

ABSTRACT
Retinoma, also referred to as retinocytoma, is a benign manifestation of biallelic retinoblastoma gene (RB1) inactivation. Genetic or epigenetic loss of retinoblastoma protein in maturing cone precursors induces genomic instability which leads to upregulation of senescence‐associated p16INK4a and p130, resulting in non‐proliferative retinoma. When senescence pathways fail and genetic instability accumulates to a critical level through altered gene copies of oncogenes and tumor suppression genes, transformation into RB1 −/‐ retinoblastoma occurs. Thus, the management of retinoma involves frequent ophthalmic examination and imaging to monitor the size and characteristics of the tumor, ensure stability, and rule out malignant transformation. Key ophthalmoscopic features of retinoma often include a translucent whitish‐gray retinal mass, calcification, retinal pigment epithelial alterations with well‐defined margins, located typically around the lesion, as well as a zone of chorioretinal atrophy. This review aims to provide a comprehensive overview of this non‐malignant tumor drawing from current understanding of its molecular genetics, clinical characteristics, diagnostic modalities, differential diagnosis, management, and prognosis. A deeper understanding of retinoma could offer valuable insights into how retinoblastoma develops and oncogenesis more broadly, paving the way for improved strategies to prevent and treat this malignant tumor.
Keywords: Ocular oncology, Retinal tumor, Retinoma, Retinoblastoma, Retinocytoma
Retinoma, a benign tumor caused by biallelic retinoblastoma gene (RB1) inactivation, offers key insights into retinoblastoma development and oncogenesis. This review explores its molecular genetics, clinical features, diagnosis, management, and potential implications for advancing the prevention and treatment of retinoblastoma.

INTRODUCTION
Retinoblastoma is the most common ocular malignancy in children, typically arising from biallelic loss of the retinoblastoma gene (RB1) within a single vulnerable developing retinal cell. 1 However, the genetic or epigenetic loss of both RB1 alleles does not inevitably lead to a malignant phenotype; instead, it results in non‐proliferative retinomas. Based on clinical, histopathologic, and genetic evidence, 2 , 3 , 4 retinoma is now considered a benign precursor to retinoblastoma, sharing the same genetic origin as most retinoblastomas and demonstrating early genomic changes that precede malignant transformation. This review aims to provide a comprehensive panorama of retinoma, encompassing current insights from molecular genetics, clinical characteristics, diagnostic modalities, differential diagnosis, contemporary management strategies, and prognosis. By synthesizing current evidence and highlighting key developments in the field, this review seeks to enhance understanding of retinoma and facilitate informed decision‐making in patient management.
TERMINOLOGY
The terms “retinoma” and “retinocytoma” are commonly used interchangeably across various studies. In 1977, Gallie et al. 5 found that individuals with pediatric eye cancer retinoblastoma displayed an inactive tumor, or “spontaneously regressed retinoblastoma”, at a frequency as high as 1%. In 1982, Gallie et al. 6 coined the term “retinoma” to describe non‐progressive retinal lesions with the characteristic appearance of translucent, gray, retinal masses with calcified nodules and disturbances of pigment epithelium, observed in individuals known to carry a heritable pathogenic variant causing retinoblastoma. The term “retinoma” emerged to replace the previously utilized term “spontaneously regressed retinoblastoma”, since these tumors lacked compelling clinical evidence of regression or involution. One year later, a histopathological investigation conducted by Margo et al. 7 revealed the benign nature of these tumors, characterizing them as a benign variant of retinoblastoma. Borrowing from the nomenclature used to classify neurogenic pineal tumors (benign ones termed pineocytomas and malignant ones termed pineoblastomas) they introduced the term “retinocytoma”. An alternative term suggested in the literature includes “retinoblastoma group 0” and “spontaneously arrested retinoblastoma”, though they are seldom utilized. 8
CLINICAL FEATURES
Retinoma typically manifests without symptoms or signs. However, when do occur, blurred vision and strabismus are the most commonly reported complaints. 9 , 10 Also, a case involving retinoma and photopsia has been reported. 11 Leukocoria, the most frequent sign of retinoblastoma, is not commonly observed in retinoma. Younger patients (under 4 years old) with retinoma are more likely than older ones to exhibit leukocoria or any other sign or symptom. 9
Retinoma can be either unilateral or bilateral, with unilateral cases being more common. 9 , 10 The key ophthalmoscopic features of retinoma comprise a translucent whitish‐gray retinal mass, calcification, retinal pigment epithelial alterations with well‐defined margins, located typically around the lesion, as well as a zone of chorioretinal atrophy (Figure 1). 6 , 9 , 10 , 12 , 13 This tetrad of features does not always coexist in every case; that occurs in only one‐quarter of patients. 9 However, one‐third of retinoma cases exhibit at least three of them. 9 Other less common features of retinoma include localized calcium deposits in the vitreous (calcified vitreous seeding), 13 , 14 , 15 , 16 , 17 , 18 retinal tractions, 13 and even intralesional cavitary lesions, 19 a feature typically associated with well‐differentiated retinoblastoma. In some patients with retinoma, phthisis bulbi has been observed in the other eye. 14 , 20 Benign cystic growth has also been noted in certain retinomas. 14 , 21
FIGURE 1.
