

Abstract
New guidelines for the definitions of steatotic liver disease have named the entity of metabolic dysfunction and alcohol-associated liver disease (MetALD) as an overlap condition of metabolic dysfunction–associated steatotic liver disease (MASLD) and alcohol-associated liver disease. There is a broad range of therapeutics in all stages of development for MASLD, but these therapeutics, in general, have not been studied in patients with significant ongoing alcohol use. In this review, we discuss the current understanding of the endogenous and exogenous risks for MASLD and MetALD. Rational strategies for therapeutic intervention in MetALD include biopsychosocial interventions, alcohol use cessation strategies, including the use of medications for alcohol use disorder, and judicious use of therapeutics for steatotic liver disease. Therapeutics with promise for MetALD include incretin-based therapies, FGF21 agonists, thyroid hormone receptor beta agonists, sodium-glucose co-transporter 2 inhibitors, and agents to modify de novo lipogenesis. Currently, glucagon-like peptide 1 receptor agonists and peroxisome proliferator–activated receptor γ agonists have the largest body of literature supporting their use in MASLD, and there is a paucity of agents in trials for alcohol-associated liver disease. From existing studies, it is not clear if unique therapeutics or a combinatorial approach are needed for MetALD. Further elucidation of the safety and benefits of MASLD-related therapies is of paramount importance for advancing therapeutics for MetALD in carefully designed inclusive clinical trials.
INTRODUCTION
Steatotic liver disease (SLD) is increasingly prevalent in the population, now estimated to affect at least 30% of the adult population, with increasing prevalence in the pediatric population as well. Meanwhile, alcohol use in amounts ranging from any to significant consumption impacts liver disease in individuals with SLD. The most recent American Association For the Study of Liver Diseases (AASLD) guidelines define the entity of metabolic dysfunction and alcohol-associated liver disease (MetALD) as a dual etiology SLD in individuals who have evidence of liver steatosis and meet cardiometabolic criteria for metabolic dysfunction–associated steatotic liver disease (MASLD) while also consuming significant amounts of alcohol defined as > 140 g/wk for females or > 210 g/wk for males. The relative contribution of alcohol is assumed to be dependent on the degree of excess alcohol, ranging from MASLD predominant to alcohol-associated liver disease (ALD) predominant. ALD is defined by alcohol intake > 350 g/wk for females and 420 g/wk for males, regardless of cardiometabolic risk factors.[1] Importantly, MetALD differs from ALD based on the quantity of alcohol consumed. Patients can have ALD without additional cardiometabolic risk factors, but MetALD requires the presence of cardiometabolic risks and alcohol consumption that does not meet the threshold for ALD. MASLD, ALD, and MetALD are chronic, progressive, and potentially lead to liver cirrhosis. However, they are vastly different from acute alcohol-associated hepatitis (AH), which is characterized by acute onset of significant inflammatory liver injury in the presence or absence of cardiometabolic factors. AH presents with significant derangements of liver function and high mortality after diagnosis.[2]
The distinction between MetALD and ALD exists because metabolic disease increases the risk of steatohepatitis and fibrosis in ALD. Conversely, any alcohol consumption increases the risk of fibrosis when MASLD or metabolic dysfunction-associated steatohepatitis (MASH) are present.[1] Etiologically, MASLD and the spectrum of ALD have significant behavioral overlap, as excess use of alcohol and excess caloric intake are key factors in the development of MetALD.[3] However, the risk for progressive disease is guided by more than just caloric excess and alcohol use. Individuals start with an inherent set of genetic risks that intersect with other factors such as diet and microbial diversity, environmental exposures, social determinants of health, and even epigenetic transmission of multigenerational risks of metabolic disease, all of which function as disease modifiers. When SLD is driven by both alcohol and metabolic dysfunction, the question arises about which medical interventions may have benefits for MetALD? It also highlights the question of whether new therapeutics are needed for MetALD as a separate entity or if a combinatorial approach may be effective. This review focuses on the elaboration of the modifiable and non-modifiable risks for adults with MetALD and how therapeutic options for MASLD may be applicable to MetALD.
RISK FACTORS AND PATHOPHYSIOLOGY
The risk modifiers that contribute to developing MetALD encompass both distinct and shared features of MASLD development and AUD. Risk factors for MASLD are primarily metabolic, whereas risk factors for alcohol consumption and AUD are often behavioral and associated with complex psychosocial factors. The primary risk factor for AUD is the consumption of alcohol in quantities and patterns that lead to health problems, which can include binge drinking and chronic heavy drinking. Genetic predisposition, psychiatric comorbidities, and social and environmental factors also contribute to the risk of AUD.[4–6] MASLD is primarily associated with cardiometabolic risk factors, including overweight or obesity, type 2 diabetes mellitus (T2DM), dyslipidemia, and hypertension These conditions contribute to the development of MASLD through mechanisms such as insulin resistance, lipotoxicity, and sterile inflammation. Other conditions that impact insulin resistance, nutrient handling, and lipotoxicity, such as hypothyroidism, polycystic ovary syndrome, hypopituitarism, hypogonadism, and pancreatoduodenal resection, have also been associated with an increased prevalence of MASLD.[7] Risk factors with less well-understood etiological contribution include psoriasis, environmental toxins, alterations in the gut microbiome, and various polygenic risks.
At the core of steatosis in MASLD is an imbalance of lipogenic and lipolytic pathways guided by hormonal and dietary control of transcription factors. A major regulator of these pathways is the conserved lipogenic transcription factor sterol regulatory element binding protein 1. Lipid oxidation genes are regulated through the master regulator peroxisome proliferator–activated receptor (PPAR)α; the fate of newly synthesized long chain fatty acids is guided by the interplay between these master regulators and are in turn regulated by peroxisome proliferator–activated gamma coactivator 1 α and β. The interplay between insulin and glucagon signal liver–related responses to starvation and energy excess through AMP-activated protein kinase, which controls malonyl CoA levels in the cell through acetyl-CoA carboxylase (ACC), the ratio of malonyl CoA to intracellular long chain fatty acid help determine if free fatty acids are directed to mitochondrial oxidation or the formation of triacylglycerol.[8,9]
ALD and steatosis secondary to alcohol are mediated by the direct and metabolic effects of alcohol. Alcohol metabolism alters the NADH:NAD ratio leading eventually to metabolic inhibition of fatty acid oxidation. Alcohol’s metabolism by alcohol dehydrogenase and the microsomal p450 system generates acetaldehyde and reactive oxygen species which are mainly quenched through glutathione.[10] When alcohol consumption is chronic and excessive, this leads to depletion of glutathione and imbalances in toxic metabolites, leading to cumulative effects of organelle damage by toxic protein adducts, lipid peroxides, and reduced mitochondrial function. Chronic alcohol-associated hepatocellular injury is sufficient to lead to immune cell activation, steatohepatitis, and liver fibrosis over time.[11]
SLD is intertwined with energy and lipid storage. Adipocyte lipolysis is essential for the mobilization of stored triacylglycerides into free fatty acids, which are transported to the liver. Insulin resistance leads to increased adipocyte lipolysis in MASLD, and the catecholamine-mediated effects of ethanol on adipocyte lipolysis contribute to steatosis in ALD. However, interrupting adipocyte lipolysis in murine models protects against hepatocyte injury and steatosis in models of MASLD and binge alcohol use. Recent studies have demonstrated hepatocyte to HSC nutrient–driven communication, which may play a role in the increased fibrosis risk observed in MetALD. Altogether, these data suggest that adipocyte lipolysis may play a conserved foundational role in SLD, particularly MetALD, and provide a potential therapeutic target.[12–14]
MetALD IN THE SPECTRUM OF MASLD AND ALD
The defining factors for MetALD are the presence of MASLD with concomitant alcohol use above the threshold of 20 grams daily or 140 g/wk for women and 30 grams daily or 210 g/wk for men. Importantly, MetALD is a diagnosis born out of consensus; studies defining thresholds for the effects of alcohol use are variable, as are recommendations about alcohol use between international societies. Longitudinal studies in multinational cohorts show that light and moderate alcohol consumption is associated with worsening liver outcomes in patients with MASLD.[15] Population-based longitudinal data in the Finnish cohort further suggest that dose-dependent increased mortality for alcohol consumption occurs at as low as 1 beverage/wk.[16] In the Japanese population, incident MASLD in women was associated with alcohol consumption in quantities > 140 g/wk.[17]. The new nomenclature recognizes many of the nuances of alcohol’s effects on SLD and, importantly, creates a defined group amenable to rational therapeutic design.[1]
The current definition of MetALD encompasses ongoing alcohol use, but patients may have variable consumption throughout their lives. Recent longitudinal studies to this effect suggest that prolonged abstinence from alcohol reduced the risk of decompensation over time. Thus, subjects who do not currently meet the criteria for MetALD may, by prior exposure, share mechanisms and risks of MetALD-related disease.[18]
Alcohol consumption itself is directly intertwined with the cardiometabolic risks that define MASLD. Excessive alcohol consumption exacerbates hypertension, dyslipidemia, insulin resistance, and obesity.[19] However, small amounts of alcohol have also been associated with favorable serum triglyceride levels.[20] Moreover, past and present alcohol use confounds the diagnosis of MASLD by directly influencing cardiometabolic risk factors, including but not limited to obesity, diabetes, hypertriglyceridemia, and hyperlipidemia; changes that may persist even after abstinence from alcohol.[21] When MASLD becomes MetALD or vice versa, and how the duration of abstinence required to reverse alcohol-associated effects will need further study.
GENETIC RISKS
Several dozen genetic loci have been attributed to increased risk of SLD in multiethnic cohorts.[22] Notable well-studied variants with clinical implications for both MASLD and ALD are polymorphisms in genes associated with lipid droplet and lipid metabolism, such as hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) and patatin-like phospholipase domain-containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2, membrane-bound O-acyltransferase domain-containing 7, and the glucokinase regulatory protein. Heterozygous mutations in the alpha 1 antitrypsin (AAT) gene may also act as disease modifiers in MASLD and ALD.[23]
Lipid droplet–associated protein HSD17B13 upregulation is strongly associated with steatotic disease and clinical outcomes. Meanwhile, polymorphisms resulting in reduced function and expression are associated with reduced risk of liver disease, suggesting that inhibition of HSD17B13 is a potential approach to ameliorate disease. Transmembrane 6 superfamily member 2 influences liver fat through the export of triglyceride-rich lipoproteins, and the inactivating variant rs58542926 has been associated with MASLD. Glucokinase regulatory protein regulates glucose storage and disposal and has been associated with SLD. Membrane-bound O-acyltransferase domain-containing 7 regulates toll-like receptors through membrane composition of inflammatory cells; loss-of-function polymorphisms are associated with inflammatory burden and MASLD. The PNPLA3 variant I148M results in loss of function and promotes the accumulation of triglycerides as well as hepatic inflammation through IL-6/STAT3-dependent pathways.[24] SERPINA1 PiZ variant is the cause of classic alpha-1 antitrypsin deficiency, which directly causes proteotoxicity and ER stress; heterozygous carriers may still be at risk for proteotoxicity, and AAT has been identified in GWAS for hepatic steatosis.
