

Abstract
BACKGROUND:
Persistent systemic thromboxane generation, predominantly from non-platelet sources, in aspirin (ASA) users with cardiovascular disease (CVD) is a mortality risk factor.
OBJECTIVES:
To determine the mortality risk associated with systemic thromboxane generation in an unselected population irrespective of ASA use.
METHODS:
Stable thromboxane metabolites (TXB2-M) were measured by ELISA in banked urine from 3044 participants (mean age 66±9 years, 53.8% women) in the Framingham Heart Study. The association of TXB2-M to survival over a median observation period of 11.9 years (IQR 10.6, 12.7) was determined by multivariable modeling.
RESULTS:
In 1363 participants (44.8%) taking ASA at the index examination, median TXB2-M was lower than in ASA non-users (1147 vs. 4179 pg/mg creatinine, P<0.0001). TXB2-M was significantly associated with all-cause and cardiovascular mortality irrespective of ASA use (HR 1.96 and 2.41, respectively, P<0.0001 for both) for TXB2-M in the highest quartile based on ASA use compared to lower quartiles, and remained significant after adjustment for mortality risk factors for similarly-aged individuals (HR 1.49 and 1.82, respectively, P≤0.005 for both). In 2353 participants without CVD, TXB2-M was associated with cardiovascular mortality in ASA non-users (adjusted HR 3.04, 95% CI, 1.29, 7.16) but not in ASA users, while ASA use was associated with all-cause mortality in those with low (adjusted HR 1.46, 95% CI 1.14, 1.87) but not elevated TXB2-M.
CONCLUSION:
Systemic thromboxane generation is an independent risk factor for all-cause and cardiovascular mortality irrespective of ASA use and its measurement may be useful for risk stratification, particularly in those without CVD.
Keywords: Thromboxane, aspirin, platelets, mortality, isoprostane
CONDENSED ABSTRACT
Persistent thromboxane generation in aspirin users with cardiovascular disease, originating predominantly from non-platelet sources, is associated with adverse outcome. In 3044 participants in the Framingham Heart Study, thromboxane generation was associated with long-term adjusted all-cause and cardiovascular mortality, irrespective of aspirin use. In participants without cardiovascular disease, thromboxane generation was associated with increased mortality in aspirin non-users whereas aspirin use was associated with increased mortality in those with low levels of thromboxane generation. These findings suggests that measurement of systemic TXA2 generation may be useful in risk stratification, particularly in those without cardiovascular disease.
INTRODUCTION
Thromboxane A2 (TXA2) is an eicosanoid with potent platelet activating and vasoconstrictor properties generated from arachidonic acid by the actions of cyclooxygenase (COX) and downstream thromboxane synthase (TXAS) enzymes.1 In healthy individuals, TXA2 generation is thought to occur mainly in platelets and is effectively inhibited by aspirin (ASA), which irreversibly inhibits the COX-1 enzyme. In over a quarter of individuals with cardiovascular disease (CVD), especially those with increased oxidative stress, substantial amounts of TXA2 is generated in non-platelet tissues that is not fully suppressed by standard ASA therapy.2–5 Several modestly-sized studies of individuals with CVD taking ASA revealed that systemic TXA2 generation was associated with adverse cardiovascular outcome and death, though the mechanism by which this occurs is not fully understood.6–10 It is unknown if systemic TXA2 generation from combined platelet and non-platelet sources in non-ASA users also predicts adverse clinical outcome or if the stimuli for systemic TXA2 generation differ between ASA users and non-users.
To address these knowledge gaps, we measured systemic non-renal TXA2 generation by quantifying stable metabolites of thromboxane B2 (TXB2-M)11 in banked urine samples from participants in the Offspring and Omni cohorts of the Framingham Heart Study and determined its relationship to long-term survival. To help identify differential stimuli for systemic TXA2 generation, we analyzed variables associated with systemic TXA2 generation in participants stratified by ASA use at the time of the index examinations.
METHODS
Study Cohort:
The Framingham Heart Study (FHS) is a longitudinal community-based study comprised of several study cohorts with serial examinations every 4 to 8 years.12 The study population for this investigation included participants in examination cycles 8 (2005–2008) of the Offspring and cycle 3 (2007 to 2008) of the Omni cohorts in whom there was an available urine sample banked at the time of the examination.13,14 At these examinations, attendees underwent a medical history, a cardiovascular-focused physical examination, anthropometric and blood pressure measurements, and laboratory assessment of standard cardiovascular disease risk factors. Written informed consent was obtained by study participants at each examination and study protocols were approved by the human subject institutional review boards of the Boston University School of Medicine and the University of Massachusetts Chan Medical School.
Biologic Assays:
TXB2-M was measured in duplicate in urine samples stored at −80ºC using the AspirinWorks® 11dhTXB2 Test Kit (Corgenix, Bromfield, CO) and normalized to urine creatinine according to the manufacturer’s instruction. The assay has a limit of detection of 156 pg/mL with a linear range up to 5000 pg/mL. Samples with values below the limit of detection were reported as 156 pg/mL while those above 5000 pg/mL were re-assayed at an additional 1:4 dilution and reported as 20,000 pg/mL if they remained above the linear range. The intra-assay coefficient of variance was 3.3%. The stability of TXB2-M measurements with this assay in urine after prolonged storage and multiple freeze-thaw cycles was verified in our laboratory.15 Urine 8-iso-PGF2α , serum C-reactive protein (CRP), interleukin-6 (IL-6), monocyte chemotactic protein (MCP), plasma P-selectin and lipoprotein-associated phospholipase A2 (Lp-PLA2) and other standard laboratory variables had previously been measured in these participants as described16,17 with biomarker measurement manuals available on the FHS website at http://www.framinghamheartstudy.org.
Statistical Analyses:
Descriptive statistics were calculated as usual with ASA use defined by review of medications at the time of the examination. Frequency of categorical variables was compared between groups using the Pearson chi-square test. Means of continuous normally distributed variables were compared using Student’s t-test, while non-normally distributed variables were compared using the Kruskal Wallis test. Multivariable modeling was used to determine variables associated with urinary TXB2-M after natural log transformation using a standard general linear model (GLM). Univariate modeling of variables by ASA use was first conducted to determine the significance of the effect on lnTXB2-M. An additional variable was added to adjust for the ASA dose (dichotomized as a weekly average of ≤ 81 versus >81 mg/day). Variables with significant effects (p ≤ 0.05) on lnTXB2-M were then included in a full GLM and backward elimination of non-significant variables (p-value >0.05) was used to achieve the final parsimonious model.
Cox proportional hazards regression was used to model the relationship between urinary TXB2-M and time to death after confirming the assumption of proportionality was satisfied and accounting for an interaction between ASA use and urinary TXB2-M where applicable. Urinary TXB2-M was considered as a binary variable of respective quartile 4 versus quartiles 1–3 for ASA users and non-users based on the Kaplan-Meier survival plots. The Fine-Gray method was used to account for competing risk when analyzing causes of death.18 Multivariable analysis was used to adjust for relevant predictors of mortality as indicated.
RESULTS
Baseline Characteristics
Of the 3021 Offspring and 298 Omni 1 Cohort participants, urine samples were available and TXB2-M measurable in 3044 (91.7%) participants, of whom 1363 (44.8%) were taking ASA at the time of the index examination. Median urinary TXB2-M was expectedly lower in ASA users compared to non-users with some participants in both groups exhibiting very high levels (Figure 1). Characteristics of participants stratified by ASA use and in those without TXB2-M determination are shown in Table 1. Compared to ASA non-users, ASA users were older, with a higher BMI, more likely to be male and have a higher prevalence of risk factors for/established CVD with associated medical therapy.
