated in the absence (control; open bars) or presence (filled bars) of NPP and CIP (see Supporting Materials and Methods). CB MNC (1 × 106 per ml) were incubated for 10 days in 70% CM (30% fresh, complete medium), with one medium change after 5 days. Aliquots of cells (1-2 × 104) were then seeded in semisolid medium, and colony growth was estimated after 2 weeks.
In conclusion, here we demonstrate that ADP-ribosyl cyclases from porifera, molluscs, and mammals produce three adenine dinucleotides from cADPR and adenine: Ap2A and two isomers thereof. Conservation of this catalytic property from lower to higher Metazoa suggests an important physiological role of these molecules. The Ap2A isomers contain unusual N-glycosidic bonds (C1′-N1 and C1′-N3 in P18 and P24, respectively), thus representing naturally occurring adenine nucleotide isomers in animal cells. These dinucleotides share the following properties: (i) they are present and metabolized in ADPRC+ but not ADPRC- cells; and (ii) in both cell types, they affect cell Ca2+ and are cytotoxic, although a causal relationship between these effects remains to be established. Synthesis of these dinucleotides requires the presence of Ade: interestingly, in A. polypoides, cyclase activity colocalizes with a strong ATP-hydrolyzing activity that produces Ade and ribose-5′-triphosphate (34). In CD38+ HeLa cells, Ade was detected at 0.8 ± 0.1 nmol/mg of protein, a concentration 16 times higher than that of cADPR (0.05 ± 0.01 nmol/mg; n = 5). An Ade/cADPR ratio of 10 or more ensures optimal dinucleotide production by the purified cyclases (see above): the presence of P18 and of P24 in acid extracts from ADPRC+ sponge and human cells indicates that their biosynthesis indeed occurs in both cell types.
The identification of these members of the family of Ca2+-active nucleotides opens a new area of investigation into their metabolism, their mechanism of action, their functional cooperation with other products of ADP-ribosyl cyclase activity (cADPR and NAADP+), and their involvement in the physiology and pathology of Ca2+-controlled cell functions.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Associazione Italiana per la Ricerca sul Cancro, the Italian Ministry of Education, University and Scientific Research, the Fondazione Cassa di Risparmio di Genova e Imperia, and the CIPE Project of the Italian Ministry of Economy to the Advanced Biotechnology Center.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ADPRC, ADP-ribosyl cyclase; Ap2A, diadenosine diphosphate; [Ca2+]i, cytosolic free calcium concentration; cADPR, cyclic ADP-ribose; CB MNC, cord blood-derived mononuclear cells; CFC, colony-forming cell; CIP, calf intestinal phosphatase; CM, conditioned medium; ER, endoplasmic reticulum; HP, hemopoietic progenitors; NPP, nucleotide pyrophosphatase.
References
- 1.Lee, H. C., Walseth, T. F., Bratt, G. T., Hayes, R. N. & Clapper, D. L. (1989) J. Biol. Chem. 264, 1608-1615. [PubMed] [Google Scholar]
- 2.Masuda, W., Takenaka, S., Tsuyama, S., Tokunaga, M., Inui, H. & Miyatake, K. (1997) FEBS Lett. 405, 104-106. [DOI] [PubMed] [Google Scholar]
- 3.Galione, A., Lee, H. C. & Busa, W. B. (1991) Science 253, 1143-1146. [DOI] [PubMed] [Google Scholar]
- 4.Zocchi, E., Carpaneto, A., Cerrano, C., Bavestrello, G., Giovine, M., Bruzzone, S., Guida, L., Franco, L. & Usai, C. (2001) Proc. Natl. Acad. Sci. USA 98, 14859-14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puce, S., Basile, G., Bavestrello, G., Bruzzone, S., Cerrano, C., Giovine, M., Arillo, A. & Zocchi, E. (2004) J. Biol. Chem. 279, 39783-39788. [DOI] [PubMed] [Google Scholar]
- 6.Leckie, C. P., McAinsh, M. R., Allen, G. J., Sanders, D. & Hetherington, A. M. (1998) Proc. Natl. Acad. Sci. USA 95, 15837-15842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Podestà, M., Pitto, A., Figari, O., Bacigalupo, A., Bruzzone, S., Guida, L., Franco, L., De Flora, A. & Zocchi, E. (2003) FASEB J. 17, 310-312. [DOI] [PubMed] [Google Scholar]
- 8.Franco, L., Bruzzone, S., Song, P., Guida, L., Zocchi, E., Walseth, T. F., Crimi, E., Usai, C., De Flora, A. & Brusasco, V. (2001) Am. J. Physiol. 280, L98-L106. [DOI] [PubMed] [Google Scholar]
- 9.Takasawa, S., Nata, K., Yonekura, H. & Okamoto, H. (1993) Science 259, 370-373. [DOI] [PubMed] [Google Scholar]
- 10.Lee, H. C. (2002) Cyclic ADP-ribose and NAADP: Structures, Metabolism, and Functions (Kluwer Academic, Dordrecht, The Netherlands).
- 11.Morita, K., Kitayama, S. & Dohi, T. (1997) J. Biol. Chem. 272, 21002-21009. [DOI] [PubMed] [Google Scholar]
- 12.Lee, H. C., Munshi, C. B. & Graeff, R. (2002) in Cyclic ADP-ribose and NAADP: Structures, Metabolism, and Functions, ed. Lee, H. C. (Kluwer Academic, Dordrecht, The Netherlands), pp. 23-43.
