

Abstract
The development of efficient and inexpensive genome sequencing methods has revolutionized the study of human bacterial pathogens and improved vaccine design. Unfortunately, the sequence of a single genome does not reflect how genetic variability drives pathogenesis within a bacterial species and also limits genome-wide screens for vaccine candidates or for antimicrobial targets. We have generated the genomic sequence of six strains representing the five major disease-causing serotypes of Streptococcus agalactiae, the main cause of neonatal infection in humans. Analysis of these genomes and those available in databases showed that the S. agalactiae species can be described by a pan-genome consisting of a core genome shared by all isolates, accounting for ≈80% of any single genome, plus a dispensable genome consisting of partially shared and strain-specific genes. Mathematical extrapolation of the data suggests that the gene reservoir available for inclusion in the S. agalactiae pan-genome is vast and that unique genes will continue to be identified even after sequencing hundreds of genomes.
Keywords: bacterial species, comparative genomics, group B Streptococcus
The most recent definition of a bacterial species comes from the pregenomic era. In 1987, it was proposed (1) that bacterial strains showing >70% DNA·DNA reassociation and sharing characteristic phenotypic traits should be considered to be strains of the same species. Today, this classical definition is being challenged by an increasing amount of genomic information, which, in theory, can more precisely describe bacterial species. Thus far, the genome sequence of one or two strains for each species has provided unprecedented information; however, the question of how many genomes are necessary to fully describe a bacterial species has yet to be asked. We have addressed this question by sequencing the genome of strains of each of the major pathogenic serotypes of Streptococcus agalactiae [group B Streptococcus (GBS)].
S. agalactiae is a leading cause of illness or death among newborn infants (2) and an emerging cause of invasive infection in the elderly (3, 4). Nine distinct capsular serotypes of GBS have been described; however, the major disease-causing isolates in the United States and Europe belong to only five serotypes: Ia, Ib, II, III, and V (5). Recently, the complete nucleotide sequences of a serotype III and a serotype V GBS isolate were reported (6, 7). To fully explore gene variability within the GBS species, we determined the complete genome sequence of the type Ia strain A909 and draft genome sequences (8× sequence coverage) of five additional strains, representing the five major serotypes. Comparative analysis of the six newly sequenced genomes and the two genomes already available in the databases suggests that a bacterial species can be described by its “pan-genome” (pan, from the Greek word π αν, meaning whole), which includes a core genome containing genes present in all strains and a dispensable genome composed of genes absent from one or more strains and genes that are unique to each strain. Surprisingly, unique genes were still detected after eight genomes were sequenced, and mathematical extrapolation predicts that new genes will still be found after sequencing many more strains. Thus, the genomes of multiple, independent isolates are required to understand the global complexity of bacterial species. Analysis of multiple GBS genomes was found to be instrumental for the development of vaccines (8) and for the functional characterization of important genetic determinants (9).
Materials and Methods
Sequenced Strains. All newly sequenced strains were deposited at American Type Culture Collection under the following accession numbers: A909, BAA-1138; CJB111, BAA-23; H36b, BAA-1174; 18RS21, BAA-1175; COH1, BAA-1176; and 515, BAA-1177. References for the eight strains are as follows: NEM316 (6); 2603V/R (7); A909, H36B, and 18RS21 (10); 515 (11); COH1 (12); and CJB111 (Carol Baker Collection, Division of Infectious Diseases, Baylor College of Medicine, Houston).
Sequencing, Annotation, and Unfinished Genomes. Genome sequences were generated by the whole-genome shotgun sequencing approach (13, 14). Draft genomes were sequenced to 8×-sequence coverage, and the sequences were assembled by using the Celera Assembler (Celera Genomics, Rockville, MD) (15). Contigs were ordered and oriented according to their alignment to strain 2603V/R by using promer (16). Ordered matching contigs were pasted together into a pseudochromosome, and nonmatching contigs were tacked on the end in random order. In the pseudochromosome, contigs were separated by the sequence NNNNNCATTCCATTCATTAATTAATTAATGAATGAATGNNNNN, which (i) generates a stop codon in all six reading frames so that no gene is predicted across junctions and (ii) provides a start site in all frames, pointing toward contigs to predict incomplete genes at their extremities. ORFs were predicted and annotated by using an automated pipeline that combines glimmer gene prediction (17, 18), ORF and non-ORF feature identification, and assignment of functional role categories to genes (14). Assembly of strain 18RS21 resulted in a higher number of contigs than for the other unfinished genomes, leading to the prediction of >3,500 genes. Many small contigs did not harbor protein-coding genes, and several were fragments of rRNAs or coded for tRNAs or structural RNAs.