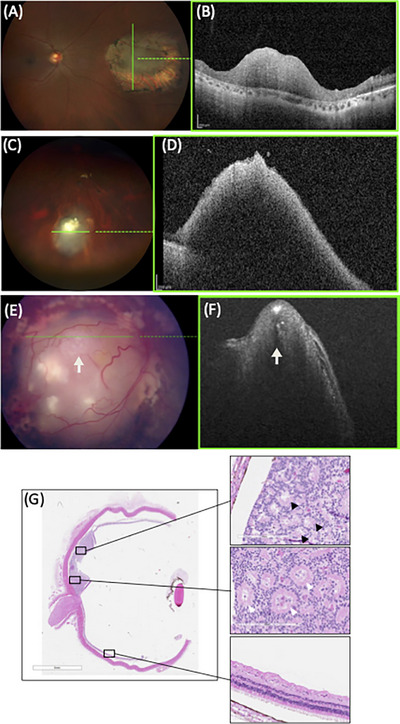
Clinical presentation of patients with retinoma. Fundus photo (A) and optical coherence tomography (OCT) image (B) of retinoma in a 43‐year‐old father identified incidentally after his son was diagnosed with retinoblastoma, showing a grey translucent elevated mass in the middle of a retinal scar. Fundus photo (C) and OCT image (D) of retinoma in a 9‐year‐old girl under surveillance, showing grey translucent elevated mass with calcification. Fundus photo (E) and OCT image (F) of a 10‐month‐old child with retinoblastoma treated by chemotherapy and enucleated for reactivation. (G) Histopathology demonstrated active retinoblastoma (white arrows) adjacent to retinoma (black arrows).
Generally, the ophthalmoscopic appearance of retinoma bears a resemblance to the array of regression patterns observed in retinoblastoma post‐irradiation, with the majority of cases being appearance‐wise compatible with a type 3 retinoblastoma regression pattern. 13 Retinoma foci do not predominantly appear in any of the three retinal zones. 13 Retinoma and retinoblastoma can co‐occur, indicating that they are not mutually exclusive conditions (Figure 1). There have been reports of instances where both conditions occur in the same family, in the same patient with retinoma in one eye and retinoblastoma in the other, or as distinct areas within the same eye. 13
The majority of retinomas retain their benign nature throughout an individual's lifetime and never progress to retinoblastoma (Figure 1). In subsequent follow‐up sessions, retinomas typically maintain their size. 13 However, this observation may reflect a referral bias, as retinomas may have reached their maximum growth potential at the time of diagnosis, and all lesions found in patients with a family history of retinoblastoma are treated under the assumption that they are retinoblastomas. Interestingly, in some retinoma cases, photographic documentation following extended follow‐up periods revealed lesion regression. 13 , 22 The precise mechanisms underlying retinoma regression remain unknown but may potentially involve apoptosis. 23 The histopathology of retinoma does not support the involvement of ischemia or immune‐mediated necrosis. 24
However, some rare cases of clinically diagnosed retinomas have undergone malignant transformation (Figure 1). 9 , 25 , 26 , 27 , 28 Recently, Shields et al. 9 reported that the transformation of retinoma into retinoblastoma occurred in 2.7% of cases by 2 years, 9.2% by 5 years, and 15.3% by 10–20 years. They also reported that the only factor predictive of malignant transformation was an increase in tumor thickness. 9 Malignant transformation is characterized by progressive opacification and enlargement of the tumor, along with the presence of irregular, fine tumor vessels. 17
EPIDEMIOLOGY
The incidence of retinoma varies in the literature. Clinical observations across various studies have shown retinoma to be present in approximately 1.8% to 3.2% of retinoblastoma cases, while pathological examinations have identified retinoma in 15.6% to 20.4% of enucleated retinoblastoma specimens. 2 , 3 , 6 , 14 This variability in incidence likely indicates a referral bias. Even though retinoma is often found in eyes diagnosed with retinoblastoma upon pathological examination, its clinical documentation is rare because it is commonly overgrown by malignant retinoblastoma, making its detection difficult. 29 Also, diagnosis often happens when examining symptomatic cases or those with a family history of retinoblastoma.