Figure 1. Thromboxane generation in aspirin users and non-users.

Systemic thromboxane generation was quantified by measuring urinary thromboxane B2 metabolites (TXB2-M) in 1681 aspirin (ASA) users and 1681 ASA non-users. Median values with interquartile rages (boxes), 5% and 95% confidence intervals (bars) and individual outliers are shown.
Table 1.
Clinical and laboratory characteristics of participants at the index examination stratified by aspirin use.
| Characteristic | ASA Users (N = 1363) | ASA Non-users (N = 1681) | P-value | Missing TXB2-M (N=275) |
|---|---|---|---|---|
| Age, mean (SD) | 68 (8) | 64 (9) | <0.0001 | 73 (10) |
| Female sex, n (%) | 604 (44.3) | 1033 (61.5) | <0.0001 | 212 (77.1) |
| Non-white race, n (%) | 84 (6.2) | 124 (7.5) | 0.1635 | 3 (1.1) |
| Hispanic ethnicity, n (%) | 34 (2.8) | 58 (3.9) | 0.1416 | 9 (3.9) |
| BMI (kg/m2), mean (SD) | 28.8 (5.4) | 28.0 (5.5) | <0.0001 | 28.4 (5.6) |
| eGFR (mL/min/1.73 m2), mean (SD) | 76.0 (16.8) | 81.0 (15.7) | <0.0001 | 71.4 (19.2) |
| Cigarette use, n (%) | 0.2472 | |||
| Current | 90 (6.6) | 138 (8.2) | 27 (9.8) | |
| Former | 47 (3.5) | 54 (3.2) | 14 (5.1) | |
| Never | 1221 (89.9) | 1489 (88.6) | 234 (85.1) | |
| LVEF (%), mean (SD) | 65.8 (7.4) | 66.5 (6.2) | 0.0103 | 65.7 (7.7) |
| Atrial fibrillation/flutter rhythm on ECG, n (%) | 28 (2.1) | 44 (2.6) | 0.3093 | 9 (3.3) |
| Medical history of: | ||||
| Hypertension, n (%) | 902 (66.2) | 628 (37.4) | <0.0001 | 169 (61.4) |
| Hyperlipidemia, n (%) | 822 (60.4) | 463 (27.6) | <0.0001 | 124 (45.3) |
| Diabetes, n (%) | 275 (20.3) | 162 (9.7) | <0.0001 | 41 (22.4) |
| Heart failure, n (%) | 39 (2.9) | 29 (1.7) | 0.0350 | 18 (6.6) |
| Myocardial infarction, n (%) | 205 (15.0) | 88 (5.2) | <0.0001 | 38 (13.8) |
| Atrial fibrillation/flutter, n (%) | 113 (8.3) | 96 (5.7) | 0.0051 | 32 (11.6) |
| Cerebrovascular disease, n (%) | 110 (8.1) | 97 (5.8) | 0.0122 | 41 (14.9) |
| Peripheral vascular disease, n (%) | 88 (6.5) | 42 (2.5) | <0.0001 | 26 (9.5) |
| Coronary revascularization, n (%) | 306 (22.5) | 83 (4.9) | <0.0001 | 38 (13.8) |
| PCI, n (%) | 203 (14.9) | 65 (3.9) | <0.0001 | 23 (8.4) |
| CABG surgery, n (%) | 155 (11.4) | 26 (1.6) | <0.0001 | 20 (7.3) |
| Valvular heart surgery, n (%) | 41 (3.0) | 19 (1.1) | 0.0002 | 10 (3.6) |
| COPD, n (%) | 104 (7.6) | 114 (6.8) | 0.3666 | 30 (10.9) |
| DVT/PE, n (%) | 21 (1.5) | 39 (2.3) | 0.1240 | 8 (2.9) |
| Cancer, n (%) | 390 (28.6) | 382 (22.7) | 0.0002 | 78 (28.4) |
| Medication use: | ||||
| Aspirin dose >81 mg/d, n (%) | 353 (25.9) | 29/129 (22.5) | ||
| NSAID, n (%) | 315 (23.1) | 453 (27.0) | 0.0154 | 63 (22.9) |
| Antihypertensive therapy n (%) | 51 (3.7) | 35 (2.1) | 0.0060 | 6 (2.2) |
| Beta-blocker, n (%) | 540 (39.6) | 275 (16.4) | <0.0001 | 110 (40.0) |
| ACEi/ARB, n (%) | 622 (45.6) | 385 (22.9) | <0.0001 | 106 (38.6) |
| Lipid therapy, n (%) | 893 (65.5) | 578 (34.4) | <0.0001 | 131 (47.6) |
| Statin, n (%) | 776 (56.9) | 429 (25.5) | <0.0001 | 115 (41.8) |
| Non-statin, n (%) | 117 (8.6) | 149 (8.9) | 0.7858 | 16 (5.8) |
| Diuretic, n (%) | 423 (31.0) | 323 (19.2) | <0.0001 | 82 (29.8) |
| Insulin, n (%) | 38 (2.8) | 11 (0.7) | <0.0001 | 12 (4.4) |
| Non-insulin diabetic therapy, n (%) | 179 (13.1) | 103 (6.1) | <0.0001 | 24 (8.7) |
| Oral anticoagulant, n (%) | 52 (3.8) | 102 (6.1) | 0.0048 | 18 (6.6) |
| Laboratory data: | ||||
| Serum creatinine (mg/dL), mean (SD) | 0.95 (0.30) | 0.88 (0.27) | <0.0001 | 1.00 (0.81) |
| Fasting plasma glucose (mg/dL) | 110 (27) | 104 (21) | <0.0001 | 105 (22) |
| Hemoglobin A1C (%), mean (SD) | 5.8 (0.8) | 5.7 (0.6) | <0.0001 | 5.8 (0.6) |
| Plasma lipid profile: | ||||
| Total cholesterol (mg/dL), mean (SD) | 175 (36) | 195 (36) | <0.0001 | 186 (38) |
| LDL cholesterol (mg/dL), mean (SD) | 97 (30) | 112 (32) | <0.0001 | 104 (31) |
| HDL (mg/dL), mean (SD) | 55 (17) | 60 (18) | <0.0001 | 56 (17) |
| Triglyceride (mg/dL), mean (SD) | 119 (75) | 116 (68) | 0.2351 | 131 (67) |
| Serum CRP (mg/L), mean (SD) | 3.2 (7.4) | 3.4 (7.3) | 0.4580 | 4.2 (7.1) |
| Serum insulin (pmol/L), mean (SD) | 83.2 (65.9) | 70.5 (47.1) | <0.0001 | 82.4 (72.2) |
| Serum MCP (pg/mL), mean (SD) | 383 (140) | 380 (136) | 0.5633 | 420 (136) |
| Serum IL-6 (pg/mL), mean (SD) | 2.74 (2.94) | 2.51 (2.93) | 0.0395 | 3.38 (3.66) |
| Plasma P-selectin (ng/mL), mean (SD) | 41.2 (13.8) | 41.3 (13.5) | 0.8124 | 40.1 (13.8) |
| Plasma Lp-PLA2 (ng/mL), mean (SD) | 195 (52) | 204 (48) | <0.0001 | 204 (55) |
| Urine 8-isoPGF2α (pg/mg creatinine), mean (SD) | 1096 (599) | 1149 (663) | 0.0203 | 949 (334) |
| Urine albumin-creatinine ratio (mg/g), median (IQR) | 61.2 (34.3, 137.8) | 59.2 (34.9, 114.8) | 0.2125 | 68.0 (42.7, 201.0) |
Abbreviations: SD, standard deviation; BMI, body mass index; eGFR, estimated glomerular filtration rate; LVEF, left ventricular ejection fraction; ECG, electrocardiogram; PCI, percutaneous coronary intervention; CABG, coronary artery bypass graft; COPD, chronic obstructive pulmonary disease; DVT, deep vein thrombosis; PE, pulmonary embolus; ACEi, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker; LDL, low-density lipoprotein; HDL, high-density lipoprotein; CRP, C-reactive protein; MCP, macrophage chemotactic factor; IL-6, interleukin-6; Lp-PLA2, lipoprotein-associated phospholipase A2; IQR, interquartile range.