Shared and Strain-Specific Genes. Each strain pair was compared by means of the following: (i) a Smith and Waterman protein search on all of the predicted proteins by using the ssearch program (version 3.4) (19, 20); (ii) a DNA search of all of the predicted ORFs of a strain against the complete DNA sequence of the other strain, by using the fasta program (version 3.4) (20); and (iii) a translated protein search of all of the predicted proteins of a strain against the complete DNA sequence of the other strain, by using the tfasty program (version 3.4) (20). A gene was considered conserved if at least one of these three methods produced an alignment with a minimum of 50% sequence conservation over 50% of the protein/gene length.
Core-Genome and Pan-Genome Extrapolation. The number of genes shared by all GBS isolates and the number of strain-specific genes depend on how many strains are taken into account. The sequential inclusion of up to eight strains was simulated in all possible combinations. The number (N) of independent measurements of the shared (see Fig. 2) and strain-specific genes (see Fig. 3) present in the nth genome is N = 8!/[(n – 1)!·(8 – n)!]. The size of the species core genome and the number of strain-specific genes for a large number of sequenced strains were extrapolated by fitting the exponential decaying functions Fc = κc exp[–n/τc] +Ω and Fs = κs exp[–n/τs] + tg(θ), respectively, to the amount of conserved genes (see Fig. 2) and of strain-specific genes (see Fig. 3), where n is the number of sequenced strains and κc, κs, τc, τs, Ω, and tg(θ) are free parameters. tg(θ) represents the extrapolated rate of growth of the pan-genome size, P(n), as a greater number of independent GBS strain sequences become available, i.e.,
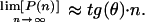 |
The Inset of Fig. 3 displays the measured size of the pan-genome as a function of n [in this case, N = 8!/(8 – n)!; points are obtained for each value of n] together with a plot of the calculated P(n) (see Supporting Text, which is published as supporting information on the PNAS web site).
Fig. 2.

GBS core genome. The number of shared genes is plotted as a function of the number n of strains sequentially added (see Materials and Methods). For each n, circles are the 8!/[(n – 1)!·(8 – n)!] values obtained for the different strain combinations. Squares are the averages of such values. The continuous curve represents the least-squares fit of the function Fc = κc exp[–n/τc] + Ω (see Eq. 1 in Supporting Text) to data. The best fit was obtained with correlation r2 = 0.990 for κc = 610 ± 38, τc = 2.16 ± 0.28, and Ω= 1,806 ± 16. The extrapolated GBS core genome size Ω is shown as a dashed line.
Fig. 3.

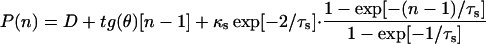 |
Synteny. Paralog clusters in each genome were generated by using the Jaccard algorithm (21), with ≥80% identity, and a Jaccard coefficient ≥0.6. Members of paralog clusters were then organized into ortholog clusters by allowing any member of a paralog cluster to contribute to the reciprocal best matches used to construct the ortholog clusters. Syntenic blocks are defined as a set of five or more consecutive pairs of genes from the same ortholog cluster. Because they do not participate in clusters, all contigs that do not contain protein-coding genes from the five draft genomes were searched against all genomes by using the nucmer program (16). Syntenic blocks and nucmer results were drawn (Fig. 1) by using sybil (http://sybil.sourceforge.net/).
Fig. 1.

Whole genome alignment of GBS strains. The eight genomes are compared to each other by using COG (41) and nucmer analyses (see Materials and Methods). Each genome (shaded strain name) is colored with a gradient that ranges from yellow (nucleotide 1) to blue (end). Differences in color between a reference sequence (the last colored line in each genome) and the other genomes indicate conserved protein-coding regions that have been rearranged. Uncolored segments denote coding regions in which no conserved genes were detected. nucmer matches for contigs that do not contain protein-coding genes are displayed by red blocks (matches within the reference strain are displayed on the line directly above it). Genomic islands of diversity are boxed and numbered “x.y,” where x is the panel or strain number where the island first appeared and y is the island location in that genome from left to right. A + indicates an island that was not identified in a previous genome. Islands that overlap by at least 50% (based on the number of shared genes) with previously identified islands receive the same number as the initial island. The gene content of the 69 islands identified is listed in Table 2, which is published as supporting information on the PNAS web site. Strain-specific regions, free of COG or nucmer matches, are displayed in black at the bottom of each panel. Portions of these regions that harbor protein-coding genes are indicated in gray below the black blocks. The curves on top of each panel represent the nucleotide composition (χ2 analysis) (see Materials and Methods) of the reference strain of the panel, and peaks indicate regions of atypical composition.