DIAGNOSTIC ASSESSMENT
The diagnosis of retinoma relies heavily on a comprehensive approach that integrates patient and family history with meticulous ophthalmic examination and adjunct imaging techniques. A multi‐faceted evaluation not only aids in confirming the presence of the tumor but also in differentiating it from more aggressive conditions, mainly retinoblastoma.
The diagnosis cannot be confirmed histopathologically unless the eye is enucleated; biopsy is contraindicated in cases of suspected retinoma due to the risk of tumor seeding if retinoblastoma is ultimately confirmed. 30 Indirect ophthalmoscopy with pharmacologically dilated pupils, performed under general anesthesia in uncooperative patients, is usually sufficient for diagnosis by an eye specialist, ideally an ocular oncologist. This diagnostic method allows for the identification of characteristic features of the retinal mass.
Ancillary examinations can provide important information. Both fluorescein and indocyanine green angiography revealed low vascular activity of retinoma. 14 Ocular ultrasonography might show hyperechoic spikes corresponding to intralesional calcification or hypoechoic areas, indicating the presence of fluid‐filled spaces (cysts). 14 , 31 Optical coherence tomography (OCT) of retinoma typically depicts a superficial retinal mass lesion with homogeneous internal reflectivity. 31 OCT is very useful since it can detect small, visually undetectable tumors in susceptible patients and evaluate any changes in their size during follow‐up appointments. 32
DIFFERENTIAL DIAGNOSIS
Although retinoma possesses distinct ophthalmoscopic features, several conditions can closely mimic its presentation, such as retinoblastoma and retinal astrocytic hamartoma (RAH) (Table 1).
TABLE 1.
Differential diagnosis of retinoma
| Clinical features | Retinoma | Retinoblastoma | Retinal astrocytic hamartoma |
|---|---|---|---|
| Age at diagnosis |
Older children (>6 years old), adults |
Younger children (<5 years old) | Typically childhood |
| Symptoms | Typically asymptomatic | Leukocoria | Typically asymptomatic |
| Associated systematic diseases | 13q deletion syndrome | 13q deletion syndrome |
Tuberous sclerosis complex Neurofibromatosis type 1 Usher syndrome Stargardt disease |
| Calcification color | Chalky white | Chalky white | Bright yellow |
| Calcification appearance | Chunky | Chunky | Spherical |
| RPE alterations | Present | Present | Absent |
| Chorioretinal atrophy | Present | Absent | Absent |
| Feeder vessels | Absent | Present | Absent |
| Retinal hard exudates | Absent | Absent | Sometimes present |
| Intralesional cavitation | Typically absent | Present | Present |
| Growth dynamic | Typically stable | Growing | Typically stable |
Abbreviation: RPE, retinal pigment epithelium.