Determinants of Systemic Thromboxane Generation
Previous studies in ASA users with CVD identified age, sex and oxidative stress as major independent determinants of non-platelet TXA2 generation, with ASA dose, race, lipid therapy, left ventricular ejection fraction and renal function identified as minor independent determinants.2,7,10,19,20 Multivariable modeling was performed to determine variables associated with TXA2 generation in this unselected population and identify differences based on ASA use. Supplemental Table 1 shows univariate regression analyses of demographic and laboratory variables available at the time of the index examination. Multivariable modeling revealed that age, sex, oxidative stress and renal function were independently associated with TXA2 generation irrespective of ASA use, as were cigarette use and the inflammatory markers IL-6 and P-selectin (Table 2). ASA dose, diabetes, proteinuria and atrial fibrillation/flutter were independently associated with TXB2-M in ASA users while NSAID use, hypertension, oral anticoagulant use, COPD and HDL were associated with TXB2-M in non-ASA users.
Table 2.
Variables independently associated with urinary TXB2-Ma by multivariable regression analysis.
| ASA Users (N=1294) |
ASA Non-users (N=1600) |
|||
|---|---|---|---|---|
| Variable | Standardized Regression Coefficient | P-value | Standardized Regression Coefficient | P-value |
| eGFR (per mL/min/1.73m2) | 0.235 | <0.0001 | 0.200 | <0.0001 |
| Age (per year) | 0.180 | <0.0001 | 0.178 | <0.0001 |
| Female sex (versus male) | 0.139 | <0.0001 | 0.139 | <0.0001 |
| Cigarette use (versus never) | ||||
| Current | 0.125 | <0.0001 | 0.046 | 0.0542 |
| Former | 0.061 | 0.0084 | ||
| Urinary 8-isoPGF2α (per pg/mg creatinine) | 0.069 | 0.0114 | 0.097 | <0.0001 |
| IL-6 (per pg/mL) | 0.066 | 0.0128 | 0.089 | 0.0003 |
| P-selectin (per pg/mL) | 0.064 | 0.0160 | 0.065 | 0.0058 |
| ASA dose (>81versus ≤81mg/day) | 0.158 | <0.0001 | ||
| Urine albumin-creatinine ratio (per ln mg/g) | 0.107 | 0.0002 | ||
| Diabetes (versus none) | 0.097 | 0.0003 | ||
| Lipid therapy (versus none) | -0.063 | 0.0162 | ||
| Atrial fibrillation/flutter (versus never) | 0.054 | 0.0409 | ||
| NSAID use (versus none) | -0.144 | <0.0001 | ||
| Oral anticoagulant use (versus none) | 0.122 | <0.0001 | ||
| HDL (per mg/dL) | -0.107 | <0.0001 | ||
| Hypertension (versus none) | 0.076 | 0.0021 | ||
| COPD (versus none) | 0.049 | 0.0399 | ||
Abbreviations: eGFR, estimated glomerular filtration rate; IL-6, interleukin-6; ASA, aspirin; NSAID, non-steroidal antiinflammatory drug; HDL, high density lipoprotein; COPD, chronic obstructive pulmonary disease.
Ln pg/mg creatinine.
Association of Systemic Thromboxane Generation with Long-term Survival
Survival data was available for 3043 (99.9%) participants in whom urinary TXB2-M was measured, 701 (23.0%) of whom died during a median observation period of 11.9 years (IQR, 10.6, 12.7 years) from the index examination. Long-term survival was significantly lower in ASA users compared to non-users (Figure 2) and was significantly associated with the degree of systemic TXA2 generation irrespective of ASA use (Figures 3). The association between urinary TXB2-M and all-cause mortality was present in a wide array of subgroups irrespective of ASA use (Figure 4 and Supplemental Figure 1). Urinary TXB2-M was significantly associated with increased risk of cardiovascular death in both ASA groups, but not with stroke in either ASA group (Table 3).
Figure 2. Aspirin use and all-cause mortality risk.

All-cause mortality risk was significantly greater in the 1363 aspirin (ASA) users (blue line) compared to the 1680 ASA non-users (red line) at the time of the index examination. Number at risk over time in each group is indicated.
Figure 3. Thromboxane generation and all-cause mortality risk.

Systemic thromboxane generation was quantified by measuring urinary thromboxane B2 metabolites (TXB2-M). In 1363 aspirin (ASA) users (A), 1680 ASA non-users (B) and their combination (C) at the time of the index examination, all-cause mortality risk was greatest in participants with TXB2-M in the highest quartile compared to lower quartiles. Number at risk over time in each group is indicated.
Figure 4. Thromboxane generation and all-cause mortality risk by participant characteristics.