In Fig. 1, genomic islands of diversity >5 kb are predicted as follows: (i) strains are inspected from the top panel and down and from left to right on each panel; (ii) regions of at least 1 kb not shared with another strain are identified; (iii) regions are merged into single islands if they are within 5 kb of each other; and (iv) resulting islands >5 kb are considered. It should be noted that some islands are composed of more than one contig. Genomic islands discussed in the text are the following: the α-galactosidase region in strain H36B, island 7.4; the prophage region in strain H36B, island 7.5; the DNA restriction/modification system in strain 515, part of island 4.5; the Tn916 regions in strains 2603V/R, 515, CJB111, and COH1, islands 1.8 and the left side of 5.3; and serine-rich protein and glycosyl-transferases flanked by cell-wall-anchored proteins and sortases in strain COH1, unnumbered region between islands 6.5 and 1.15. Fig. 1 reveals many non-protein-coding regions in strain 18RS21 that display nucmer matches elsewhere in the 18RS21 genome. Most of these regions correspond to fragments of rRNAs, tRNAs, or structural RNAs, all of which exhibit an expected atypical nucleotide composition.
χ2 Analysis. Regions of atypical nucleotide composition were identified by the χ2 analysis; the distribution of all 64 trinucleotides (3mers) was computed for the complete genome in all six reading frames, followed by the 3mer distribution in 5,000-bp windows. Windows overlapped by 500 bp. For each window, the χ2 statistic on the difference between its 3mer content and that of the whole genome was computed. Peaks in Fig. 1 indicate regions of atypical nucleotide composition.
Results
General Features of GBS Genomes. Draft genome sequences (8× coverage) were obtained for strains 515, H36B, 18RS21, COH1, and CJB111, belonging, respectively, to serotypes Ia, Ib, II, III, and V, which are responsible for >90% of human infections in the United States. The full genome sequence was obtained for strain A909 of serotype Ia. With the exception of NEM316 and 515, both belonging to ST23, the other isolates also belong to different sequence types, as determined from recent multilocus sequence typing (MLST) studies (22) and likely represent the genetic diversity of the GBS species (see Table 1, which is published as supporting information on the PNAS web site). The six newly sequenced genomes and the genomes of strains 2603V/R and NEM316 already available in the databases were used for subsequent analysis. The eight strains revealed similar genome sizes and a similar number of predicted genes. The entire nucleotide sequences (pseudochromosomes) from all of the GBS strains were compared in all possible pairwise combinations with nucmer. The overall percent identity between pairs ranged from 85% to 95%, with no particular bias between coding and noncoding regions.
Fig. 1 summarizes the information derived from the comparison of the eight genomes. The isolates share a high degree of gene synteny interrupted by 69 genomic islands that are absent in at least one of the genomes (see Table 2, which is published as supporting information on the PNAS web site). Some of the genomic islands are characterized by an atypical nucleotide composition, suggesting possible acquisition by lateral exchange. Gene comparisons indicate that orthologs are highly conserved. Of the genes that have orthologs in each of the sequenced strains, 95% display sequence identity >90%.
GBS Core Genome. To estimate the number of genes present in every GBS strain (core genome), the number of shared genes found on sequential addition of each new genome sequence was extrapolated by fitting an exponential decaying function to the data (Fig. 2). The results of all permutations of the order of addition for each of the eight genomes are shown. As expected, the number of shared genes initially decreased with addition of each new sequence. Nevertheless, extrapolation of the curve indicates that the core genome reaches a minimum of 1,806 genes (95% confidence interval = 1,750–1,841) and will remain relatively constant, even as many more genomes are added (see Tables 3 and 4, which are published as supporting information on the PNAS web site). The actual number of shared genes in each genome varies because of duplicated genes and paralogs.