Each of these genes has been found to be associated with both ALD and MASLD. Alcohol use disorder itself has also been associated with various genetic traits, including polymorphisms in variants of alcohol dehydrogenase; however, these variants have not been clearly associated with SLD or alcohol-associated cirrhosis on their own.[25,26] Genetic variants represent real-world opportunities for personalized medicine in MASLD and MetALD. PNPLA3, HSD17B13, and SERPINA1 gene variants have been shown to have clinically significant roles in alcohol-associated disease through their respective mechanisms, and they all have emerging therapeutics in clinical trials.[27] While no personalized therapies currently exist for MetALD, it is likely that understanding of the genetic underpinnings of liver disease will take on an increasingly important role for therapeutic intervention as drug development progresses. This review does not encompass the entire breadth of genetic diversity driving SLD, and many permutations of risk for both nonalcohol and alcohol-associated liver disease also exist.
THE MICROBIOME IN MASLD, ALD, AND MetALD
The microbiome is a significant risk modifier for MASLD and ALD. The gut microbiota is involved in the complex transformation of carbohydrates, lipids, and bile acids in the gut, and microbiota-derived metabolites such as short-chain fatty acids contribute to the balance of proinflammatory and anti-inflammatory effectors in the liver. Changes in gut permeability and dysbiosis, which frequently co-exist, can circumvent normal protective mechanisms which control the flow of substances such as ethanol, lipopolysaccharides, short-chain fatty acids, and other microbial products through the portal venous circulation.[28,29]
Across the spectrum of liver disease, including MASLD, MASH, and cirrhosis, there are complex relationships between both changes in the types and localization of microbial communities.[30] Alcohol use itself has a profound impact on the diversity of microbes in the gut, directly contributes to impaired barrier function, and provides a substrate for the production of microbially derived acetaldehyde. Moreover, the microbiome itself likely plays a direct role in the gut-brain axis and influences emotional processing and cravings for both food and alcohol.[31] We reviewed trials listed for exploration of the microbiome on ClinicalTrials.gov as of December 2023. There have been nearly 2 dozen trials of probiotic and prebiotic diets and nutraceuticals completed in MASLD and MASH, but evidence for improvement is limited, and most studies have not reported results. There are several ongoing trials in various stages of recruitment to assess the efficacy of fecal microbiota transplantation (FMT).
Although FMT has not been studied in MetALD, some of the best evidence for modifying the microbiome comes from studies in severe AH; these limited studies in patients with acute and severe disease suggested that FMT is a safe event in very ill individuals and that there were favorable reductions in cravings and alcohol-associated adverse events compared to patients who received placebo.[32] There are 2 additional phase 2 and 3 studies ongoing for FMT in AH and several additional phase 1 studies in progress. While the insights from these studies cannot be directly applied to MetALD, the effects on alcohol use and the potential for improved gut barrier function and metabolic profile make FMT an important ongoing area of therapeutic investigation.
The virome refers to the diverse collection of viral elements found in the human body. The human intestinal tract harbors viruses with tropism for eukaryotic and prokaryotic cells. The virome, and in particular, bacteriophages (phages), can also impact liver health. Phages are viruses that infect bacteria and can alter the composition of the bacterial microbiota. Changes in the gut virome have been associated with metabolic syndrome and fatty liver.[33,34] Emergent evidence also suggests that there are major differences in the intestinal virome in AUD and AH. In AH, virome diversity was associated with the severity of AH and mortality, as well as the microbial communities in fecal samples.[35,36] Phage therapy, which uses phages to target specific bacterial species, has shown potential in the treatment of liver diseases, including MASLD, through the reduction in alcohol-producing klebsiella species.[37] Together, these studies suggest that the intestinal virome may offer an additional therapeutic target.
THE ROLE OF USE DISORDERS IN ALD AND MetALD
The spectrum of alcohol use, misuse, dependence, and alcohol addiction are nuanced aspects of care that reach across socioeconomic divides and affect the physical and mental health of millions worldwide. In the United States alone, > 100 million people have alcohol use disorders. Per DSM-V criteria for alcohol use disorders, patients must experience 2 of 11 criteria defining use disorders, with mild, moderate, and severe use disorders based on the number of disease-defining symptoms that a person experiences. These criteria are notably separate from the amount that a person drinks. Thus, in some cases, MetALD may be a disease-defining diagnosis for alcohol use disorder. This is especially true for patients who meet additional criteria for disease and are unable to stop alcohol use despite the knowledge of ongoing liver injury.[38]
Defining alcohol use disorder requires appropriate screening and can be challenging to treat. However, psychosocial and pharmacologic therapies targeting cravings and recurrent use have demonstrated varying degrees of efficacy in the treatment of use disorders. Greater than 70% of people with alcohol use disorder will continue to go back to drinking despite attempts at treatment. Despite these sobering statistics, treatment of alcohol use disorder, if successful, is a cornerstone of treatment for MetALD because of the removal of direct effects on liver parenchyma and reduced consumption of excessive calories.[39] Replacement of alcohol with excess calories in the form of palatable foods and the use of food as a reward stimulus raises the question if there are definable and actionable maladaptive consumption behaviors that can be intervened upon in the case of MetALD.
Recognition of excess alcohol consumption presents a unique challenge for clinicians as self-reported alcohol use does not always accurately reflect intake. The reasons for this can range from recall bias, denial, fear of judgment, and cognitive effects of alcohol itself. These effects are compounded in patients with active AUD. The US Preventative Services Task Force recommends screening for AUD with the Alcohol Use Disorders Identification Test (AUDIT). However, these screening tools do not necessarily help distinguish MetALD from ALD when recall bias is involved.[40,41].
Biomarkers for alcohol use can help overcome denial and can help reveal inaccuracies in self-reported consumption. Assessing serum phosphatidylethanol can detect chronic exposure to alcohol; ethylglucuronides in hair give a measure of alcohol exposure over a month, while serum ethylglucuronides are useful for consumption over the preceding 3–4 days. Serum studies such as elevated gamma-glutamyl transpeptidase, liver enzyme ratios, and mean corpuscular volume lack specificity but can suggest patterns of alcohol consumption.[42,43] From a therapeutic perspective, underestimated alcohol use can be very problematic, and the development and use of biomarkers will likely be very important in the course of trials for MetALD.
THERAPEUTIC STRATEGY IN MetALD
There have been over 1200 trials for SLD, most focusing on MASLD and MASH, encompassing diagnostics, behavioral interventions, dietary supplements, devices, and drugs. Recently, resmetirom (Madrigal Pharmaceuticals) became the first FDA-approved therapy for MASH with moderate to severe liver fibrosis after meeting endpoints for fibrosis regression.[44] There are over 500 active or completed drug trials for MASLD and around 90 trials for ALD. The vast majority of drug trials for ALD have targeted AH due to its significant morbidity and mortality. Meanwhile, therapeutic trials for MASLD and MASH include targets across the spectrum of liver disease and have traditionally excluded patients with significant alcohol intake. There are 157 ongoing trials in phase 2 or higher for MASLD and MASH in various stages of recruitment and analysis (Table 1).
TABLE 1.
Current phase 2+ trials for MASLD/MASH
Abbreviations: FXR, Farnesoid X receptor; HSD17B13, hydroxysteroid 17-beta dehydrogenase 13; MASH, metabolic dysfunction-associated steatohepatitis; MASLD, metabolic dysfunction–associated steatotic liver disease; PPAR, peroxisome proliferator–activated receptor; SGLT2, sodium-glucose–linked transporter 2; THR-β, thyroid hormone receptor beta.
Part of the nomenclature change in SLD is aimed at defining MetALD as a specific entity, but no current agents are in development for MetALD as a separate disease.[1] The development of therapeutics for overlap conditions is complicated because predominant alcohol use carries with it the risk of poor nutritional intake, hypoglycemia, nutritional deficiency, and significant compliance issues, whereas MASLD is often more associated with increased calorie consumption. The presence of significant alcohol consumption also affects drug metabolism through cytochrome p450 enzymes and can make drug effects less predictable. In addition, there are significant logistical concerns, including compliance and follow-up for patients who are using excessive amounts of alcohol. The National Institute on Alcohol Abuse and Alcoholism has specific guidance for research in substance use disorders, which require both additional oversight and resources to ensure trials are completed with appropriate scientific and ethical rigor in vulnerable populations who have severe physical and psychiatric diseases. Moreover, the risks of significant alcohol use are broad, and studies including people with alcohol use disorder generally require cessation of alcohol use as part of management, which if successful, can confound the results of studies themselves. Having a formal definition of MetALD is a major step toward establishing endpoints to judge the efficacy of drug trials, but at this time, these have not been established.
Given these caveats, an ideal approach to evaluating drugs for MetALD would be looking for agents that have efficacy in treating MASH and promoting cessation of alcohol use. In addition, medications for alcohol use disorder (MAUD), antidiabetic and anti-obesity drugs, including incretins which act on the GLP-1 receptor, glucagon inhibitory peptide (GIP), and glucagon axis, thyroid hormone receptor beta (THR-β) agonists, sodium-glucose co-transporter 2 (SGLT2) inhibitors, PPAR and peroxisome proliferator–activated gamma coactivator 1-axis modifiers and insulin therapies as well as combination therapies are in various stages of clinical development for MASLD. Liver-targeted therapies such as FGF21 analogs, Farnesoid X receptor (FXR) agonists, agents targeting de novo lipogenesis (DNL), and agents targeting genetic risk modifiers such as PNPLA3 mutations and HSD17B13, SERPINA1 are all in clinical development. However, the safety of each agent and their relationship with alcohol use disorders are largely unclear; some agents show promise for alcohol use disorder, and some have been associated with significant adverse effects when paired with alcohol. Rational therapy incorporates comprehensive psychosocial interventions and the potential use of known and novel therapeutics to help patients with evidence of MetALD achieve disease regression (Figure 1).