The patient's medical history can provide valuable clues to aid in diagnosis. The first clue is the patient's age. Retinoblastomas are commonly present in early childhood, typically affecting children under the age of 5, while retinomas are often diagnosed in older children (after the age of 6) or adults. 13 Another key point is the presence or absence of symptoms, along with the timeline of symptom onset and progression. RAH is usually asymptomatic and may be discovered incidentally or during screening of patients suspected of having phakomatoses. Inquiring about family history is crucial, as a family history of retinoblastoma directs the diagnostic reasoning toward retinoma or retinoblastoma. Another aspect to consider is the presence of associated systemic diseases. Patients diagnosed with 13q deletion syndrome face a heightened risk of developing retinoblastoma due to the deletion of the RB1 gene, situated within the chromosomal band 13q14.2. 33 Given that retinoma requires RB1 loss, it can similarly be linked to 13q deletion syndrome. RAH is strongly associated with tuberous sclerosis complex (TSC) as well as with neurofibromatosis type 1, Usher syndrome, and Stargardt disease. 34
Regarding clinical features, calcification in RAH is glistening yellow with an appearance like fish eggs, whereas in retinomas and retinoblastomas, it is chalky white. 34 Areas of chorioretinal atrophy and associated retinal pigment epithelial changes observed in retinomas are uncommon in astrocytic hamartomas and untreated retinoblastomas. 17 , 31 , 34 Although uncommon, hard exudates may occasionally be found around astrocytic hamartomas, whereas they are generally absent in (untreated) retinomas and retinoblastomas. 34 Using OCT imaging, RAH typically initiates as a flat tumor located in the nerve fiber layer that gradually grows into a nodular, full‐thickness retinal mass, demonstrating “moth‐eaten” intralesional, optically empty spaces, related to intrinsic calcification or cavitation. 35 On the other hand, retinomas and retinoblastomas in OCT exhibit a homogenous appearance with the retina. 31 , 36 Regarding growth dynamics, RAH typically follows a stable course; however, rare cases of tumor growth or even regression have been reported. 37 Retinoblastoma exhibits gradual growth, specifically will show growth within 4–6 weeks, while retinoma typically appears unchanged. 17 The only reliable method for distinguishing retinomas from retinoblastomas is through ongoing follow‐up to monitor for any progressive changes in the lesions. 31
MANAGEMENT CONSIDERATIONS
The management of retinoma involves conservative approaches due to its benign nature and low risk of progression to malignancy. Retinomas show little to no response to chemoreduction. 38 Observations with regular follow‐up visits are often adequate. Periodic imaging should be conducted to monitor the size and characteristics of the tumor, ensure stability, and rule out malignant transformation. However, in cases where the diagnosis is uncertain, ophthalmologists may opt to treat the tumor assuming it to be retinoblastoma, especially if treatment is unlikely to result in significant visual impairment, or if the clinician is concerned about patient loss‐to‐follow‐up.
Genetic testing and counseling are integral parts of the management of patients with retinoma. A family history of retinoblastoma can imply a germline RB1 pathogenic variant. Detection of a germline RB1 pathogenic variant informs the screening strategy of relatives and subsequent generations. Somatic RB1 variants in retinoma have only been detected in eyes enucleated for subsequent retinoblastoma, 29 as enucleation for retinoma is contraindicated; it is possible that liquid biopsy of aqueous humor to assess cell‐free tumor DNA, which is increasingly being used for retinoblastoma, 39 , 40 may hold promise for retinoma patients as well. However, given that inactive retinoblastoma is typically associated with low tumor fraction, 41 it could be presumed that quiescent retinoma would similarly yield little‐to‐no cell‐free tumor DNA, precluding analysis. Regarding genetic counseling, healthcare providers should prioritize educating and supporting patients and their families about the importance of regular monitoring of retinoma. These practices help alleviate anxiety and promote compliance with follow‐up visits.
Long‐term considerations for individuals with retinoma include the risk of developing a second malignant neoplasm (SMN). Patients with the hereditary form of retinoblastoma are at a significantly increased risk of SMNs. 42 Consequently, it is plausible that individuals with retinoma and germline RB1 pathogenic variant may also face a substantial risk of SMNs. However, such cases have rarely been reported, and the majority of them also suffer from retinoblastoma. 43
HISTOPATHOLOGY
Early evidence of retinoma histopathology was reported by Margo et al. 7 in 1983, who described this tumor as a small, placoid, non‐invasive lesion composed entirely of benign‐appearing cells that exhibited photoreceptor differentiation and formed bouquet‐like arrangements (“fleurettes”). Additionally, they did not observe any necrosis or mitotic activity. 7 In further studies, histopathological examination of eyes enucleated due to retinoblastoma showed regions with characteristics of retinoma, adjacent to both normal retina and retinoblastoma. 2 , 3 , 21 The fact that retinoma and retinoblastoma do not arise from separate areas of the retina supports the idea that retinoblastoma represents the ultimate malignant stage in a series of clonal developments from normal to benign to malignant cells in the context of biallelic RB1 loss.