The GBS Pan-Genome Concept. To determine the global gene repertoire of the GBS bacterial species (GBS pan-genome), the number of new genes added by each genomic sequence was estimated (Fig. 3). As with the shared genes, the plot of the numbers of new genes was well fitted by a decaying exponential. The average number of new genes added by a novel sequence was 161 when a second genome was added, and this number decreased to 54 after five genomes; but, even the eighth genome continued to add new genes. Remarkably, the extrapolated curve reaches a nonzero asymptotic value of 33 new genes (95% confidence interval = 22–42) with increasing numbers of genomes (see Tables 5 and 6, which are published as supporting information on the PNAS web site). In other words, the model predicts that for every new GBS genome sequenced, an average of 33 new strain-specific genes will be identified and added to the pan-genome. Although the confidence interval is rather large, the probability that this average number would be zero is smaller than 6 × 10–4. This finding suggests that the GBS pan-genome is open and that its size grows with the number of independent strains sequenced (Fig. 3 Inset). To verify whether an open pan-genome model is unique to GBS, we repeated the analysis by using the complete sequence of five strains of Streptococcus pyogenes [group A Streptococcus (GAS)] and eight strains of Bacillus anthracis, which are known to have different levels of genomic diversity (data not shown). As in the case of GBS, each additional GAS genome added an average of 27 new genes to the pool, leading to an open pan-genome. In the case of B. anthracis, the number of specific genes added to the pan-genome dropped to zero after the addition of a fourth strain. This result probably reflects the fact that B. anthracis is a highly clonal, recently evolved species in which genome variability is associated only with virulence plasmids (23, 24). Alternatively, the sequenced strains may belong to the same evolutionary clade and may not adequately represent the B. anthracis species.
Genome Diversity Is Independent of Capsular Serotype. Convenient phenotypic traits, such as agglutination by specific antisera against the capsular polysaccharide surrounding bacterial cells, have been widely used to classify bacteria within the same species, and this information has been used for epidemiology, vaccine design, and therapy. Recently, MLST analysis based on fragments of seven conserved core genes indicated that the GBS serotype does not fully correlate with actual evolutionary relationships (7, 25). To characterize the genetic relationship between the eight genomes of GBS isolates, a dendrogram was drawn according to the distribution of genes across the strains (Fig. 4). Among the genomes compared, two belong to serotype Ia (515 and A909), two are serotype III (NEM316 and COH1), and two are serotype V (2603 and CJB111). Furthermore, strains 515 and NEM316 belong to the same ST type (ST23). Comparative analysis of the strains' gene content (see Table 7, which is published as supporting information on the PNAS web site) showed that strains of different serotypes and different MLST type often share a higher number of genes than strains of the same serotype, resulting in a serotype-independent clustering of the eight strains. In support of this conclusion, global genome comparisons at the nucleotide level indicate that strains from two different but related serotypes, type Ia strain 515 and type Ib strain H36B, were the least conserved, with 85% identity over 90% of the genome, whereas the two most conserved strains, 2603V/R and COH1 (95% identity over 96% of the genome), belong to two distinct serotypes (type V and III, respectively) and to two different MLST types.
Fig. 4.

Dendrogram of the eight GBS genomes. Shared gene information was used to cluster proteins into groups by using the single-linkage method of the program cluster (http://rana.lbl.gov). Groups were then converted into profiles of presence or absence of each gene (0 or 1) in the eight GBS strains and used as input to paup* 4.0b10 (Sinauer, Sunderland, MA) for dendrogram drawing and bootstrapping. Numbers at the nodes indicate bootstrap values. Serotypes and MLST types of each strain are within parentheses.
Functional Classification of the Core and Dispensable Genes. Genes belonging to core and dispensable genomes have been classified according to their predicted functional role (see Fig. 5, which is published as supporting information on the PNAS web site). As expected, the vast majority of genes making up the core genome belong to the groups of housekeeping functions, the cell envelope, regulatory functions, and transport and binding proteins. About one-third of the shared genes fall into the class of hypothetical proteins and proteins of unknown function, however, suggesting that many aspects of basic GBS biology still need to be explored. Although genes associated with housekeeping functions are also found within the dispensable genome, they are not as well represented there, whereas hypothetical genes and genes of unknown function represent the vast majority of the dispensable genome. Furthermore, genes associated with mobile and extrachromosomal elements are particularly abundant in this group, supporting the hypothesis that the majority of specific traits depend on lateral gene transfer events. Nevertheless, this class of genes is poorly represented within the core genome, indicating that only a few rearrangements have remained stable during the evolution of GBS.
Origin of Genomic Islands and Strain-Specific Genes. The eight sequenced GBS genomes revealed 358 genes found only in a single strain. Of these genes, 137 belong to NEM316, 61 to H36B, 47 to 2603V/R, 35 to COH1, 31 to 515, 20 to CJB111, 14 to A909, and 13 to 18RS21 (see Table 8, which is published as supporting information on the PNAS web site). Many of the strain-specific genes are in genomic islands, which, although they do not have the classical features of pathogenicity islands, are often flanked by insertion elements and display an atypical nucleotide composition, suggesting possible acquisition through horizontal transfer (Fig. 1).