FIGURE 1.
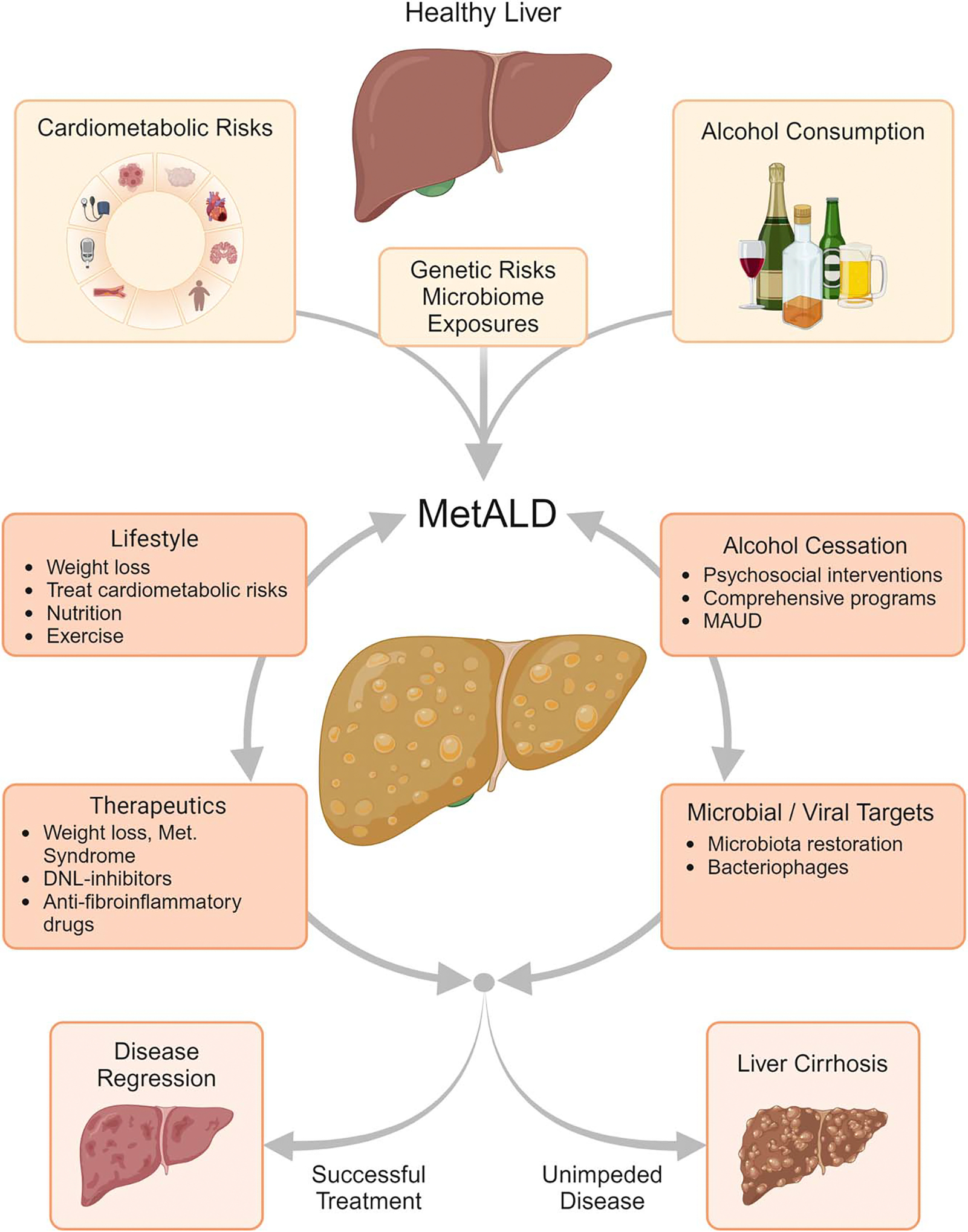
Dynamic risks for MetALD: alcohol consumption and cardiometabolic disease with genetic and environmental modifiers cause MetALD. Other interventions, including therapeutics, can contribute to the resolution of the disease or the development of liver fibrosis. Abbreviations: DNL, de novo lipogenesis; MAUD, medications for alcohol use disorder; MetALD, metabolic dysfunction and alcohol-associated liver disease.
MAUD AND PSYCHOSOCIAL TREATMENTS
The first target for counseling and treatment of MetALD and the entire spectrum of alcohol-associated disease is alcohol cessation. This approach has variable efficacy, and a subset of patients with MetALD may continue to drink even with knowledge of the excess alcohol-associated risk. Several psychosocial therapeutic modalities have been studied in AUD. Cognitive behavioral therapies, couples therapy, 12-step facilitation programs, and additional psychosocial approaches can all be effective options with or without pharmacotherapy for alcohol use disorder, and patients should be referred to appropriate practitioners as soon as a diagnosis of AUD or MetALD is identified.
MAUD refers to the use of pharmacotherapy to aid in cessation, reduce cravings, and relapse of disease. The US Food and Drug Administration (FDA) has approved 3 medications for the treatment of AUD: disulfiram, naltrexone (oral and long-acting injectable), and acamprosate.[38] Disulfiram inhibits aldehyde dehydrogenase; it aids in alcohol cessation by producing unpleasant adverse effects such as flushing, nausea, and palpitations if alcohol is consumed. It is used in patients who are highly motivated to abstain from alcohol and can adhere to the medication regimen.[38] Naltrexone, an opioid antagonist, is available in both oral and long-acting injectable forms. It is thought to reduce the rewarding effects of alcohol, thereby decreasing the desire to drink. Acamprosate is a small molecule drug that is thought to modulate GABA and NMDA neurotransmission. Acamprosate is used as a means of maintaining sobriety in patients who have already achieved abstinence. The mechanisms for acamprosate are not entirely known but likely aid in rebalancing neurotransmitter pathways in the brain.[38]
In addition to these FDA-approved medications, there are several other drugs that have been studied for the treatment of AUD, although they are not currently FDA-approved for this indication. These include the anticonvulsants topiramate and gabapentin, the smoking cessation drug varenicline, and the muscle relaxant baclofen. The European Medicines Agency has also approved nalmefene for the treatment of AUD. Nalmefene, like naltrexone, is an opioid antagonist, but it is used to reduce heavy drinking rather than to promote abstinence.[45] Efficacy of psychosocial intervention and MAUD is variable and thought to be most effective when combined as part of comprehensive care. For the patient with MetALD, the approach to this may be variable as individuals may not have had prior characterizing events for AUD. Each patient faces individual circumstances, and comprehensive tailored care likely has the best chance of being efficacious. There is a paucity of direct drug trials for pure ALD. Currently, phase 1 trials include interleukin 23 inhibitor Guselkumab (NCT04736966), proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab (NCT04781322), simvastatin (NCT04971577). In addition, there are a few additional ongoing phase 1 studies of probiotic therapies (NCT05007470 and NCT05178069) and dietary supplements (NCT03503708 and NCT05623501) (Table 2).
TABLE 2.
Ongoing trials for ALD and AUD
| NCT Number | Class | Interventions | Phases | Locations |
|---|---|---|---|---|
| NCT03503708 | Supplement | Livitol-70 | NA | India |
| NCT04781322 | PCSK9 inhibitor | Alirocumab | placebo | PHASE 1 | MD, USA |
| NCT06175507 | MAUD | Naltrexon | baclofen | NA | India |
| NCT05159830 | MAUD | Cannabidiol | placebo | PHASE 2 | France |
| NCT04736966 | IL23 inhibitor | Guselkumab | PHASE 1 | CA, USA |
| NCT05007470 | Probiotic | VSL #3 Capsule | placebo | NA | CA, USA |
| NCT05623501 | Supplement | Dihydromyricetin | PHASE 1 | CA, USA |
| NCT05178069 | Probiotic | Placebo | Lactobacillus rhamnosus GG | PHASE 2 | KY, USA |
Abbreviations: ALD, alcohol-associated liver disease; AUD, alcohol use disorder; MAUD, medications for alcohol use disorder; NA, not applicable; PCSK9, proprotein convertase subtilisin/kexin type 9.
DIETARY, LIFESTYLE, AND BARIATRIC INTERVENTIONS
Dietary and lifestyle interventions are the first-line treatment for SLD. AASLD recommends weight loss as a primary goal, with a target of at least 5% and preferably ≥ 10% of body weight, as the magnitude of weight loss is associated with greater liver histologic and cardiometabolic benefits. Dietary modifications should include a reduction of macronutrient content to induce an energy deficit, with restriction of saturated fat, starch, and added sugar, and adoption of healthier eating patterns, such as the Mediterranean diet. The proportions of energy intake from carbohydrates and lipids are often restricted to 50%–60% and 20%–25%, respectively.[46–48]
Exercise therapy is another useful lifestyle-related intervention for MASLD/MASH. Consistent aerobic exercise in 30- to 60-minute sessions held 3–4 times weekly for 4–12 weeks in patients with MASLD complicated by obesity has been shown to improve liver fat content, even without accompanying body weight reduction.
Bariatric surgery can be considered in otherwise eligible obese individuals with MASLD or MASH, but it is not yet established as a specific treatment for MetALD. The safety and efficacy of bariatric surgery in obese individuals with established cirrhosis attributed to MetALD is not established, and it may be considered on a case-by-case basis by an experienced bariatric surgery program.[49] Patients may be at greater risk of alcohol-induced liver injury post-bariatric surgery, and alcohol cessation should be an important pillar of pre-bariatric surgery patient optimization. The increased risk is attributed to a reduction in gastric metabolism of alcohol, leading to increased delivery of alcohol to the liver.[50] Newer endobariatric procedures do not have enough data to understand their effects on alcohol use at this time and should be approached with caution.