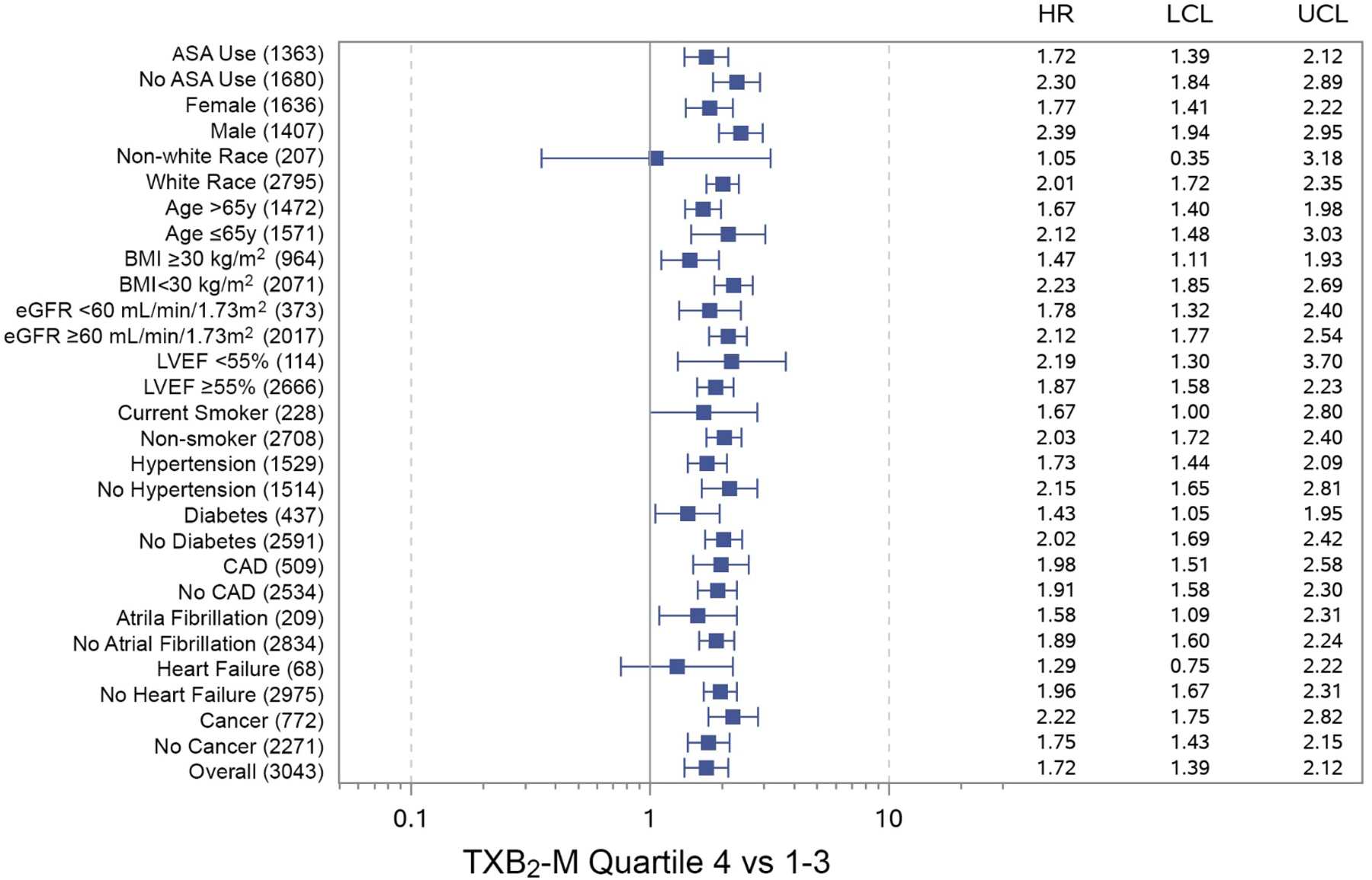
Systemic thromboxane generation was quantified by measuring urinary thromboxane B2 metabolites (TXB2-M) and participant subgroups were stratified by respective quartile 4 versus quartiles 1–3 based on aspirin (ASA) use at the time of the index examination. Urinary TXB2-M in the highest quartile was associated with increased all-cause mortality risk in a wide array of participant subgroups, irrespective of ASA use. Number of participants at risk shown in parentheses. HR=unadjusted hazard ratio; UCL=upper 95% confidence limit; LCL=lower 95% confidence limit. ASA=aspirin; BMI=body-mass index; CAD=coronary artery disease; eGFR=estimated glomerular filtration rate; LVEF=left ventricular ejection fraction.
Table 3.
Association of urinary TXB2-Ma with unadjusted mortality risk by cause of death.
| Cause of Death | ASA User (N=1363) | ASA Non-user (N=1680) | All (N=3043)b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N (%) | HR | 95% CI | P-value | N (%) | HR | 95% CI | P-value | N (%) | HR | 95% CI | P-value | |
| Any | 389 (28.5) | 1.72 | 1.39–2.13 | <0.0001 | 312 (18.6) | 2.30 | 1.83–2.89 | <0.0001 | 701 (23.0) | 1.96 | 1.68–2.29 | <0.0001 |
| CVD | 88 (6.5) | 1.87 | 1.22–2.88 | 0.0043 | 45 (2.7) | 3.87 | 2.15–6.97 | <0.0001 | 133 (4.4) | 2.41 | 1.71–3.39 | <0.0001 |
| Stroke | 22 (1.6) | 0.91 | 0.34, 2.45 | 0.8448 | 7 (0.4) | 1.22 | 0.23–6.29 | 0.8166 | 29 (1.0) | 0.97 | 0.41–2.27 | 0.9431 |
| Cancer | 113 (8.3) | 1.28 | 0.85–1.91 | 0.2388 | 117 (7.0) | 2.02 | 1.39–2.92 | 0.0002 | 230 (7.6) | 1.63 | 1.24–2.13 | 0.0005 |
| Other | 136 (10.0) | 1.61 | 1.13–2.30 | 0.0087 | 125 (7.4) | 1.97 | 1.37–2.83 | 0.0002 | 261 (8.6) | 1.78 | 1.38–2.29 | <0.0001 |
| Unknown | 30 (2.2) | 2.37 | 1.15–4.87 | 0.0192 | 18 (1.1) | 1.17 | 0.42–3.25 | 0.7708 | 48 (1.6) | 1.83 | 1.02–3.28 | 0.0418 |
Abbreviations: HR, hazard ratio; CI, confidence interval; CVD, cardiovascular disease.
Quartile 4 versus quartiles 1–3.
P-value not significant for interaction between ASA use and urinary TXB2-M in all categories of death.
To understand the strength of association between systemic TXA2 generation and mortality, multivariable modeling was performed to adjust for known predictors of all-cause mortality in individuals of similar age (Table 4). Urinary TXB2-M remained significantly associated with all-cause mortality irrespective of ASA use when adjusted for age and sex (Model 2), oxidative stress and inflammation (Model 3), modifiable CVD risk factors (Model 4), or a combination of known predictors of death from heart disease, stroke, diabetes and kidney disease (Model 5). Urinary TXB2-M was also was significantly associated with cardiovascular mortality risk when adjusted for the same variables (Models 1–4, Table 5). When adjusted for known predictors of cardiovascular death (Model 5), urinary TXB2-M was associated with cardiovascular death in ASA non-users but not in ASA users.
Table 4.
Association of urinary TXB2-Ma with all-cause mortality risk when adjusted for relevant predictors of survival.
| Model | ASA User (N=1363) | ASA Non-user (N=1680) | All (N=3043)b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Model 1c | 1.72 | 1.39, 2.12 | <0.0001 | 2.30 | 1.84, 2.89 | <0.0001 | 1.96 | 1.68, 2.29 | <0.0001 |
| Model 2d | 1.58 | 1.28, 1.96 | <0.0001 | 1.68 | 1.34, 2.12 | <0.0001 | 1.63 | 1.40, 1.91 | <0.0001 |
| Model 3e | 1.67 | 1.34, 2.08 | <0.0001 | 1.87 | 1.47, 2.38 | <0.0001 | 1.75 | 1.49, 2.06 | <0.0001 |
| Model 4f | 1.58 | 1.27, 1.97 | <0.0001 | 2.13 | 1.69, 2.70 | <0.0001 | 1.79 | 1.53, 2.10 | <0.0001 |
| Model 5g | 1.32 | 1.03, 1.70 | 0.0288 | 1.60 | 1.22, 2.10 | 0.0007 | 1.43 | 1.19, 1.71 | 0.0001 |
Abbreviations: ASA, aspirin; HR, hazard ratio; CI, confidence interval; BMI, body mass index; IL-6, interleukin 6; LVEF, left ventricular ejection fraction; eGFR, estimated glomerular filtration rate.
Quartile 4 versus quartiles 1–3.
P-value not significant for interaction between ASA use and urinary TXB2-M in all models.
Unadjusted.
Adjusted for age and sex.
Adjusted for 8-isoPGF2α, IL-6 and P-selectin. (N=1299 for ASA and N=1600 for non-ASA groups.)
Adjusted for systolic blood pressure, total cholesterol/HDL ratio, current smoking status, BMI and hemoglobin A1C. (N =1346 for ASA and N=1664 for non-ASA groups.)
Adjusted for age, sex, systolic blood pressure, total cholesterol/HDL ratio, current smoking status, atrial fibrillation, LVEF, hemoglobin A1C, eGFR and IL-6. (N =1186 for ASA and N=1467 for non-ASA groups.)
Table 5.