Acquisition of traits from other pathogens may contribute to the virulence of GBS strains. For example, a strain-specific locus in type III strain COH1, encoding the preprotein translocase SecA and SecY subunits, three glycosyltransferases, and a highly repetitive Ser-rich cell-wall-anchored protein, displays remarkable similarity to a genomic island present in Streptococcus pneumoniae TIGR4 (SP1757–SP1772). A second COH1-specific island encodes sortases and three cell-wall-anchored proteins. Of these proteins, COH1_ORF01523 corresponds to adhesin Spb1, a serotype III-specific protein implicated in adhesion and invasion of epithelial cells (26), whereas the other two (COH1_ORF01521 and COH1_ORF01524) are similar to a fimbrial subunit and to internalin A of Listeria monocytogenes, respectively. Another region shared by strain H36B and S. pneumoniae contains an α-galactosidase (H36B_ORF00495) and a system for transport and metabolism of sugar that may allow H36B to degrade and transport host α-galactosides.
As in the case of S. pyogenes, phage-associated genes account for 10% of all strain-specific genes in GBS. For example, type Ib isolate H36B contains a 41-kb prophage element (H36B_ORF00576–H36B_ORF00630) that displays strong mosaicism and contains a protein (H36B_ORF00630) similar to the S. pyogenes phage-associated pyrogenic exotoxin C. Homology searches revealed that, besides the streptococci, other more distant species are involved in exchanging genetic material with GBS strains. One intact copy of the 18-kb conjugative plasmid Tn916 of Enterococcus faecium (27), which encodes tetracycline resistance as well as determinants necessary for its own movement, was detected at >95% identity in GBS isolates 2603V/R, 515, CJB111, and COH1, suggesting a recent acquisition of this trait.
Genomic Variation in Gene Expression. Phase variation is an important mechanism by which bacteria can modulate their life style and virulence in response to external stimuli, stress conditions, and adaptation to different niches (28–30). Such variation occurs by altering the length of short, repeated DNA tracts within or immediately upstream of coding regions (contingency genes), resulting in frame-shifts and affecting protein synthesis. A recent study (31) indicated that an important virulence-associated gene in GBS is regulated by phase variation. With the availability of the genome sequence of multiple strains of GBS, it is possible to identify DNA tracts likely associated with contingency genes and to determine how these repeats vary in the other genomes, allowing direct evaluation of their potential role in phase variation.
Potential phase variation-associated repeats in the form of homopolymeric tracts of nucleotides, as well as dinucleotide, trinucleotide, and polynucleotide repeats, were searched in the 2603V/R genome (32). Among the 602 repeats identified, 17 were divergent in at least one of the other GBS genomes (see Table 9, which is published as supporting information on the PNAS web site). Of the 17 proteins potentially affected by the presence of phase-variable repeats, 11 are predicted to be surface-associated.
Discussion
After sequencing multiple strains of GBS, we found that eight genomes are not enough to identify all genes present in this species, and mathematical modeling made the surprising prediction that even hundreds of genomes might not be sufficient. These findings have implications for pathogenesis, vaccine design, evolution, and the concept of species and suggest that the research strategies for microbial genomes may need to be reconsidered.
The Bacterial Pan-Genome. Regression analysis showed that in the case of GBS and GAS, the bacterial pan-genome is vast because new genes continue to be added to the gene pool of the species any time a new strain is sequenced. In this view, the core genome would then represent only a small fraction of the pan-genome. This theory challenges our concept of limited variability within a bacterial species, as has been suggested recently (33), and raises the question of whether such large numbers of genes are actually available. More accurate estimates of the size of the pan-genome should be possible once the sequences of a much greater number of GBS genomes become available.
Nevertheless, the prediction of an open pan-genome is not surprising if we consider that sequencing a few hundred liters of water from the Sargasso Sea identified 1.2 million previously unknown genes from 1,800 predicted genomic species (34) and that gastrointestinal microbial flora contains almost 400 different bacterial phylotypes (35). These results suggest that the environmental gene pool available for inclusion by mechanisms such as horizontal transfer, transposition, and transformation is much larger than previously estimated. Finally, if we consider that there are 1031 bacteriophages on earth (36), which infect 1024 bacteria per second, we can imagine that a continuous flow of genetic material occurs between bacteria sharing the same environments. In contrast, species living in restricted environments and lacking mechanisms of gene exchange may have evolved with considerably less variation. An example is the obligate intracellular endosymbiont of aphids, Buchnera aphidicola, in which no chromosome rearrangements or gene acquisitions have occurred in the past 50–70 million years (37). Other species may have closed pan-genomes because they occupy an isolated niche or have a low capacity to acquire foreign genes. For instance, the pan-genome of B. anthracis can be fully described by four genomes only. Hence, analysis of the pan-genome structure of a pathogen may give important insights into the biology of the species and open new avenues to cure disease.