INCRETINS (GLP-1ra, GIP, and GLUCAGON)
Glucagon-like peptide 1 receptor agonists (GLP-1ra), such as exenatide, liraglutide, and semaglutide, were the initial prototypes for GLP-1ra along with dual GIP and GLP-1 (GIP/GLP) agonist therapy tirzepatide as well as triple GIP/GLP-1ra and glucagon therapy retapatide have arisen as powerful antidiabetic and weight loss therapeutics. They target GLP-1 receptors as an agonist and have broad effects on gut motility, satiety, and the gut-brain neurohormonal axis. In the liver, these agents broadly affect protein kinase A and phospholipase A phosphorylation leading to increased mitochondrial function and reduced oxidative stress as well as decreased lipogenesis and stellate cell activation.[51] Semaglutide and liraglutide therapy have both been shown to resolve MASH without increases in fibrosis, but these therapies have not shown a clear signal for reversing fibrosis. Dual Incretin therapy with tirzepatide acting as dual GIP and GLP-1ra was associated with significant liver fat reduction at 1 year; additional studies of dual and triple therapies are ongoing. Notably, excessive alcohol use has been excluded from enrollment in these trials. These agents are also likely to directly affect reward responses and GLP-1 function in the hypothalamus.[52,53]. Current practice guidelines have supported GLP-1ra therapy for SLD for the treatment of obesity and steatosis since 2022.
GLP-1ra liraglutide and semaglutide have the potential to attenuate the reinforcing qualities of alcohol; in animal models and limited human studies, GLP-1ra therapies have been shown to favorably affect parameters such as cravings and total alcohol consumption.[54–56]. Together, these data suggest that incretins may have favorable therapeutic effects for people with MetALD, but the effects on alcohol use remain unproven in humans.[57]. In a randomized placebo-controlled study of patients with comorbid psychiatric disease, liraglutide was not shown to decrease alcohol consumption.
Mortality in SLD is partially driven by etiology. Risk factors for MASLD also predispose to major adverse cardiac events in addition to liver-related mortality. While ALD itself has higher mortality from cirrhosis and HCC.[58,59] The predominant drivers for mortality in MetALD are not clearly defined at this time. However, individuals with MetALD are likely at risk from both major adverse cardiac event and earlier liver disease. Incretin therapies have significant benefits for major adverse cardiac event, which suggests they would have more favorable effects in both MASLD and MetALD.[60]
Despite their many benefits, GLP-1ra has significant GI adverse effects and some evidence of gallbladder-related events due to rapid weight loss. While meta-analyses have not indicated an increased risk of acute pancreatitis, pancreatic cancer, or other malignancies with GLP-1ra, a phase 2 trial reported malignant neoplasms in 1% of patients receiving semaglutide. Overall, neoplasms (benign, malignant, or unspecified) were reported in 15% of patients in the semaglutide groups compared to 8% in the placebo group, although no specific pattern of occurrence in particular organs was observed.[61–63] Recent retrospective studies have demonstrated a decrease in colorectal cancer risk in patients treated with GLP-1ra.[64] Thus, the summative effects of increased risk of rare cancers versus decreased risk of a common cancer should guide individual recommendations for patients.
The difficulty with these studies is that it is unclear if using these medications with ongoing heavy alcohol use is safe or effective. Furthermore, comorbid substance use disorder with alcohol could potentially complicate the GI adverse effect profiles and safety of these drugs.
FXR AGONISTS
FXR is a nuclear receptor that regulates bile acid, lipid, and glucose metabolism, and inflammation, making it a promising target for MASH and MetALD treatment.
Clinical trials have demonstrated the effectiveness of FXR agonists in reducing liver fat content and improving liver enzymes in patients with MASH. For instance, the FXR agonist MET409, in a 12-week randomized controlled trial, showed significant reductions in liver fat content with mean relative reductions of 55% at an 80 mg dose and 38% at a 50 mg dose compared to 6% in the placebo group. In addition, vonafexor, another non-bile acid FXR agonist, was evaluated in the LIVIFY trial and demonstrated potent liver fat reduction, improvement in liver enzymes, weight loss, and a possible renal benefit.
The role of FXR in the management of ALD is an area of active research. Studies have shown that activation of FXR can attenuate hepatic injury in murine models of ALD by modulating lipid homeostasis, reducing oxidative stress, and decreasing inflammation. However, all the insights into FXR and alcohol-associated disease are preclinical, and in trials of FXR agonists in MASH individuals who reported significant alcohol use were excluded.
For instance, the use of FXR agonists such as WAY-362450 protects mice from the development of ALD by rescuing FXR activity, suppressing ethanol-induced Cyp2e1 upregulation, and attenuating oxidative stress in the liver. Another study found that yangonin, an FXR agonist, improved lipid homeostasis, ameliorated cholestasis, and reduced cellular senescence and inflammation in ALD.[65]. In vitro examination of FXR agonist GW4064 shows FXR agonists increase expression of class I alcohol dehydrogenases; this could have benefits by shunting alcohol metabolism away from ROS generating CYP2E1.
These preclinical findings suggest that FXR agonists could potentially be used as a therapeutic strategy for ALD. However, it is important to note that these studies are based on animal models, and the efficacy and safety of FXR agonists for ALD in humans have not been established. Clinical trials would be necessary to determine the appropriate dosages and to evaluate the therapeutic potential of FXR agonists in the treatment of ALD in humans.
While these results are promising, the use of FXR agonists is not without adverse effects. Pruritus, an increase in low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) decreases have been reported with FXR agonist therapy. These adverse effects are important to consider when evaluating the risk-benefit profile of FXR agonists for individual patients.
Currently, there are no United States guidelines that specifically recommend FXR agonists for the treatment of MASH. Obeticholic acid did not receive regulatory approval for MASH due to the moderate benefits and significant safety risks. However, the evidence from clinical trials supports further investigation into the long-term efficacy and safety of newer FXR agonists in larger phase 3 trials and potentially for MetALD.
PPAR agonists
Thiazolidinediones, particularly pioglitazone, have been studied for the treatment of MASH. Randomized controlled trials have demonstrated that pioglitazone improves hepatic steatosis, lobular inflammation, and hepatocellular ballooning, with modest effects on fibrosis in both patients with diabetes and without diabetes with MASH. A meta-analysis reported that pioglitazone therapy was associated with improved advanced fibrosis, fibrosis of any stage, and MASH resolution. The typical dosage used in these studies was 45 mg daily.
However, the use of pioglitazone is associated with adverse effects, including significant weight gain (mean, 4.4 kg) and lower limb edema. There have also been concerns about bone loss in women and controversial findings regarding the risk of bladder cancer.[66]. Furthermore, cessation of therapy is associated with disease recurrence. Given the favorable profile of pioglitazone on the liver, the question remains if additional PPARγ-specific agonists with more favorable adverse effect profiles may be viable therapeutic agents.
The AASLD and the American Association of Clinical Endocrinology recommend pioglitazone for persons with T2DM and biopsy-proven MASH, with a high strength of evidence. Clinicians are advised to consider treating diabetes with pioglitazone and/or GLP-1ra when there is an elevated probability of having MASH based on elevated plasma aminotransferase levels and noninvasive tests.[1]
PPARs enable adaptation to metabolic, nutritional, and inflammatory stress through transcriptional networks. They are intimately involved in hepatocyte carbohydrate and lipid biology and are directly involved in MASH-associated liver pathogenesis. Beyond thiozolidinediones, PPAR agonists targeting varied aspects of PPAR function show promise for metabolic liver disease in preclinical and clinical trials. The targets of PPAR-based therapy include single γ agonists, α/γ agonists, α/δ, and pan-α/δ/γ agonists.
Two dual PPAR-α/γ agonists, saroglitazar and aleglitazar, both have benefits for experimental MASH. Saroglitazar demonstrated overall improvement in experimental MASH models, with effects appearing superior to those of the pure PPARα agonist fenofibrate and the PPARγ agonist pioglitazone. In the phase 2 clinical trial, saroglitazar met the primary endpoints of ALT and liver fat reduction and is currently in phase 3 and 4 studies.
Aleglitazar improved noninvasive tests of liver steatosis and fibrosis in a randomized, double-blind, placebo-controlled trial involving patients with T2DM and recent coronary artery disease as a post hoc analysis. Aleglitazar has not progressed with further MASH-target clinical trials.
Elafibranor, a PPAR α/δ agonist, was studied in preclinical and reached phase 2 randomized, double-blind placebo-controlled trial where patients with MASH without cirrhosis received elafibranor 80 mg or 120 mg daily for 52 weeks. The primary endpoint of resolution of MASH without fibrosis worsening was not met in the intention-to-treat analysis. However, a post hoc analysis using a modified definition showed that the 120 mg dose of elafibranor resolved MASH. Elanafbranor was weight-neutral and improved cardiometabolic risk factors but also carried with it reversible effects on creatinine clearance. Additional phase 2 and 3 studies were subsequently terminated.
Lanifibronor, a pan-PPAR agonist, has been evaluated in a phase 2b trial where patients with non-cirrhotic active MASH were treated with either 1200 mg or 800 mg of lanifibronor or placebo once daily for 24 weeks. The trial demonstrated that the 1200 mg dose has a higher proportional decrease in steatosis activity and fibrosis compared to placebo. In addition, lanifibronor showed resolution of MASH without worsening of fibrosis, improvement in fibrosis stage, and reductions in liver enzyme levels. The addition of γ agonist activity came with additional adverse effects, including diarrhea, nausea, edema, anemia, and weight gain.[67] Lanifibranor is currently still under investigation with further phase 2 and 3 studies, including the study of lanifibranor in combination with SGLT2 inhibitor empagliflozin for T2DM NCT05232071.
Preclinical evaluation suggests that PPARα/γ but not pan-PPAR or selective PPARγ reduced ethanol intake in mouse models of alcohol dependence.[68] PPAR α dysregulation has been demonstrated in ALD, and agents, such as dual agonists, have direct effects on fibrogenesis caused by alcohol. Separate studies show improvements in alcohol-induced steatosis in rats with pioglitazone. From an alcohol use perspective, there is little evidence to suggest and association between pioglitazone and alcohol use or cessation. One observational study suggests reduced alcohol consumption after starting pioglitazone. A separate randomized placebo-controlled study of patients with alcohol dependence and withdrawal was stopped early because of safety concerns about myopathy and increased rather than reduced alcohol cravings in a group of 16 subjects. Together, this suggests pioglitazone should be used with caution in patients with active alcohol use. Dual agonist therapies have not been studied in the context of significant alcohol consumption, but there is modest preclinical evidence suggesting that they could be beneficial.