Association of urinary TXB2-Ma with cardiovascular mortality risk when adjusted for relevant predictors of survival.
| Model | ASA User (N=1363) | ASA Non-user (N=1680) | All (N=3043)b | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Model 1c | 1.87 | 1.22, 2.88 | 0.0043 | 3.87 | 2.15, 6.97 | <0.0001 | 2.41 | 1.71, 3.39 | <0.0001 |
| Model 2d | 1.69 | 1.08, 2.64 | 0.0219 | 2.69 | 1.46, 4.96 | 0.0015 | 1.93 | 1.35, 2.76 | 0.0003 |
| Model 3e | 1.68 | 1.06, 2.65 | 0.0259 | 3.69 | 1.95, 6.96 | <0.0001 | 2.11 | 1.46, 3.06 | <0.0001 |
| Model 4f | 1.65 | 1.02, 2.65 | 0.0397 | 3.56 | 1.92, 6.62 | <0.0001 | 2.13 | 1.47, 3.09 | <0.0001 |
| Model 5g | 1.36 | 0.77, 2.38 | 0.2898 | 2.82 | 1.39, 5.66 | 0.0041 | 1.70 | 1.10, 2.62 | 0.0163 |
Abbreviations: ASA, aspirin; HR, hazard ratio; CI, confidence interval; BMI, body mass index; IL-6, interleukin 6; LVEF, left ventricular ejection fraction; eGFR, estimated glomerular filtration rate.
Quartile 4 versus quartiles 1–3.
P-value not significant for interaction between ASA use and urinary TXB2-M in all models.
Unadjusted.
Adjusted for age and sex.
Adjusted for 8-isoPGF2α, IL-6 and P-selectin. (N=1299 for ASA and N=1600 for non-ASA groups.)
Adjusted for systolic blood pressure, total cholesterol/HDL ratio, current smoking status, BMI and hemoglobin A1C. (N =1346 for ASA and N=1664 for non-ASA groups.)
Adjusted for age, sex, systolic blood pressure, total cholesterol/HDL ratio, current smoking status, LVEF, hemoglobin A1C, eGFR and IL-6. (N =1186 for ASA and N=1467 for non-ASA groups.)
Given that ASA was frequently used for primary prevention during the time-period of the index examinations (2005–2008), we investigated the association of urinary TXB2-M with all-cause and cardiovascular mortality in the 2353 participants without established CVD. The risk of all-cause but not cardiovascular mortality was greater in the 898 (38.2%) participants taking ASA compared to the 1455 (61.8%) not taking ASA at the index examination (Figure 5). Survival plots of ASA users and non-users stratified by urinary TXB2-M quartile grouping are shown in Figure 6. Analyses accounting for the interaction between urinary TXB2-M and ASA use revealed the former to be robustly associated with all-cause and cardiovascular mortality in participants not taking ASA but not in those taking ASA at the index examination, associations that remained significant after adjustment for modifiable CVD risk factors (Figure 7A). ASA use was associated with increased risk of all-cause and cardiovascular mortality in participants with low levels (quartiles 1–3) but not with high levels (quartile 4) of urinary TXB2-M (Figure 7B).
Figure 5. Aspirin use and mortality risk in participants without cardiovascular disease.
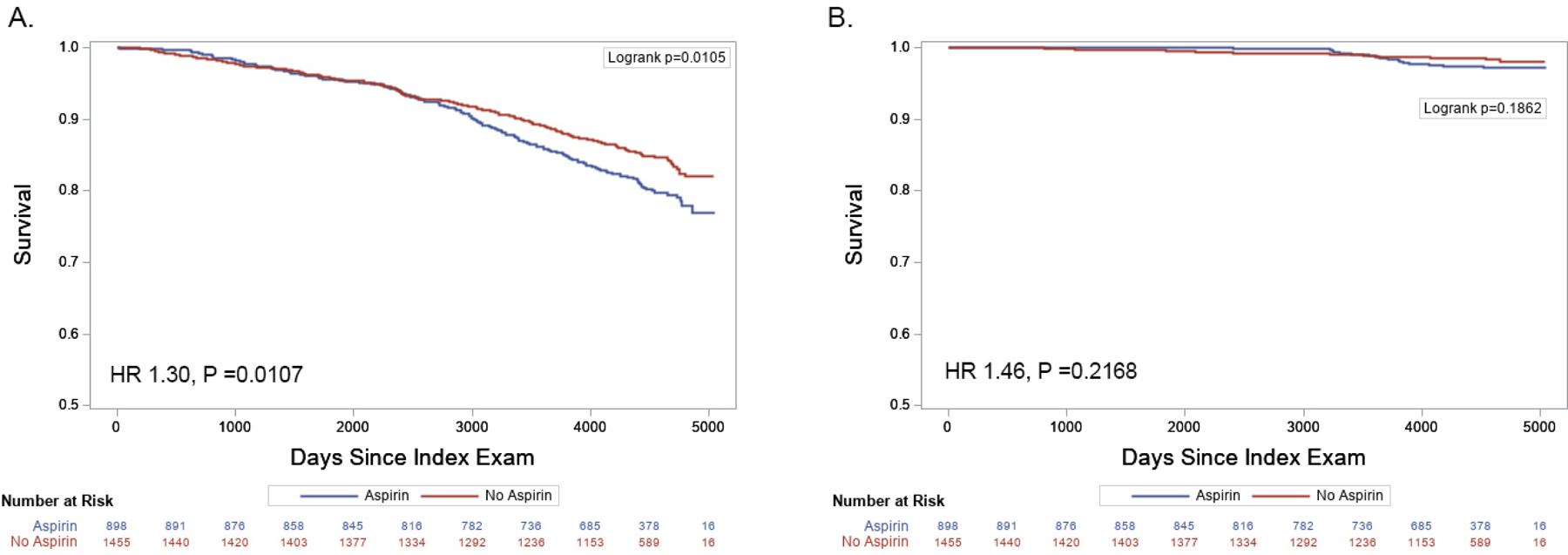
All-cause mortality (A) but not cardiovascular mortality (B) risk was greater in the 898 participants taking ASA (blue line) compared to the 1455 not taking ASA (red line) at the time of the index examination. Number at risk over time in each group is indicated.
Figure 6. Thromboxane generation and mortality risk in participants without cardiovascular disease.

Systemic thromboxane generation was quantified by measuring urinary thromboxane B2 metabolites (TXB2-M) in 2353 participant stratified by aspirin (ASA) use and without cardiovascular disease at the index examination. Urinary TXB2-M in the highest quartile (blue line) was associated with grater all-cause mortality risk compared to lower quartiles (red line) in 898 ASA users (A) and 1455 ASA non-users (B) and was associated with increased cardiovascular mortality risk in ASA non-users (D) but not in ASA users (C). Number at risk over time in each group is indicated.
Figure 7. Interaction of aspirin use and thromboxane generation with mortality risk.

Systemic thromboxane generation was quantified by measuring urinary thromboxane B2 metabolites (TXB2-M) in 2353 participant without established cardiovascular disease stratified by aspirin (ASA) use at the index examination. Interaction analyses revealed all-cause and cardiovascular mortality risk was greater in ASA non-users, but not ASA users with urinary TXB2-M in the highest quartile (A) as well as in ASA users with urinary TXB2-M in the lowest quartiles (B), even after adjustment for modifiable cardiovascular risk factors. Numbers of participants at risk shown in parentheses. HR=unadjusted hazard ratio; UCL=upper 95% confidence limit; LCL=lower 95% confidence limit. ASA=aspirin; BMI=body mass index. aUnadjusted. bAdjusted for systolic blood pressure, total cholesterol/HDL ratio, smoking status, BMI and hemoglobin A1C.