Serotype Classification Does Not Reflect Genetic Diversity. By comparative analysis of the eight GBS isolates, we conclude that the classical and convenient typing of bacteria on the basis of their capsular polysaccharide composition does not reflect the genetic diversity of the species. In fact, strains belonging to different serotypes can be more closely related than strains of the same serotype. The genetic selection and maintenance of strongly conserved structural motifs in the polysaccharide-repeat units of all nine capsule serotypes, such as the αNeupNAc-(2→3)-Galp, that are required for evasion of host-mediated immune responses are selected independently of other factors driving GBS diversity (38). Indeed, it has been shown that the horizontal transfer of as little as one gene between two GBS serotypes can lead to seroconversion of the polysaccharide capsule (39). More surprising is the fact that even other commonly used strain classification methods, such as MLST, do not reflect the real genetic diversity described by the whole genome analysis. These observations may suggest that the attempts to identify more or less virulent lineages on the basis of serotype or core-genome-based methods fail to take into account the variable genome in which many of the virulence-related genes might reside.
Implications for Bacterial Taxonomy. Methods commonly used to define bacterial species (DNA·DNA reassociation, 16S rRNA typing, MLST, etc.) rely mostly on features associated with the core genome (40). Our work confirms that the essence of the species is linked to the core genome. However, the majority of the genetic traits linked to virulence, capsular serotype, adaptation, and antibiotic resistance pertain to the dispensable genome. Therefore, sequencing of multiple strains is necessary to understand the virulence of pathogenic bacteria and to provide a more consistent definition of the species itself. We identified species with an open pan-genome, such as GBS and GAS, and species with a closed pan-genome, such as B. anthracis. Nevertheless, a different interpretation of the same data may lead to the conclusion that the present definition of bacterial species is inconsistent because, in reality, only species with an open pan-genome are species, whereas B. anthracis is not a true genetic species on its own, but only a clone of Bacillus cereus, with very distinctive phenotypic traits provided by the acquisition of the virulence plasmid coding for the anthrax toxin.
Concluding Comment. Our data clearly show that the strategy to sequence one or two genomes per species, which has been used during the first decade of the genomic era, is not sufficient and that multiple strains need to be sequenced to understand the basics of bacterial species. The methods presently used to evaluate the species diversity, such as complete genome hybridization and MLST, can explain only the presence, absence, and variability of the genetic loci that are already known and do not provide information on the genes that are not present in the reference genome. Our work provides a clear demonstration that, by these approaches, we fail to include in the analysis the entire dispensable genome, the size of which can be vastly larger than the core genome. Our work on the protein-based vaccine against GBS has shown that this is not just a theoretical disadvantage but has very important practical consequences because a universal vaccine is possible only by including dispensable genes (8).
Supplementary Material
Acknowledgments
We thank Antonello Covacci for help with the pan-genome concept, Robert Janulczyk for help with the phase-variable repeats analysis, and David Rasko and Jacques Ravel for providing access to their unpublished Bacillus sequence data. We also thank Hean Koo, Seth Schobel, and Martin Shumway for sequence data management; Martin Wu for help with phylogeny; and The Institute for Genomic Research (TIGR) information technology and database server groups led by Vadim Sapiro and Michael Heaney, respectively. This work was supported by Chiron Corporation, National Institutes of Health Grants U01-AI50909 (to H.T., A.L.J., and C.E.R.), AI42940 (to M.R.W.), and AI38424 (to L.C.M.), and the Charles Hood Foundation (M.J.C.).
Author contributions: R.R. designed research; H.T., V.M., M.J.C., C.D., D.M., N.L.W., S.V.A., J.C., A.L.J., A.S.D., R.T.D., T.M.D., M.M., M.S., I.M.y.R., J.D.P., C.R.H., J.P.S, W.C.N., R.M., L.M.B., R.J.D., M.J.R., S.A.S., S.C.D., D.H.H., J.S., M.L.G., L.Z., N.Z., H.K., D.R., G.D., K.W., K.J.B.O., S.S., T.R.U., C.E.R., and G.G. performed research; H.T., V.M., C.D., D.M., O.W., L.C.M., D.L.K., and M.R.W. analyzed data; and H.T., V.M., M.J.C., C.D., D.M., N.L.W., J.L.T., C.M.F., and R.R. wrote the paper.