SODIUM-GLUCOSE CO-TRANSPORTER 2
SGLT2 inhibitors are glucose-lowering agents that have shown potential in the treatment of MASLD in patients with T2DM. These agents improve glucose control, promote weight loss, and lower serum uric acid levels, which may contribute to their beneficial effects on MASLD. SGLT2 inhibitor’s primary mechanism of action is through glucose excretion, which drives an additional diuretic effect.[69]
Recent findings from both randomized controlled trials and open-label studies have shown that SGLT2 inhibitors can reduce fatty liver content and decrease low-grade inflammation, oxidative stress, and fibrosis in people with fatty liver and T2DM.[69]
SGLT2 inhibitors significantly reduced indicators of liver fibrosis, including liver stiffness measurement, controlled attenuation parameter, serum ferritin, serum type 4 collagen 7s, and FIB-4 index. Ipragliflozin significantly decreased hepatic fibrosis and resolved MASH in a higher proportion of patients compared to those managed without SGLT2 inhibitors.[69–71] Several additional trials are underway for SGLT2 in MASLD and cirrhosis alone and in combination with other drugs.
In people with active heavy alcohol use, SGLT2 inhibitors are not recommended due to an increased risk of life-threatening ketoacidosis. Although they may be beneficial for the metabolic complications of MetALD and comorbid T2DM, these medications would likely be best suited for use in patients who have achieved durable abstinence from alcohol.
FGF21 analogs
FGF21 is a hormone-like member of the FGF superfamily; it has reduced plasma binding, which allows it to function in an endocrine manner as well as through autocrine and paracrine signaling. FGF21 is produced by multiple organs it signals through FGF receptors, requiring a cofactor, β-Klotho.
FGF21 has multiple metabolic actions. Initially, it was discovered as a regulator of glucose uptake in adipocytes, but it is now known to stimulate many other pathways and display multiple pharmacological effects in metabolically compromised animals and humans. FGF21 promotes lipid catabolism, including lipolysis, fatty acid oxidation, mitochondrial oxidative activity, and thermogenic energy dissipation, rather than directly regulating insulin and appetite. It also plays a key role in eliciting and coordinating the adaptive starvation response.
FGF21 has been shown to have beneficial effects on dyslipidemia, body weight, fasting insulin, and adiponectin levels in clinical trials, suggesting potential therapeutic applications for metabolic disorders. Furthermore, FGF21 has been found to protect against hepatic lipotoxicity, inflammation, and fibrogenesis, highlighting its potential role in the treatment of NASH.
The prototype drug for this class is efruxifermin, which is a hybrid Fc-FGF21 fusion protein engineered for its stability and potential role as an FGF21 pathway agonist. Initial phase 2a data suggest efficacy for reducing hepatic fat fraction in MASH, with the caveat of gastrointestinal adverse effects in up to 46% of the participants. In a separate study of patients with MASH cirrhosis, efruxifermin treatment resulted in 25% MASH resolution and improvement in fibrosis by at least 1 stage.[72]
From an alcohol use perspective, FGF21 is more complicated; alcohol use itself was associated with increased levels of circulating FGF21 in people with alcohol use disorder. However, FGF21 itself reduced alcohol consumption through action on the amygdala-striatal circuit, suggesting that increases in FGF21 activity may be beneficial for suppressing alcohol use, and the increase in FGF21 has been shown to reduce fibrosis in experimental models of liver fibrosis.[73]. It is not clear if any cerebral effects of FGF21 would extend to Fc-fusion proteins such as efruxifermin given exogenously. Certainly, additional studies, including evaluation of the effects of alcohol cessation and MetALD, may be valuable.
THYROID HORMONE RECEPTOR BETA AGONISTS
THR-β agonists have emerged as a promising therapeutic strategy for MASLD. Thyroid hormones play a crucial role in regulating metabolic activities in the liver, including lipid export and oxidation, DNL, hepatic insulin sensitivity, and suppression of hepatic gluconeogenesis. Hypothyroidism, characterized by low thyroid hormone levels, has been identified as a risk factor for MASLD.
THR-β is primarily expressed in the liver and plays a significant role in lipid metabolism. Activation of THR-β has been shown to inhibit hepatic triglyceride synthesis, increase hepatic cholesterol clearance, reduce lipid deposition, partly increase insulin sensitivity, promote glucose metabolism, and improve inflammation.[74]
Resmetirom, also known as MGL-3196, is an orally active, selective THR-β agonist that has been shown to reduce liver fat content and improve NASH. Recently, resmetirom became the first drug approved by the FDA to treat MASH. In a randomized, double-blind, placebo-controlled, phase 3 trial, resmetirom treatment resulted in a significant reduction in MASH and fibrosis, meeting endpoints for early FDA approval.[44] Other THR-β agonists, such as TG68, have also demonstrated beneficial effects on fatty liver accumulation and liver injury in animal models.[74,75] There are currently 6 ongoing trials for THR-β agents in at least phase 2.
It is unclear if there would be any direct overlap between THR agonist therapy and alcohol-associated disease. Alcohol use disorder itself suppresses the central and peripheral effects of thyroid hormone and can cause aberrant readings in the evaluation of thyroid function.
DRUGS TARGETING DE NOVO LIPOGENESIS
DNL refers to the process of converting excess intracellular carbohydrate molecules into fatty acids. DNL inhibitors have the potential to play an important role in controlling the development of steatosis due to metabolic syndrome.[76]
The therapeutic targets for DNL are major enzymes such as ACC, ATP citrate lyase, fatty acid synthase and diacylglycerol acyltransferases (DGATs) which catalyze the esterification of diacylglycerol to triacylglycerol. Ketohexokinase inhibitors suppress the lipogenic effects of fructose and stearoyl-CoA desaturase modulators.
ACC inhibition in animals reduces hepatic steatosis.[76] In humans, the prototype drug firsocostat reduced hepatic steatosis in humans and has been studied alone and in combination with apoptosis signal–regulating kinase 1 inhibitor selonsertib and FXR agonist cilofexor. The trial was exploratory in nature and evaluated mono and combination therapy. The data suggested improvements in some parameters on MASH, and numerically (21%) of people in the firsocostat/cilofexor group achieved this, but the differences between groups and placebo were not statistically significant after 48 weeks. Notable adverse effects were pruritus and nausea in up to a quarter of participants.[77,78]
Diacylglycerol acyltransferase 2 (DGAT2) is a liver and adipose-expressed DGATs that converts DNL-derived fatty acids to triacylglycerol. Theoretically, DGAT2 inhibition would lead to diacylglycerol accumulation; however, in rodents, suppression of DGAT2 activity with antisense oligonucleotide therapy leads to adaptive decreased expression of multiple lipogenic and increased oxidative pathways.[79]
Two DGAT2 inhibitors and 2 antisense oligonucleotides have been developed clinically. PF-06427878 was studied in a short 2-week study and showed improvement in liver fat but had a suboptimal pharmacokinetic profile. PF-06865571 (ervogastat) has progressed to short phase 2 studies and showed improvement in liver fat as monotherapy or with coadministration of ACC1 inhibitor.[80]
Fatty acid synthase inhibition has reached phase 2a trials, which have demonstrated improvements in liver fat and biochemical signatures. However, no additional studies are currently ongoing.[81] In addition, ketohexokinase inhibitors and stearoyl-CoA desaturase modulators have shown modest improvements in hepatic steatosis in limited trials, but these agents have required some reformulation for pharmacodynamics and have not yet been shown to match the efficacy of other DNL inhibitors.[82,83]
In the context of MetALD and alcohol use in general, there is no specific evidence to suggest safety or efficacy with active alcohol use disorder. Given the shared pathways of alcohol metabolism through acetyl-CoA, it is possible that DNL agents, especially DGAT2 inhibitors, may be able to combat steatosis due to alcohol use. However, the effects on alcohol metabolism are unknown, and it is unclear if these agents would exacerbate or alleviate cellular reactive oxygen species due to alcohol metabolism. There is no evidence to suggest any effects on craving or alcohol use with DNL inhibitors to date. Given the overall lack of clear effects on alcohol metabolism, DNL inhibitors may initially have limited roles for people with MetALD or be more useful in patients who have achieved deeper remission.
GENETIC TARGETS OF SLD
The PNPLA3 gene encodes a protein that is involved in lipid metabolism within hepatocytes. The I148M variant of PNPLA3, resulting from a single-nucleotide polymorphism rs738409 C > G, is strongly associated with the development and progression of MASLD. This variant leads to a loss of function of the PNPLA3 enzyme, which normally has triglyceride hydrolase activity. The I148M substitution impairs this activity, promoting triglyceride accumulation in hepatocytes and contributing to hepatic steatosis.[24]
The presence of the I148M variant has been linked to increased hepatic fat levels, inflammation, and a higher risk of progression to more severe liver disease, including fibrosis, cirrhosis, and HCC. The variant is also associated with differences in hepatic fat content among different ancestry groups, with the G allele being most common in Hispanic populations, who have a higher susceptibility to MASLD.
The I148M variant’s impact on liver disease is further influenced by dietary factors, with an interaction between carbohydrate intake and the G allele of PNPLA3 rs738409 being associated with more severe steatosis. Notably, knockout mice attenuate the effect of I148M. This suggests that the PNPLA3 I148M variant not only contributes to the pathophysiology of SLD but also interacts with environmental factors to modulate disease severity.
HSD17B13 plays a significant role in the pathogenesis and progression of MASLD. HSD17B13 is a liver-specific, lipid droplet–associated protein involved in enzymatic pathways related to steroids, proinflammatory lipid mediators, and retinol.[84]
Inactivating variants of the HSD17B13 gene are associated with a reduced risk of chronic liver diseases, including MASLD. The most well-characterized variant, rs72613567, a loss-of-function variant in HSD17B13, is protective against the progression of MASLD to MASH, liver fibrosis, cirrhosis, and HCC.
Furthermore, carriers of the HSD17B13 gene variant (rs72613567:TA) have been found to have decreased fibrosis and expression of inflammation-related genes but increased phospholipids in the liver. These changes are not secondary to steatosis, DNL, adipose tissue lipolysis, or hepatic insulin sensitivity and place HSD17B13 as a novel but also less-understood modifier of liver disease.