DISCUSSION
The major findings of this study (Central Illustration) include: 1) Systemic TXA2 generation is significantly associated with long-term all-cause and cardiovascular mortality in a large unselected population irrespective of ASA use and remains so after adjustment for known risk factors for CVD and mortality, and; 2) In participants without CVD, systemic TXA2 generation is associated with all-cause and cardiovascular mortality after adjustment for modifiable CVD risk factors in ASA non-users, but not in ASA users, while ASA use is associated with increased mortality in those with lower levels, but not in those with high levels, of systemic TXA2 generation.
Central Illustration. Schematic of systemic thromboxane generation and association with long-term mortality.

Aspirin (ASA) effectively suppresses thromboxane A2 (TXA2) generation in platelets but not in endothelial cells under oxidative stress or activated inflammatory cells. TXA2 generation and signaling through cellular thromboxane-prostanoid receptors has been implicated in multiple disease processes and increased systemic TXA2 generation, as evidenced by elevated levels of stable urinary thromboxane B2 metabolites (TXB2-M), is associated with reduced long-term survival irrespective of ASA use. In individuals without established cardiovascular disease, measurement of urinary TXB2-M may be useful to identify those who would benefit from or be harmed by ASA therapy for primary prevention.
TXA2 generation is most recognizably associated with platelet activation, both as an intracellular amplifier of signaling in response to multiple physiologic agonists and as a potent extracellular agonist via binding to surface thromboxane-prostanoid receptors (TPr).1 While persistent systemic TXA2 generation despite ASA use was initially thought to be due to incomplete ASA-mediated suppression of platelet COX-1, it is now appreciated that daily ASA doses of 81–325 mg are effective at inhibiting platelet TXA2 generation in the vast majority of individuals and that residual TXA2 generation in compliant ASA users originates predominantly from non-platelet tissues.2–5,21,22 The circulating half-life of ASA is ~20 minutes with negligible blood levels 6–8 hours after ingestion of a single 81mg dose.23 Unlike platelets that lack capacity to regenerate COX-1 irreversibly acetylated by ASA, nucleated cells can produce TXA2 following regeneration of COX-1 or by an inducible COX-2-mediated pathway that is not effectively inhibited by standard doses of ASA.24 Though standard ASA therapy does not completely suppress TXA2 generation in non-platelet tissue, the inverse association between TXB2-M and ASA dose observed in ours and other studies suggest that there is at least some degree of suppression.2,7
Prior studies identifying an association of systemic TXA2 generation with adverse outcome were mostly performed in ASA users with established CAD.6–10 To our knowledge, ours is the largest study to measure systemic TXA2 generation in an unselected population and the first to identify strong associations with all-cause and cardiovascular mortality risk in both ASA users and non-users. Importantly, the association of systemic TXA2 generation with mortality remained strong after adjustment for age, sex and oxidative stress, previously identified risk factors for non-platelet TXA2 generation, as well as markers of inflammation, cardiovascular disease, renal disease and diabetes that are recognized predictors of survival in middle-aged and older individuals. Association of systemic TXA2 generation with mortality risk was further observed in a diverse array of subgroups irrespective of ASA use, including those without established CVD at the time of the index examination.
Particularly intriguing was the observed interaction between ASA use and systemic TXA2 generation in participants without CVD and their respective association with mortality risk. ASA use alters systemic TXA2 generation, an effect that was accounted for by using quartile groupings of urinary TXB2-M based on ASA use. While we did not find significant interactions between ASA use and urinary TXB2-M in the full study cohort, we did observe a significant interaction in the subgroup of participants without CVD. Accounting for that interaction, the mortality risk associated with urinary TXB2-M and ASA use were interdependent and remained significant after adjustment for modifiable CVD risk factors: Elevated urinary TXB2-M was associated with increased mortality risk only in ASA non-users and ASA use was associated with increased mortality risk only in those with lower levels of urinary TXB2-M. While the indications for ASA use in individuals without CVD have evolved substantially over the last few years, uncertainty remains regarding its potential benefit in certain high-risk groups.25 Our data raise the possibility that measurement of urine TXB2-M in individuals without established CVD might potentially help identify those who could benefit or be harmed by ASA therapy for primary prevention, a premise that warrants evaluation in future studies.
Major in vivo sources of non-platelet TXA2 generation have yet to be identified but likely differ among various patient populations. Like previous studies, we found a strong association of urinary TXB2-M irrespective of ASA use with oxidative stress as measured by urinary 8-iso-PGF2α.2,19,26,27 Isoprostanes, such as 8-iso-PGF2α, are formed non-enzymatically from arachidonic acid under conditions of oxidative stress. 8-iso-PGF2α is not only a marker of oxidative stress but it is also a biologically active stimulatory ligand of TPr, though of lower affinity than TXA2 28 Oxidative stress is intimately involved in the pathobiology of a myriad of human diseases and is the sine qua non of endothelial dysfunction.29 In vitro studies by our group and others revealed that macro- and microvascular endothelial cells exposed to oxidative stress generate both TXA2 and 8-isoPGF2α, and that 8-isoPGF2α can directly stimulate TXA2 generation via activation of the TPr.2,30,31 We also observed an independent association between urinary TXA2-M and IL-6, an inflammatory cytokine produced by activated monoctes/macrophages and activated endothelial cells that has been associated with adverse cardiovascular outcome.32 TXA2 is generated in monocytes via an inducible COX-2 pathway where it influences acquired immunity.33 In a study of cardiac bypass graft surgery patients in whom platelet TXA2 was documented to be completely suppressed, we found non-platelet TXA2 generation in the early post-operative period to be 3-fold greater and predominantly associated with the degree of inflammation compared to 6 months later when the intense post-operative inflammation had subsided and urinary TXB2-M was more strongly associated with oxidative stress.9 In a study of hypertensive individuals not taking ASA the administration of a selective COX-2 inhibitor did not suppress systemic TXA2 generation, suggesting that mononuclear cells may only be a significant source of systemic TXA2 generation in situations of intense inflammation.34
While TXA2 generation in platelets is clearly a key process in the development of atherothrombosis, it is not clear how TXA2 generation in non-platelet tissue adversely influences outcome. Evidence does not suggest that TXA2 generated in non-platelet tissue causes pathologic thrombosis by stimulating platelet activation. The integrin P-selectin is highly expressed in both endothelial cells and platelets where it mediates adhesion of neutrophils and platelets to the endothelium and its soluble form is thought to predominantly originate from activated platelets.35 Median plasma P-selectin levels in our cohort did not differ between ASA users and non-users, suggesting similar degrees of platelet reactivity in both groups, despite a more than three-fold higher median urinary TXB2-M in the latter. Though non-platelet TXA2 generation was associated with an increased risk of early graft thrombosis and death after coronary artery bypass graft surgery, that risk was independent of measures of platelet reactivity and the risk of adverse outcome associated with increased systemic TXA2 generation in ASA users with CVD was not attenuated by the addition of the ADP-receptor antagonist clopidogrel.7,8 TPr is expressed to variable degrees in nearly all tissues of the body and its activation is known to elicits a myriad of biological effects in non-platelet tissue, including vasoconstriction, atherogenesis, fibrosis, inflammation, immune modulation and cancer progression.36–38 Increased systemic TXA2 generation has been observed in several conditions where these non-platelet TPr signaling effects are known to play prominent pathologic roles. For example, TXA2 is a potent systemic/intrarenal vasoconstrictor and urinary TXB2-M has been shown to be elevated in patients with heart failure and proportional to its severity.39,40 TPr activation is arrhythmogenic in cardiomyocytes by altering intracellular calcium signaling and urinary TXB2-M is elevated in atrial fibrillation where it associates with adverse outcomes.41–43 TPr expression is increase in a variety of cancer types and increased urine TXB2-M has been observed in patients with bladder, prostate, breast and colorectal cancer.44–47 Our study adds to the body of evidence linking TXA2 generation and TPr signaling with malignancy by showing a strong association of systemic TXA2 generation to cancer mortality.