Abbreviations: GBS, group B Streptococcus; MLST, multilocus sequence typing; GAS, group A Streptococcus.
Data deposition: The sequences reported in this paper have been deposited in the DDBJ/EMBL/GenBank database [accession nos. AAJO01000000 (18RS21), AAJP01000000 (515), AAJQ01000000 (CJB111), AAJR01000000 (COH1), AAJS01000000 (H36B), and CP000114 (A909)].
References
- 1.Wayne, L., Brenner, D., Colwell, R., Grimont, P., Kandler, O., Krichevsky, L., Moore, L., Moore, W., Murray, R., Stackebrandt, E., et al. (1987) Int. J. Syst. Bacteriol. 37, 463–464. [Google Scholar]
- 2.Schuchat, A. & Wenger, J. D. (1994) Epidemiol. Rev. 16, 374–402. [DOI] [PubMed] [Google Scholar]
- 3.Tyrrell, G. J., Senzilet, L. D., Spika, J. S., Kertesz, D. A., Alagaratnam, M., Lovgren, M. & Talbot, J. A. (2000) J. Infect. Dis. 182, 168–173. [DOI] [PubMed] [Google Scholar]
- 4.Harrison, L. H., Elliott, J. A., Dwyer, D. M., Libonati, J. P., Ferrieri, P., Billmann, L. & Schuchat, A. (1998) J. Infect. Dis. 177, 998–1002. [DOI] [PubMed] [Google Scholar]
- 5.Lin, F. Y., Clemens, J. D., Azimi, P. H., Regan, J. A., Weisman, L. E., Philips, J. B., III, Rhoads, G. G., Clark, P., Brenner, R. A. & Ferrieri, P. (1998) J. Infect. Dis. 177, 790–792. [DOI] [PubMed] [Google Scholar]
- 6.Glaser, P., Rusniok, C., Buchrieser, C., Chevalier, F., Frangeul, L., Msadek, T., Zouine, M., Couve, E., Lalioui, L., Poyart, C., et al. (2002) Mol. Microbiol. 45, 1499–1513. [DOI] [PubMed] [Google Scholar]
- 7.Tettelin, H., Masignani, V., Cieslewicz, M. J., Eisen, J. A., Peterson, S., Wessels, M. R., Paulsen, I. T., Nelson, K. E., Margarit, I., Read, T. D., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 12391–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maione, D., Margarit, I., Rinaudo, C. D., Masignani, V., Mora, M., Scarselli, M., Tettelin, H., Brettoni, C., Iacobini, E. T., Rosini, R., et al. (2005) Science 309, 148–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lauer, P., Rinaudo, C. D., Soriani, M., Margarit, I., Maione, D., Rosini, R., Taddei, A. R., Mora, M., Rappuoli, R., Grandi, G., et al. (2005) Science 309, 105. [DOI] [PubMed] [Google Scholar]
- 10.Lancefield, R. C., McCarty, M. & Everly W. N. (1975) J. Exp. Med. 142, 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wessels, M. R., Paoletti, L. C., Rodewald, A. K., Michon, F., DiFabio, J., Jennings, H. J. & Kasper, D. L. (1993) Infect. Immun. 61, 4760–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson, C. B. & Weaver, W. M. (1985) J. Infect. Dis. 152, 323–329. [DOI] [PubMed] [Google Scholar]
- 13.Fleischmann, R. D., Adams, M. D., White, O., Clayton, R. A., Kirkness, E. F., Kerlavage, A. R., Bult, C. J., Tomb, J. F., Dougherty, B. A., Merrick, J. M., et al. (1995) Science 269, 496–512. [DOI] [PubMed] [Google Scholar]
- 14.Tettelin, H. & Feldblyum, T. V. (2004) in Genomics, Proteomics and Vaccines, ed. Grandi, G. (Wiley, London), pp. 45–73.