Both HSD17B13 and PNPLA3 variants are modifiers of alcohol-associated disease. In AUD, the risk of cirrhosis is significantly attenuated by the presence of HSD17B13 variants, whereas the inactivating PNPLA3 I148M variant was associated with an increased risk of cirrhosis with AUD. Together, the carriage of both variants resulted in the attenuation of the risk conferred by PNPLA3 I148M.[27] These data suggest that HSD17B13 inactivation and restoration of PNPLA3 activity would represent the most favorable biology to avoid the consequences of MetALD. A phase 1 clinical trial of HSD17B13 knockdown by siRNA is currently recruiting, but it excludes people with alcohol use disorders. An additional study of a PNPLA3 knockdown is also in phase 2 evaluation NCT05809934.
The alpha-1 antitrypsin (AAT) Z-allele, which has alpha-1 antitrypsin deficiency through co-dominant inheritance and proteotoxic stress, is present in at least 4% of the US population. Recent GWAS studies suggest that the Z-allele is associated with both metabolic and alcohol-associated SLD.[22,85] Moreover, carriage of one Z-allele appears to confer additional risk for progression of ALD and MASH to cirrhosis.[86,87] Fazisiran is an RNA-interference drug that has recently been investigated for reducing the expression of proteotoxic Z-AAT and is currently in trials for people with homozygous disease.[88] It is unclear if there would be a clear benefit for MASLD and MetALD in the larger population, and the relative contribution of proteotoxic stress may be different between individuals. The availability of these therapeutics may allow for additional options for personalized medicine in the future.
microRNA THERAPEUTICS
microRNAs (miRNAs) are short sequences of messenger RNA that are ubiquitous in cells and mediate varied roles in transcriptional regulation. There are distinct but also overlapping roles for miRNA in ALD and MASLD. Notably, in ALD, miR-155 and miR-21 regulate liver injury through TNF-alpha and response to bacterial lipopolysaccharides in Kupffer cells, hepatocytes, and stellate cells.[89] Inhibition of miR-33b and miR-21 has also been helpful for MASH in murine models, suggesting a potential dual therapeutic role.[90,91]
In both metabolic and alcohol-associated diseases, miRNAs appear to modify fatty acid oxidation through SIRT1 inhibition. In MASH, miR-34a is upregulated in MASH and miR-217 is upregulated in ASH. Targeting of miRNA can be accomplished through synthetic antisense miRNA (AntagomiR). Adeno-associated virus constructs can also confer some degree of tissue specificity to miRNA delivery.[92] miRNA therapeutics may play a role in the future with respect to liver disease, but these therapeutics have not reached clinical trials in liver disease at this time. It is unclear if overlapping therapeutics in MetALD would be clearly beneficial compared to other therapeutics under current development.
RATIONAL COMBINATION THERAPIES
Putative therapies for MetALD, which have been explored in this review, are broad and nuanced. Many agents show promise in reversing SLD but are unable to meet primary endpoints to justify their clinical use, or the mechanism of therapy may have unwanted adverse effects. For people with MetALD, inducing and maintaining alcohol use remission is paramount. Thus, at baseline, psychosocial and MAUD therapy can likely be added in combination with most pharmacotherapy for MASLD.
For select populations, personalization of treatment with therapeutics targeting susceptibility alleles, paired with additional pharmacotherapy such as incretins, may be a valuable step toward increasing therapeutic efficacy. Several combination therapies have been trialed or are in trials at this time. DNL inhibitors, such as ACC inhibitors as a class, have been variably trialed with therapies such as FXR, DGAT2 inhibitors, and incretins. More data and extended studies are needed to determine if the benefits outweigh the adverse effects of these combinations.
Several studies have been conducted with combination therapy and there are additional trials in process (Table 3). Like most monotherapies, combination therapies for MASLD have generally not included patients with significant alcohol consumption in the initial clinical studies; thus, safety in patients with ongoing alcohol use is a significant concern.
TABLE 3.
Current trials of combination interventions
| NCT number | Interventions | Phases |
|---|---|---|
| NCT05942963 | Empagliflozin 10 mg | pioglitazone 15 mg | metformin NNC0194 0499 50 mg/mL | placebo (NNC0194-0499) | semaglutide 3 mg/mL | semaglutide placebo | NNC0174 0833 10 mg/mL | NNC0174 0833 placebo |
PHASE 4 |
| NCT04976283 | Pioglitazone | empagliflozin | pioglitazone + empagliflozin | PHASE 4 |
| NCT04971785 | Semaglutide | cilofexor/firsocostat | PHASE 2 |
| NCT05623189 | HTD1801 | placebo | PHASE 2 |
| NCT04639414 | Empagliflozin 10 mg oral tablet/semaglutide 1 mg pen injector | empagliflozin 10 mg oral tablet and placebo| Placebo | PHASE 4 |
WEIGHING CONSIDERATIONS FOR TREATMENT
SLD now affects more than 40% of the global population and represents one of the largest disease burden on the worldwide population. Alcohol use disorders and heavy alcohol use simultaneous represent an expanding global crisis. Overall, the greatest risk factor for all SLD is obesity and other cardiometabolic risk factors.[93]
The sheer number of people who have obesity-associated metabolic diseases has led to shortages in incretin-based therapies for diabetes and obesity, and the significant cost of these medications impacts resource-constrained populations disproportionately.[94] As more patients are diagnosed with MetALD and there is interest in GLP-1ra, cost and availability may become more acute.
Treating MetALD with drugs that potentially reverse the progression of disease in the liver also raises important questions for providers caring for patients with heavy alcohol use, relapse, or inability to achieve cessation in terms of their safety, efficacy, and if the use of medications for SLD may give patients false security in their alcohol consumption.
Ultimately, therapeutic interventions in MetALD will need careful scrutiny from regulatory bodies around the world. And the choice, duration, and modification of therapeutics will need to be managed within the physician-patient relationship. We discussed several treatment strategies which are currently in at least phase 2 clinical development. Most MASLD and MASH directed strategies have the potential to treat MetALD as well. However, safety data are lacking in virtually all agents with regard to heavy alcohol consumption, and certain agents such as SGLT2 inhibitors and PPARy agonists such as pioglitazone have demonstrated safety concerns in active alcohol users (Table 4). Until further safety and efficacy data are available for alcohol users, therapeutics for MetALD will require careful monitoring, and many agents may be limited to individuals who have attained some remission from their alcohol use disorder.
TABLE 4.
Therapeutic targets for MetALD
| MASLD target | MetALD target | Safety with active AUD? | |
|---|---|---|---|
| Incretin | Recommended | Probable | Likely safe, nutritional concerns |
| SGLT2 | Likely | Possible | DKA risk |
| FGF21 | Possible | Possible | Unknown |
| FXR agonists | Possible | Possible | Unknown |
| THR-B | Possible | Possible | Unknown |
| DNL agent | Possible | Possible | Unknown |
| PPARy | Recommended | Safety concerns | Myopathy, increased AUD |
| PPARad | Possible | Possible | Unknown |
| PPARayd | Possible | Possible | Unknown |
| PNPLA3 | Possible | Possible | Unknown |
| HSD17B13 | Possible | Possible | Unknown |
| microRNA | Possible | Possible | Unknown |
| Microbiome replacement | Possible | Possible | Yes |
| MAUD | Not likely | Recommended | Yes |
| Psychosocial/lifestyle interventions | Recommended | Recommended | Yes |
| Bariatric surgery | Recommended | Safety concerns | Increased AUD, cirrhosis |
Abbreviations: AUD, alcohol use disorder; DNL, de novo lipogenesis; FXR, Farnesoid X receptor; HSD17B13, hydroxysteroid 17-beta dehydrogenase 13; MASLD, metabolic dysfunction–associated steatotic liver disease; MAUD, medications for alcohol use disorder; MetALD, metabolic dysfunction and alcohol-associated liver disease; PPAR, peroxisome proliferator–activated receptor; SGLT2, sodium-glucose–linked transporter 2; THR-β, thyroid hormone receptor beta.
CONCLUSIONS
New terminology, including MetALD, poses challenges and opportunities for general practitioners and hepatologists. In general, not enough is known about the safety, symptom profile, or benefits of emerging therapeutics for MASLD because, in most studies, patients who consume sufficient alcohol to be diagnosed with MetALD have been excluded from clinical trials. Initial focus should be placed on cessation through comprehensive treatment of alcohol use disorder when it is present along with lifestyle and dietary interventions to optimize metabolic risk factors. Along with alcohol cessation and conservative approaches to metabolic disease, incretin-based therapies are promising for both alcohol cessation and metabolic disease. Many other agents evaluated for MASLD have feasible efficacy for MetALD but have not been clearly studied when alcohol use and SLD were intertwined. Further phase 3 and 4 level studies will be needed to better understand the dynamics and efficacy of these agents for MetALD. Given the wealth of potential but untested candidates for treating MetALD, efforts to develop novel therapeutics should be balanced with understanding the use and efficacy of drugs already in clinical development.
Supplementary Material
FUNDING INFORMATION
Harmeet Malhi is supported by NIH Grants DK111378 and AA21788, and the Mayo Foundation.
Abbreviations:
- AASLD
American Association For the Study of Liver Diseases
- ACC
acetyl-CoA carboxylase
- AH
alcohol-associated hepatitis
- ALD
alcohol-associated liver disease
- AUD
alcohol use disorder
- AUDIT
Alcohol Use Disorders Identification Test
- DGATs
diacylglycerol acyltransferases
- DNL
de novo lipogenesis
- FMT
fecal microbiota transplantation
- FXR
Farnesoid X-receptor
- GIP
glucagon inhibitory peptide
- GLP-1ra
glucagon-like peptide 1 receptor agonists
- HSD17B13
hydroxysteroid 17-beta dehydrogenase 13
- MASH
metabolic dysfunction-associated steatohepatitis
- MASLD
metabolic dysfunction–associated steatotic liver disease
- MAUD
medications for alcohol use disorder
- MetALD
metabolic dysfunction and alcohol-associated liver disease
- miRNA
microRNA
- PNPLA3
patatin-like phospholipase domain-containing protein 3
- PPAR
peroxisome proliferator–activated receptor
- SGLT2
sodium-glucose co-transporter 2
- SLD
steatotic liver disease
- T2DM
type 2 diabetes mellitus
- THR-β
thyroid hormone receptor beta
Footnotes
CONFLICTS OF INTEREST
Harmeet Malhi has consulted for Merck. The remaining author has no conflicts to report.