The robust association between systemic TXA2 generation and mortality observed in our study provides a rationale to investigate whether inhibiting non-platelet TXA2 generation or blocking its biological effects could improve survival in patients with CVD and potentially other diseases. Extended-release ASA could theoretically achieve more persistent inhibition of non-platelet COX-1 activity than standard once daily ASA therapy. A micronized formulation that leads to sustained blood levels of acetylsalicylic acid has been shown to effectively reduce systemic TXA2 generation in healthy individuals, though it has not been evaluated in populations with high degrees of non-platelet TXA2 generation.23 Thromboxane synthase inhibitors and TPr antagonists also have the potential to suppress both platelet and non-platelet TXA2 generation without impacting other prostaglandin pathways. While initially developed as antiplatelet agents, TPr antagonists are currently being evaluated in human clinical trials to improve endothelial function, reduce cardiac and pulmonary fibrosis as well as suppress cancer metastases.48–51
Study Limitations
There are several acknowledged limitations of our study. ASA use was determined at the time of the index examination and used to phenotypically stratify participants into those whose urine TXB2-M reflects predominantly non-platelet-TXA2 generation (ASA users) and those in whom it reflects both platelet and non-platelet TXA2 generation (ASA non-users) at a given point in time. We could not account for changes in use of ASA or other medications over time that could potentially impact survival. Changes in the magnitude of systemic TXA2 generation overtime could also potentially alter risk. This study and others revealed that urinary TXB2-M exhibits an inverse relationship to renal function.2 This is unlikely to be a significant confounder because it is small in magnitude (e.g., for every 10 mL/min/1.73m2 decrease in eGRF urinary TXB2-M increases by 1.1 pg/mg creatinine) and would tend to diminish the correlation of urinary TXB2-M to death. The Offspring cohort of the Framingham Heart Study enrolled almost exclusively white individuals of European ancestry. While 298 participants enrolled in the minority Omni cohort were included in our analysis, non-white participants constituted <10% of the entire study population. Although previous studies have indicated that non-whites have higher levels of non-platelet TXA2 generation than whites, we could not adequately investigate the impact of race on outcome due to this underrepresentation of minorities in the study cohort.
Conclusions
Systemic TXA2 generation is an independent risk factor for long-term all-cause and cardiovascular mortality in an unselected cohort of older individuals irrespective of ASA use. Measuring TXA2 generation in individuals without CVD may be useful in guiding the aggressiveness of primary prevention strategies, including use of ASA. These findings provide a rationale for investigating whether inhibiting TXA2 generation or blocking its effects could impact survival in individuals with CVD and potentially other conditions.
Supplementary Material
PERSPECTIVES.
Competency in Medical Knowledge:
Aspirin reduces cardiovascular risk by inhibiting thromboxane generation in platelets, though it is much less effective at inhibiting thromboxane generation in non-platelet tissues. Systemic thromboxane generation is robustly associated with all-cause and cardiovascular mortality in a general population of both aspirin users and non-users.
Translational Outlook 1:
Measurement of systemic thromboxane generation may help to better risk stratify individuals irrespective of aspirin use, especially in those without established cardiovascular disease who may benefit from aggressive primary prevention.
Translational Outlook 2:
Inhibiting or blocking the effects of non-platelet thromboxane generation may be a novel strategy to improve outcomes in individuals with high levels of systemic thromboxane generation.
Acknowledgements:
We also acknowledge the dedication of the FHS study participants without whom this research would not be possible.
Funding:
This study was supported by a grant from American Heart Association (17GRNT3360007 to JJR). The parent FHS was supported by contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute. Dr. Vasan is supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine. The authors had sole control of the design of the study, collection, analyses and dissemination of the data.
ABREVIATIONS
- ASA
aspirin
- BMI
body mass index
- COPD
chronic obstructive pulmonary disease
- COX
cyclooxygenase
- CVD
cardiovascular disease
- eGFR
estimated glomerular filtration rate
- FHS
Framingham Heart Study
- LVEF
left ventricular ejection fraction
- TPr
thromboxane-prostanoid receptor
- TXA2
thromboxane A2
- TXAS
thromboxane synthase
- TXB2-M
thromboxane B2-metabolites
Footnotes
Disclosures: None of the authors have any disclosures.
REFERENCES
- 1.Patrono C, Ciabattoni G, Davi G. Thromboxane biosynthesis in cardiovascular diseases. Stroke 1990;21:IV130–IV133. [PubMed] [Google Scholar]
- 2.Kakouros N, Nazarian SM, Stadler PB, Kickler TS, Rade JJ. Risk factors for non-platelet thromboxane generation after coronary artery bypass graft surgery. Journal of the American Heart Association 2016;5:e002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faraday N, Becker DM, Yanek LR et al. Relation between atherosclerosis risk factors and aspirin resistance in a primary prevention population. Am J Cardiol 2006;98:774–779. [DOI] [PubMed] [Google Scholar]
- 4.Tantry US, Bliden KP, Gurbel PA. Overestimation of platelet aspirin resistance detection by thrombelastograph platelet mapping and validation by conventional aggregometry using arachidonic acid stimulation. J Am Coll Cardiol 2005;46:1705–1709. [DOI] [PubMed] [Google Scholar]
- 5.Homorodi N, Kovacs EG, Lee S et al. The lack of aspirin resistance in patients with coronary artery disease. J Transl Med 2016;14:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002;105:1650–1655. [DOI] [PubMed] [Google Scholar]
- 7.Eikelboom JW, Hankey GJ, Thom J et al. Incomplete Inhibition of Thromboxane Biosynthesis by Acetylsalicylic Acid. Determinants and Effect on Cardiovascular Risk. Circulation 2008;118:1690. [DOI] [PubMed] [Google Scholar]
- 8.Gluckman TJ, McLean RC, Schulman SP et al. Effects of aspirin responsiveness and platelet reactivity on early vein graft thrombosis after coronary artery bypass graft surgery. J Am Coll Cardiol 2011;57:1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kakouros N, Gluckman TJ, J.V. C et al. Differential impact of serial measurment of non-platelet thromboxane generation on long-term outcome after cardiac surgery. J Am Heart Assoc 2017;6:e007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCullough PA, Vasudevan A, Sathyamoorthy M et al. Urinary 11-Dehydro-Thromboxane B2 and Mortality in Patients With Stable Coronary Artery Disease. Am J Cardiol 2017;119:972–977. [DOI] [PubMed] [Google Scholar]
- 11.Catella F, Nowak J, FitzGerald GA. Measurement of renal and non-renal eicosanoid synthesis. Am J Med 1986;81:23–29. [DOI] [PubMed] [Google Scholar]
- 12.Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet 2014;383:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study. Design and preliminary data. Prev Med 1975;4:518–525. [DOI] [PubMed] [Google Scholar]