- 15.Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., Kravitz, S. A., Mobarry, C. M., Reinert, K. H., Remington, K. A., et al. (2000) Science 287, 2196–2204. [DOI] [PubMed] [Google Scholar]
- 16.Delcher, A. L., Phillippy, A., Carlton, J. & Salzberg, S. L. (2002) Nucleic Acids Res. 30, 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delcher, A. L., Harmon, D., Kasif, S., White, O. & Salzberg, S. L. (1999) Nucleic Acids Res. 27, 4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salzberg, S. L., Delcher, A. L., Kasif, S. & White, O. (1998) Nucleic Acids Res. 26, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith, T. F. & Waterman, M. S. (1981) J. Mol. Biol. 147, 195–197. [DOI] [PubMed] [Google Scholar]
- 20.Pearson, W. R. (1999) in Bioinformatics Methods and Protocols., eds. Misener, S. & Krawetz, S. A. (Humana, Totowa, NJ), pp. 185–219.
- 21.Jaccard, P. (1908) Bull. Soc. Vaudoise Sci. Nat. 44, 223–270. [Google Scholar]
- 22.Jones, N., Bohnsack J. F., Takahashi, S., Oliver, K. A., Chan, M. S., Kunst, F., Glaser, P., Rusniok, C., Crook, D. W., Harding, R. M., et al. (2003) J. Clin. Microbiol. 41, 2530–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keim, P., Price, L. B., Klevytska, A. M., Smith, K. L., Schupp, J. M., Okinaka, R., Jackson, P. J. & Hugh-Jones, M. E. (2000) J. Bacteriol. 182, 2928–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacchi, C. T., Whitney, A. M., Mayer, L. W., Morey, R., Steigerwalt, A., Boras, A., Weyant, R. S. & Popovic, T. (2002) Emerging Infect. Dis. 8, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies, H. D., Jones, N., Whittam, T. S., Elsayed, S., Bisharat, N. & Baker, C. J. (2004) J. Infect. Dis. 189, 1097–1102. [DOI] [PubMed] [Google Scholar]
- 26.Adderson, E. E., Takahashi, S., Wang, Y., Armstrong, J., Miller, D. V. & Bohnsack, J. F. (2003) Infect. Immun. 71, 6857–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flannagan, S. E., Zitzow, L. A., Su, Y. A. & Clewell, D. B. (1994) Plasmid 32, 350–354. [DOI] [PubMed] [Google Scholar]
- 28.Henderson, I. R., Owen, P. & Nataro, J. P. (1999) Mol. Microbiol. 33, 919–932. [DOI] [PubMed] [Google Scholar]
- 29.van der Woude, M. W. & Baumler, A. J. (2004) Clin. Microbiol. Rev. 17, 581–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wren, B. W. (2000) Nat. Rev. Genet. 1, 30–39. [DOI] [PubMed] [Google Scholar]
- 31.Puopolo, K. M. & Madoff, L. C. (2003) Mol. Microbiol. 50, 977–991. [DOI] [PubMed] [Google Scholar]
- 32.Tettelin, H., Nelson, K. E., Paulsen, I. T., Eisen, J. A., Read, T. D., Peterson, S., Heidelberg, J., DeBoy, R. T., Haft, D. H., Dodson, R. J., et al. (2001) Science. 293, 498–506. [DOI] [PubMed] [Google Scholar]
- 33.Konstantinidis, K. T. & Tiedje, J. M. (2005) Proc. Natl. Acad. Sci. USA 102, 2567–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., et al. (2004) Science 304, 66–74. [DOI] [PubMed] [Google Scholar]
- 35.Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., Gill, S. R., Nelson, K. E. & Relman, D. A. (2005) Science 308, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendrix, R. W. (2003) Curr. Opin. Microbiol. 6, 506–511. [DOI] [PubMed] [Google Scholar]
- 37.Tamas, I., Klasson, L., Canback, B., Naslund, A. K., Eriksson, A. S., Wernegreen, J. J., Sandstrom, J. P., Moran, N. A. & Andersson, S. G. (2002) Science 296, 2376–2379. [DOI] [PubMed] [Google Scholar]
- 38.Cieslewicz, M. J., Chaffin, D., Glusman, G., Kasper, D., Madan, A., Rodrigues, S., Fahey, J., Wessels, M. R. & Rubens, C. E. (2005) Infect. Immun. 73, 3096–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaffin, D. O., Beres, S. B., Yim, H. H. & Rubens, C. E. (2000) J. Bacteriol. 182, 4466–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stackebrandt, E., Frederiksen, W., Garrity, G. M., Grimont, P. A., Kampfer, P., Maiden, M. C., Nesme, X., Rossello-Mora, R., Swings, J., Truper, H. G., et al. (2002) Int. J. Syst. Evol. Microbiol. 52, 1043–1047. [DOI] [PubMed] [Google Scholar]
- 41.Tatusov, R. L., Koonin, E. V. & Lipman, D. J. (1997) Science 278, 631–637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.