REFERENCES
- 1.Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79:1542–56. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R, Arab JP, Shah VH. Alcohol-associated hepatitis. N Engl J Med. 2022;387:2436–48. [DOI] [PubMed] [Google Scholar]
- 3.Grant BF, Chou SP, Saha TD, Pickering RP, Kerridge BT, Ruan WJ, et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. 2017;74:911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craciun A, Lackner C, Cortez-Pinto H. Nonalcoholic fatty liver disease versus alcohol-related liver disease: Is it really so different? Curr Pharm Des. 2020;26:1093–109. [DOI] [PubMed] [Google Scholar]
- 5.Völzke H Multicausality in fatty liver disease: Is there a rationale to distinguish between alcoholic and non-alcoholic origin? World J Gastroenterol. 2012;18:3492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Åberg F, Färkkilä M. Drinking and obesity: Alcoholic liver disease/nonalcoholic fatty liver disease interactions. Semin Liver Dis. 2020;40:154–62. [DOI] [PubMed] [Google Scholar]
- 7.Kanwal F, Shubrook JH, Adams LA, Pfotenhauer K, Wai-Sun Wong V, Wright E, et al. Clinical care pathway for the risk stratification and management of patients with nonalcoholic fatty liver disease. Gastroenterology. 2021;161:1657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cook GA, Nielsen RC, Hawkins RA, Mehlman MA, Lakshmanan M, Veech RL. Effect of glucagon on hepatic malonyl coenzyme A concentration and on lipid synthesis. J Biol Chem. 1977;252:4421–4. [PubMed] [Google Scholar]
- 9.McGarry J, Foster DW. In support of the roles of malonyl-CoA and carnitine acyltransferase I in the regulation of hepatic fatty acid oxidation andketogenesis. J Biol Chem. 1979;254:8163–8. [PubMed] [Google Scholar]
- 10.Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’shea RS, Dasarathy S, McCullough AJ. Diseases PGCotAAft-SoL, Gastroenterology tPPCotACo. Alcoholic liver disease. Hepatology. 2010;51:307–28. [DOI] [PubMed] [Google Scholar]
- 12.Mathur M, Yeh YT, Arya RK, Jiang L, Pornour M, Chen W, et al. Adipose lipolysis is important for ethanol to induce fatty liver in the National Institute on Alcohol Abuse and Alcoholism murine model of chronic and binge ethanol feeding. Hepatology. 2023;77:1688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S, Williams KJ, Verlande-Ferrero A, Chan AP, Su GB, Kershaw EE, et al. Acute activation of adipocyte lipolysis reveals dynamic lipid remodeling of the hepatic lipidome. J Lipid Res. 2024;65:100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee E, Korf H, Vidal-Puig A. An adipocentric perspective on the development and progression of non-alcoholic fatty liver disease. J Hepatol. 2023;78:1048–62. [DOI] [PubMed] [Google Scholar]
- 15.Jarvis H, O’Keefe H, Craig D, Stow D, Hanratty B, Anstee QM. Does moderate alcohol consumption accelerate the progression of liver disease in NAFLD? A systematic review and narrative synthesis. BMJ Open. 2022;12:e049767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Åberg F, Puukka P, Salomaa V, Männistö S, Lundqvist A, Valsta L, et al. Risks of light and moderate alcohol use in fatty liver disease: Follow-up of population cohorts. Hepatology. 2020;71:835–48. [DOI] [PubMed] [Google Scholar]
- 17.Sogabe M, Okahisa T, Kagawa M, Ueda H, Kagemoto K, Tanaka H, et al. Influence of alcohol on newly developed metabolic dysfunction-associated fatty liver disease in both sexes: A longitudinal study. Clin Nutr. 2023;42:810–6. [DOI] [PubMed] [Google Scholar]
- 18.Israelsen M, Torp N, Johansen S, Hansen CD, Hansen ED, Thorhauge K, et al. Validation of the new nomenclature of steatotic liver disease in patients with a history of excessive alcohol intake: An analysis of data from a prospective cohort study. Lancet Gastroenterol Hepatol. 2024;9:218–28. [DOI] [PubMed] [Google Scholar]
- 19.Hernández-Rubio A, Sanvisens A, Bolao F, Cachón-Suárez I, Garcia-Martín C, Short A, et al. Prevalence and associations of metabolic syndrome in patients with alcohol use disorder. Sci Rep. 2022;12:2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circulation. 2011;123:2292–333. [DOI] [PubMed] [Google Scholar]
- 21.Díaz LA, Arab JP, Louvet A, Bataller R, Arrese M. The intersection between alcohol-related liver disease and non-alcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2023;20:764–83. [DOI] [PubMed] [Google Scholar]
- 22.Vujkovic M, Ramdas S, Lorenz KM, Guo X, Darlay R, Cordell HJ, et al. A multiancestry genome-wide association study of unexplained chronic ALT elevation as a proxy for nonalcoholic fatty liver disease with histological and radiological validation. Nat Genet. 2022;54:761–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trépo E, Valenti L. Update on NAFLD genetics: From new variants to the clinic. J Hepatol. 2020;72:1196–209. [DOI] [PubMed] [Google Scholar]
- 24.Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. 2021;75:1476–84. [DOI] [PubMed] [Google Scholar]
- 25.Buch S, Stickel F, Trépo E, Way M, Herrmann A, Nischalke HD, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47:1443–8. [DOI] [PubMed] [Google Scholar]
- 26.Zintzaras E, Stefanidis I, Santos M, Vidal F. Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology. 2006;43:352–61. [DOI] [PubMed] [Google Scholar]
- 27.Stickel F, Lutz P, Buch S, Nischalke HD, Silva I, Rausch V, et al. Genetic variation in HSD17B13 reduces the risk of developing cirrhosis and hepatocellular carcinoma in alcohol misusers. Hepatology. 2020;72:88–102. [DOI] [PubMed] [Google Scholar]
- 28.Carpino G, Del Ben M, Pastori D, Carnevale R, Baratta F, Overi D, et al. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology. 2020;72:470–85. [DOI] [PubMed] [Google Scholar]
- 29.Wu M-Y, Fan J-G. Gut microbiome and nonalcoholic fatty liver disease. Hepatobiliary Pancreat Dis Int. 2023;22:444–51. [DOI] [PubMed] [Google Scholar]
- 30.Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol. 2020;17:279–97. [DOI] [PubMed] [Google Scholar]
- 31.Carbia C, Bastiaanssen TFS, Iannone LF, García-Cabrerizo R, Boscaini S, Berding K, et al. The microbiome-gut-brain axis regulates social cognition & craving in young binge drinkers. eBioMedicine. 2023;89:104442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bajaj JS, Gavis EA, Fagan A, Wade JB, Thacker LR, Fuchs M, et al. A randomized clinical trial of fecal microbiota transplant for alcohol use disorder. Hepatology. 2021;73:1688–700. [DOI] [PubMed] [Google Scholar]
- 33.Hsu CL, Duan Y, Fouts DE, Schnabl B. Intestinal virome and therapeutic potential of bacteriophages in liver disease. J Hepatol. 2021;75:1465–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Jonge PA, Wortelboer K, Scheithauer TPM, van den Born BH, Zwinderman AH, Nobrega FL, et al. Gut virome profiling identifies a widespread bacteriophage family associated with metabolic syndrome. Nat Commun. 2022;13:3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang L, Lang S, Duan Y, Zhang X, Gao B, Chopyk J, et al. Intestinal virome in patients with alcoholic hepatitis. Hepatology. 2020;72:2182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu CL, Zhang X, Jiang L, Lang S, Hartmann P, Pride D, et al. Intestinal virome in patients with alcohol use disorder and after abstinence. Hepatol Commun. 2022;6:2058–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan L, Feng Y, Du B, Fu H, Tian Z, Xue G, et al. Bacteriophage targeting microbiota alleviates non-alcoholic fatty liver disease induced by high alcohol-producing Klebsiella pneumoniae. Nat Commun. 2023;14:3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kranzler HR, Soyka M. Diagnosis and pharmacotherapy of alcohol use disorder: A review. Jama. 2018;320:815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McPheeters M, O’Connor EA, Riley S, Kennedy SM, Voisin C, Kuznacic K, et al. Pharmacotherapy for alcohol use disorder: A systematic review and meta-analysis. JAMA. 2023;330:1653–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curry SJ, Krist AH, Owens DK, Barry MJ, Caughey AB, Davidson KW, et al. Screening and behavioral counseling interventions to reduce unhealthy alcohol use in adolescents and adults: US Preventive Services Task Force Recommendation Statement. JAMA. 2018;320:1899–909. [DOI] [PubMed] [Google Scholar]
- 41.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–28. [DOI] [PubMed] [Google Scholar]
- 42.Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG Clinical Guideline: Alcoholic liver disease. Am J Gastroenterol. 2018;113:175–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staufer K, Huber-Schönauer U, Strebinger G, Pimingstorfer P, Suesse S, Scherzer TM, et al. Ethyl glucuronide in hair detects a high rate of harmful alcohol consumption in presumed non-alcoholic fatty liver disease. J Hepatol. 2022;77:918–30. [DOI] [PubMed] [Google Scholar]
- 44.Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R, et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. 2024;390:497–509. [DOI] [PubMed] [Google Scholar]
- 45.Thursz M, Gual A, Lackner C, Mathurin P, Moreno C, Spahr L. EASL Clinical Practice Guidelines: Management of alcohol-related liver disease. J Hepatol. 2018;69:154–81. [DOI] [PubMed] [Google Scholar]
- 46.Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67:829–46. [DOI] [PubMed] [Google Scholar]
- 47.Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: A systematic review. J Hepatol. 2012;56:255–66. [DOI] [PubMed] [Google Scholar]
- 48.Younossi ZM, Corey KE, Lim JK. AGA clinical practice update on lifestyle modification using diet and exercise to achieve weight loss in the management of nonalcoholic fatty liver disease: Expert review. Gastroenterology. 2021;160:912–8. [DOI] [PubMed] [Google Scholar]
- 49.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–57. [DOI] [PubMed] [Google Scholar]