- 14.Splansky GL, Corey D, Yang Q et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 2007;165:1328–35. [DOI] [PubMed] [Google Scholar]
- 15.Olson MT, Kickler TS, Lawson JA et al. Effect of assay specificity on the association of urine 11-dehydro thromboxane B2 determination with cardiovascular risk. J Thromb Haemost 2012;10:2462–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keaney JF Jr., Larson MG, Vasan RS et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003;23:434–439. [DOI] [PubMed] [Google Scholar]
- 17.Fontes JD, Yamamoto JF, Larson MG et al. Clinical correlates of change in inflammatory biomarkers: The Framingham Heart Study. Atherosclerosis 2013;228:217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fine JP, Gray RA. A proportional hazards model for the subdistribution of a competing risk. . J Am Stat Assoc 1999;94:496–509. [Google Scholar]
- 19.McCullough PA, Vasudevan A, Lopez LR et al. Oxidative stress reflected by increased F2-isoprostanes is associated with increasing urinary 11-dehydro thromboxane B2 levels in patients with coronary artery disease. Thromb Res 2016;148:85–88. [DOI] [PubMed] [Google Scholar]
- 20.Szczeklik W, Stodolkiewicz E, Rzeszutko M et al. Urinary 11-Dehydro-Thromboxane B2 as a Predictor of Acute Myocardial Infarction Outcomes: Results of Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) Study. J Am Heart Assoc 2016;5:e003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frelinger AL III, Furman MI, Linden MD et al. Residual arachidonic acid-induced platelet activation via an adenosine diphosphate-dependent but cyclooxygenase-1- and cyclooxygenase-2-independent pathway: a 700-patient study of aspirin resistance. Circulation 2006;113:2888–2896. [DOI] [PubMed] [Google Scholar]
- 22.Patrignani P, Tacconelli S, Piazuelo E et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J Thromb Haemost 2014;12:1320–1330. [DOI] [PubMed] [Google Scholar]
- 23.Patrick J, Dillaha L, Armas D, Sessa WC. A randomized trial to assess the pharmacodynamics and pharmacokinetics of a single dose of an extended-release aspirin formulation. Postgrad Med 2015;127:573–580. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A 1993;90:11693–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raber I, McCarthy CP, Vaduganathan M et al. The rise and fall of aspirin in the primary prevention of cardiovascular disease. Lancet 2019;393:2155–2167. [DOI] [PubMed] [Google Scholar]
- 26.Vasudevan A, Bottiglieri T, Tecson KM et al. Residual thromboxane activity and oxidative stress: influence on mortality in patients with stable coronary artery disease. Coron Artery Dis 2016. [DOI] [PubMed] [Google Scholar]
- 27.Ames PR, Batuca JR, Muncy IJ et al. Aspirin insensitive thromboxane generation is associated with oxidative stress in type 2 diabetes mellitus. Thromb Res 2012;130:350–354. [DOI] [PubMed] [Google Scholar]
- 28.Hou X, Roberts LJ, Taber DF et al. 2,3-Dinor-5,6-dihydro-15-F(2t)-isoprostane: a bioactive prostanoid metabolite. Am J Physiol Regul Integr Comp Physiol 2001;281:R391–R400. [DOI] [PubMed] [Google Scholar]
- 29.Widlansky ME, Gokce N, Keaney JF, Jr., Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 2003;42:1149–1160. [DOI] [PubMed] [Google Scholar]
- 30.Lahaie I, Hardy P, Hou X et al. A novel mechanism for vasoconstrictor action of 8-isoprostaglandin F2 alpha on retinal vessels. Am J Physiol 1998;274:R1406–R1416. [DOI] [PubMed] [Google Scholar]
- 31.Hou X, Gobeil F Jr., Peri K et al. Augmented vasoconstriction and thromboxane formation by 15-F(2t)-isoprostane (8-iso-prostaglandin F(2alpha)) in immature pig periventricular brain microvessels. Stroke 2000;31:516–524. [DOI] [PubMed] [Google Scholar]
- 32.Ridker PM, Rane M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ Res 2021;128:1728–1746. [DOI] [PubMed] [Google Scholar]
- 33.Kabashima K, Murata T, Tanaka H et al. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat Immunol 2003;4:694–701. [DOI] [PubMed] [Google Scholar]
- 34.Widlansky ME, Price DT, Gokce N et al. Short- and long-term COX-2 inhibition reverses endothelial dysfunction in patients with hypertension. Hypertension 2003;42:310–315. [DOI] [PubMed] [Google Scholar]
- 35.Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J 2003;24:2166–79. [DOI] [PubMed] [Google Scholar]
- 36.Nakahata N Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther 2008;118:18–35. [DOI] [PubMed] [Google Scholar]
- 37.Feletou M, Vanhoutte PM, Verbeuren TJ. The thromboxane/endoperoxide receptor (TP): the common villain. J Cardiovasc Pharmacol 2010;55:317–332. [DOI] [PubMed] [Google Scholar]
- 38.Ekambaram P, Lambiv W, Cazzolli R, Ashton AW, Honn KV. The thromboxane synthase and receptor signaling pathway in cancer: an emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev 2011;30:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castellani S, Paladini B, Paniccia R et al. Increased renal formation of thromboxane A2 and prostaglandin F2 alpha in heart failure. Am Heart J 1997;133:94–100. [DOI] [PubMed] [Google Scholar]
- 40.Santilli F, Davi G, Basili S et al. Thromboxane and prostacyclin biosynthesis in heart failure of ischemic origin: effects of disease severity and aspirin treatment. J Thromb Haemost 2010;8:914–922. [DOI] [PubMed] [Google Scholar]
- 41.Wacker MJ, Best SR, Kosloski LM et al. Thromboxane A2-induced arrhythmias in the anesthetized rabbit. Am J Physiol Heart Circ Physiol 2006;290:H1353–61. [DOI] [PubMed] [Google Scholar]
- 42.Wacker MJ, Kosloski LM, Gilbert WJ et al. Inhibition of thromboxane A2-induced arrhythmias and intracellular calcium changes in cardiac myocytes by blockade of the inositol trisphosphate pathway. J Pharmacol Exp Ther 2009;331:917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pastori D, Pignatelli P, Farcomeni A et al. Age-related increase of thromboxane B2 and risk of cardiovascular disease in atrial fibrillation. Oncotarget 2016;7:39143–39147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiely M, Milne GL, Minas TZ et al. Urinary Thromboxane B2 and Lethal Prostate Cancer in African American Men. J Natl Cancer Inst 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moussa O, Ciupek A, Watson DK, Halushka PV. Urinary thromboxane B2 and thromboxane receptors in bladder cancer: opportunity for detection and monitoring. Prostaglandins Other Lipid Mediat 2011;96:41–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferroni P, Santilli F, Cavaliere F et al. Oxidant stress as a major determinant of platelet activation in invasive breast cancer. Int J Cancer 2017;140:696–704. [DOI] [PubMed] [Google Scholar]
- 47.Sciulli MG, Filabozzi P, Tacconelli S et al. Platelet activation in patients with colorectal cancer. Prostaglandins Leukot Essent Fatty Acids 2005;72:79–83. [DOI] [PubMed] [Google Scholar]
- 48.https://ClinicalTrials.gov/show/NCT03962855. Thromboxane Receptor Antagonist to Improve Endothelial Cell Function. ClinicalTrials.gov, 2019.
- 49.https://ClinicalTrials.gov/show/NCT03340675. Oral Ifetroban in Subjects With Duchenne Muscular Dystrophy. https://ClinicalTrials.gov/show/NCT03340675, 2017.
- 50.https://ClinicalTrials.gov/show/NCT02682511. Oral Ifetroban to Treat Diffuse Cutaneous Systemic Sclerosis (SSc) or SSc-associated Pulmonary Arterial Hypertension. ClincialTrials.gov, 2016.
- 51.https://ClinicalTrials.gov/show/NCT03694249. Ifetroban in Treating Patients With Malignant Solid Tumors at High Risk of Metastatic Recurrence. 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.