- 13.Perraud, A. L., Fleig, A., Dunn, C. A., Bagley, L. A., Launay, P., Schmitz, C., Stokes, A. J., Bessman, M. J., Penner, R., Kinet, J. P. & Scharenberg, A. M. (2001) Nature 411, 595-600. [DOI] [PubMed] [Google Scholar]
- 14.Aarhus, R., Graeff, R. M., Dickey, D. M., Walseth, T. F. & Lee, H. C. (1995) J. Biol. Chem. 270, 30327-30333. [DOI] [PubMed] [Google Scholar]
- 15.Churchill, G. C., Patel, S., Thomas, J. M. & Galione, A. (2002) in Cyclic ADP-ribose and NAADP: Structures, Metabolism, and Functions, ed. Lee, H. C. (Kluwer Academic, Dordrecht, The Netherlands), pp. 199-215.
- 16.Jankowski, J., Hagemann, J., Tepel, M., Van der Giet, M., Stephan, N., Henning, L., Gouni-Berth, S., Sachinidis, A., Zideck, W. & Schluter, H. (2001) J. Biol. Chem. 23, 8904-8909. [DOI] [PubMed] [Google Scholar]
- 17.Luo, J., Jankowski, J., Knobloch, M., Van der Giet, M., Gardanis, K., Russ, T., Vahlensiek, U., Neumann, J., Schmitz, W., Tepel, M., et al. (1999) FASEB J. 13, 695-705. [DOI] [PubMed] [Google Scholar]
- 18.Podestà, M., Benvenuto, F., Pitto, A., Figari, O., Bacigalupo, A., Bruzzone, S., Guida, L., Franco, L., Paleari, L., Bodrato, N., et al. (2005) J. Biol. Chem. 280, 5343-5349. [DOI] [PubMed] [Google Scholar]
- 19.Itoh, M., Ishihara, K., Tomizawa, H., Tanaka, H., Kobune, Y., Ishikawa, J., Kaisho, T. & Hirano, T. (1994) Biochem. Biophys. Res. Commun. 203, 309-317. [DOI] [PubMed] [Google Scholar]
- 20.Zocchi, E., Daga, A., Usai, C., Franco, L., Guida, L., Bruzzone, S., Costa, A., Marchetti, C. & De Flora, A. (1998) J. Biol. Chem. 273, 8017-8024. [DOI] [PubMed] [Google Scholar]
- 21.Zocchi, E., Podestà, M., Pitto, A., Usai, C., Bruzzone, S., Franco, L., Guida, L., Bacigalupo, A. & De Flora, A. (2001) FASEB J. 15, 1610-1612. [DOI] [PubMed] [Google Scholar]
- 22.Mosmann, T. (1983) J. Immunol. Methods 65, 55-63. [DOI] [PubMed] [Google Scholar]
- 23.Fujii, T. & Itaya, T. (1999) Heterocycles 51, 2255-2277. [Google Scholar]
- 24.Tindall, C. G., Robins, R. K., Tolman, R. L. & Hutzenlaub, W. (1972) J. Org. Chem. 37, 3985-3989. [DOI] [PubMed] [Google Scholar]
- 25.Nair, V., Buenger, G. S., Leonard, N. J., Balzarini, J. & De Clercq, E. (1991) J. Chem. Soc. Chem. Commun. 26, 1650-1651. [Google Scholar]
- 26.Miyaki, M. & Shimizu, B. (1970) Chem. Pharm. Bull. 18, 1446-1456. [Google Scholar]
- 27.Graeff, R. & Lee, H. C. (2002) Biochem. J. 361, 379-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torti, M., Festetics, E. T., Bretoni, A., Sinigaglia, F. & Balduini, C. (1998) FEBS Lett. 428, 200-204. [DOI] [PubMed] [Google Scholar]
- 29.Higashida, H., Egorova, A., Higashida, C., Zhong, Z. G., Yokoyama, S., Noda, M. & Zhang, J. S. (1999) J. Biol. Chem. 274, 33348-33354. [DOI] [PubMed] [Google Scholar]
- 30.Lytton, J., Westlin, M. & Hanley, M. R. (1991) J. Biol. Chem. 266, 17067-17071. [PubMed] [Google Scholar]
- 31.Holden, C. P., Padua, R. A. & Geiger, J. D. (1996) J. Neurochem. 67, 574-580. [DOI] [PubMed] [Google Scholar]
- 32.Podestà, M., Zocchi, E., Pitto, A., Usai, C., Franco, L., Bruzzone, S., Guida, L., Bacigalupo, A., Scadden, D. T., Walseth, T. F., et al. (2000) FASEB J. 14, 680-690. [DOI] [PubMed] [Google Scholar]
- 33.Young, N. S. (2000) Semin. Hematol. 37, 3-14. [DOI] [PubMed] [Google Scholar]
- 34.Reintamm, T., Lopp, A., Kuusksalu, A., Pehk, T. & Kelve, M. (2003) Eur. J. Biochem. 270, 4122-4132. [DOI] [PubMed] [Google Scholar]
- 35.Bradford, M. (1976) Anal. Biochem. 72, 248-252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.