- 50.Patton H, Heimbach J, McCullough A. AGA clinical practice update on bariatric surgery in cirrhosis: Expert review. Clin Gastroenterol Hepatol. 2021;19:436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newsome PN, Ambery P. Incretins (GLP-1 receptor agonists and dual/triple agonists) and the liver. J Hepatol. 2023;79:1557–65. [DOI] [PubMed] [Google Scholar]
- 52.Ten Kulve JS, van Bloemendaal L, Balesar R, IJzerman RG, Swaab DF, Diamant M, et al. Decreased hypothalamic glucagon-like peptide-1 receptor expression in type 2 diabetes patients. J Clin Endocrinol Metab. 2016;101:2122–9. [DOI] [PubMed] [Google Scholar]
- 53.van Bloemendaal L, Veltman DJ, ten Kulve JS, Groot PF, Ruhé HG, Barkhof F, et al. Brain reward-system activation in response to anticipation and consumption of palatable food is altered by glucagon-like peptide-1 receptor activation in humans. Diabetes Obes Metab. 2015;17:878–86. [DOI] [PubMed] [Google Scholar]
- 54.Fink-Jensen A, Vilsbøll T. Glucagon-like peptide-1 (GLP-1) analogues: A potential new treatment for alcohol use disorder? Nord J Psychiatry. 2016;70:561–2. [DOI] [PubMed] [Google Scholar]
- 55.Suchankova P, Yan J, Schwandt ML, Stangl BL, Caparelli EC, Momenan R, et al. The glucagon-like peptide-1 receptor as a potential treatment target in alcohol use disorder: Evidence from human genetic association studies and a mouse model of alcohol dependence. Transl Psychiatry. 2015;5:e583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallöf D, Maccioni P, Colombo G, Mandrapa M, Jörnulf JW, Egecioglu E, et al. The glucagon-like peptide 1 receptor agonist liraglutide attenuates the reinforcing properties of alcohol in rodents. Addict Biol. 2016;21:422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leggio L, Hendershot CS, Farokhnia M, Fink-Jensen A, Klausen MK, Schacht JP, et al. GLP-1 receptor agonists are promising but unproven treatments for alcohol and substance use disorders. Nat Med. 2023;29:2993–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Younossi ZM, Stepanova M, Ong J, Yilmaz Y, Duseja A, Eguchi Y, et al. Effects of alcohol consumption and metabolic syndrome on mortality in patients with nonalcoholic and alcohol-related fatty liver disease. Clin Gastroenterol Hepatol. 2019;17:1625–633.e1621. [DOI] [PubMed] [Google Scholar]
- 59.Younossi ZM, Otgonsuren M, Venkatesan C, Mishra A. In patients with non-alcoholic fatty liver disease, metabolically abnormal individuals are at a higher risk for mortality while metabolically normal individuals are not. Metabolism. 2013;62:352–60. [DOI] [PubMed] [Google Scholar]
- 60.Marx N, Husain M, Lehrke M, Verma S, Sattar N. GLP-1 receptor agonists for the reduction of atherosclerotic cardiovascular risk in patients with type 2 diabetes. Circulation. 2022;146:1882–94. [DOI] [PubMed] [Google Scholar]
- 61.Dankner R, Murad H, Agay N, Olmer L, Freedman LS. Glucagon-like peptide-1 receptor agonists and pancreatic cancer risk in patients with type 2 diabetes. JAMA Netw Open. 2024;7:e2350408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Azoulay L, Filion KB, Platt RW, Dahl M, Dormuth CR, Clemens KK, et al. Incretin based drugs and the risk of pancreatic cancer: International multicentre cohort study. BMJ. 2016;352:i581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knapen LM, van Dalem J, Keulemans YC, van Erp NP, Bazelier MT, De Bruin ML, et al. Use of incretin agents and risk of pancreatic cancer: A population-based cohort study. Diabetes Obes Metab. 2016;18:258–65. [DOI] [PubMed] [Google Scholar]
- 64.Wang L, Wang W, Kaelber DC, Xu R, Berger NA. GLP-1 receptor agonists and colorectal cancer risk in drug-naive patients with type 2 diabetes, with and without overweight/obesity. JAMA Oncol. 2023;10:256–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kong L, Dong R, Huang K, Wang X, Wang D, Yue N, et al. Yangonin modulates lipid homeostasis, ameliorates cholestasis and cellular senescence in alcoholic liver disease via activating nuclear receptor FXR. Phytomedicine. 2021;90:153629. [DOI] [PubMed] [Google Scholar]
- 66.Arab JP, Dirchwolf M, Álvares-da-Silva MR, Barrera F, Benítez C, Castellanos-Fernandez M, et al. Latin American Association for the study of the liver (ALEH) practice guidance for the diagnosis and treatment of non-alcoholic fatty liver disease. Ann Hepatol. 2020;19:674–90. [DOI] [PubMed] [Google Scholar]
- 67.Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547–58. [DOI] [PubMed] [Google Scholar]
- 68.Blednov YA, Benavidez JM, Black M, Ferguson LB, Schoenhard GL, Goate AM, et al. Peroxisome proliferator-activated receptors α and γ are linked with alcohol consumption in mice and withdrawal and dependence in humans. Alcohol Clin Exp Res. 2015;39:136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheen AJ. Beneficial effects of SGLT2 inhibitors on fatty liver in type 2 diabetes: A common comorbidity associated with severe complications. Diabetes Metab. 2019;45:213–23. [DOI] [PubMed] [Google Scholar]
- 70.Takahashi H, Kessoku T, Kawanaka M, Nonaka M, Hyogo H, Fujii H, et al. Ipragliflozin improves the hepatic outcomes of patients with diabetes with NAFLD. Hepatol Commun. 2022;6:120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin Z, Yuan Y, Zheng C, Liu S, Weng H. Effects of sodium-glucose co-transporter 2 inhibitors on liver fibrosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: An updated meta-analysis of randomized controlled trials. J Diabetes Complications. 2023;37:108558. [DOI] [PubMed] [Google Scholar]
- 72.Harrison SA, Ruane PJ, Freilich B, Neff G, Patil R, Behling C, et al. A randomized, double-blind, placebo-controlled phase IIa trial of efruxifermin for patients with compensated NASH cirrhosis. JHEP Rep. 2023;5:100563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Desai BN, Singhal G, Watanabe M, Stevanovic D, Lundasen T, Mather ML, et al. Fibroblast growth factor 21 (FGF21) is robustly induced by ethanol and has a protective role in ethanol associated liver injury. Mol Metab. 2017;6:1395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Caddeo A, Kowalik MA, Serra M, Runfola M, Bacci A, Rapposelli S, et al. TG68, a novel thyroid hormone receptor-β agonist for the treatment of NAFLD. Int J Mol Sci. 2021;22:13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24. [DOI] [PubMed] [Google Scholar]
- 76.Esler WP, Cohen DE. Pharmacologic inhibition of lipogenesis for the treatment of NAFLD. J Hepatol. 2024;80:362–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625–43. [DOI] [PubMed] [Google Scholar]
- 78.Neokosmidis G, Cholongitas E, Tziomalos K. Acetyl-CoA carboxylase inhibitors in non-alcoholic steatohepatitis: Is there a benefit? World J Gastroenterol. 2021;27:6522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu XX, Murray SF, Pandey SK, Booten SL, Bao D, Song XZ, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42:362–71. [DOI] [PubMed] [Google Scholar]
- 80.Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials. Nat Med. 2021;27:1836–48. [DOI] [PubMed] [Google Scholar]
- 81.Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN Inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled phase 2a trial. Gastroenterology. 2021;161:1475–86. [DOI] [PubMed] [Google Scholar]
- 82.Ratziu V, De Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat Med. 2021;27:1825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kazierad DJ, Chidsey K, Somayaji VR, Bergman AJ, Birnbaum MJ, Calle RA. Inhibition of ketohexokinase in adults with NAFLD reduces liver fat and inflammatory markers: A randomized phase 2 trial. Med. 2021;2:800–813. e803. [DOI] [PubMed] [Google Scholar]
- 84.Amangurbanova M, Huang DQ, Loomba R. Review article: The role of HSD17B13 on global epidemiology, natural history, pathogenesis and treatment of NAFLD. Aliment Pharmacol Ther. 2023;57:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schwantes-An TH, Darlay R, Mathurin P, Masson S, Liangpunsakul S, Mueller S, et al. Genome-wide association study and meta-analysis on alcohol-associated liver cirrhosis identifies genetic risk factors. Hepatology. 2021;73:1920–31. [DOI] [PubMed] [Google Scholar]
- 86.Cacciottolo TM, Gelson WT, Maguire G, Davies SE, Griffiths WJ. Pi*Z heterozygous alpha-1 antitrypsin states accelerate parenchymal but not biliary cirrhosis. Eur J Gastroenterol Hepatol. 2014;26:412–7. [DOI] [PubMed] [Google Scholar]
- 87.Balcar L, Scheiner B, Urheu M, Weinberger P, Paternostro R, Simbrunner B, et al. Alpha-1 antitrypsin Pi∗Z allele is an independent risk factor for liver transplantation and death in patients with advanced chronic liver disease. JHEP Rep. 2022;4:100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strnad P, Mandorfer M, Choudhury G, Griffiths W, Trautwein C, Loomba R, et al. Fazirsiran for liver disease associated with alpha(1)-antitrypsin deficiency. N Engl J Med. 2022;387:514–24. [DOI] [PubMed] [Google Scholar]
- 89.McDaniel K, Herrera L, Zhou T, Francis H, Han Y, Levine P, et al. The functional role of microRNAs in alcoholic liver injury. J Cell Mol Med. 2014;18:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyagawa S, Horie T, Nishino T, Koyama S, Watanabe T, Baba O, et al. Inhibition of microRNA-33b in humanized mice ameliorates nonalcoholic steatohepatitis. Life Sci Alliance. 2023;6:e202301902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hochreuter MY, Dall M, Treebak JT, Barrès R. MicroRNAs in non-alcoholic fatty liver disease: Progress and perspectives. Mol Metab. 2022;65:101581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qiao C, Yuan Z, Li J, He B, Zheng H, Mayer C, et al. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 2011;18:403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wong VW- S, Ekstedt M, Wong GL- H, Hagström H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J Hepatol. 2023;79:842–52. [DOI] [PubMed] [Google Scholar]
- 94.Whitley HP, Trujillo JM, Neumiller JJ. Special report: Potential strategies for addressing GLP-1 and dual GLP-1/GIP receptor agonist shortages. Clin Diabetes. 2023;41:467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.