Light microscopy examination of retinoma sections, using hematoxylin and eosin staining, enables the observation of benign cells with abundant eosinophilic cytoplasm, nuclei of normal size with evenly dispersed chromatin, and absence of pleomorphism or mitotic activity. 2 , 44 Additionally, there are well‐differentiated, prominent photoreceptors, forming fleurettes. 2 , 44 The absence of scarring or necrosis in pathological analysis of retinomas refutes the longstanding belief that retinomas represent spontaneous regression of retinoblastomas. 29 Retinoma lacks the typical features of retinoblastoma, namely densely packed cells with little cytoplasm, nuclear molding, frequent mitoses, as well as the characteristic Flexner‐Wintersteiner and Homer Wright rosettes. 2 , 45 It should be highlighted that nuclear morphometric analysis indicated that there were no significant differences in the nuclei of retinoma and tumors showing mild anaplasia since both exhibit small, round, and band nuclei. 44 The distinguishing factor lies only in the larger amount of cytoplasm and the presence of fleurettes. 44
PATHOGENESIS
Genetic basis of retinoma
Nearly all retinoblastomas initiate in response to biallelic inactivation of tumor suppressor gene RB1 (RB1−/− ). 46 , 47 Approximately 1.4% of all retinoblastoma cases are caused by oncogene MYCN amplification (MYCNA ) with wild‐type RB1 (RB1+/+MYCNA ). 48 Interestingly, another small subset of retinoblastoma (1.5%) remains unexplained since they have normal RB1 and MYCN genes. 48 Investigations to date into the genetic status of retinomas showed that retinomas are homozygous null for RB1 −/−. 2 Retinoma has not been associated with MYCNA retinoblastoma, but the rarity of this subtype may mean that such cases may exist but have not yet been documented.
The RB1 gene is located on chromosome 13, more specifically, 13q14.2, and codes for retinoblastoma protein (pRB), a regulator of cellular replication. Numerous cellular functions have been ascribed to pRB, but a key one is to prevent progression from the G1 (first gap phase) to S (synthesis phase) phase of the cell division cycle; pRB inhibits E2F‐activated transcription of genes necessary for G1‐to‐S phase progression, while phosphorylation of pRB by cyclin‐dependent kinase (CDK) releases E2F, activating the cell cycle (Figure 2). 49 This kinase activity can be regulated by the CDK4/6 inhibitor p16INK4a. 50 , 51 Hence, pathogenic variants in the RB1 gene that result in the dysfunction or absence of pRB lead to the perpetual activation of E2Fs and persistent stimulation of the cell cycle, significantly contributing to tumor development.
FIGURE 2.
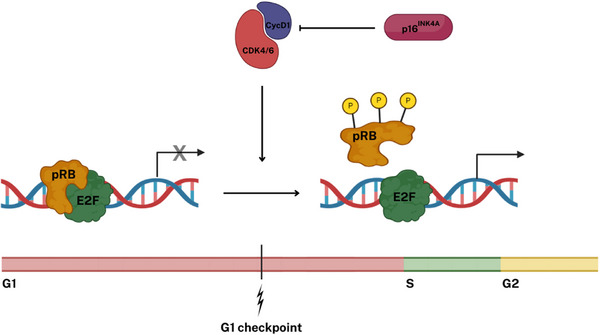
Function of the retinoblastoma protein (pRB) in the control of the G1 checkpoint. A core function of pRB is to prevent the progression from the G1 to the S phase of the cell cycle by binding to and inhibiting E2F family transcription factors, which drive the cell into the S phase. Cell cycle progression occurs through the inactivation of pRB, catalyzed by cyclin‐dependent kinases (CDKs). Specifically, CDK4 and CDK6 are activated in response to the accumulation of D‐type cyclins (CycD) due to mitogenic signaling, initiating the phosphorylation (P) of pRB. This kinase activity is counterbalanced by factors including the CDK4/6 inhibitor p16ΙΝΚ4Α. Typically, p16ΙΝΚ4Α is present at low levels in cells, but it can be upregulated in response to oncogenic stress or DNA damage to suppress CDK4/6 activity.
Knudson's two‐hit hypothesis of oncogenesis describes the tumorigenesis of retinoblastoma as a consequence of two consecutive pathogenic variants of the RB1 gene (Figure 3). 46 In particular, the initial ‘hit’ (M1) can occur in either germline or somatic cells. Germline pathogenic variants may be inherited from parents or arise de novo in individuals. The second ‘hit’ (M2) is only acquired and never inherited, often due to physical, chemical, or biological factors. Both hits (M1 and M2) that deactivate the RB1 gene have been shown to occur in retinomas. 2 Also, RB1 pathogenic variants found in retinomas are indistinguishable from those found in retinoblastomas. 2 , 52 Consistent with the genotype of RB1 −/−, no functional pRB is detectable in retinomas. 2
FIGURE 3.
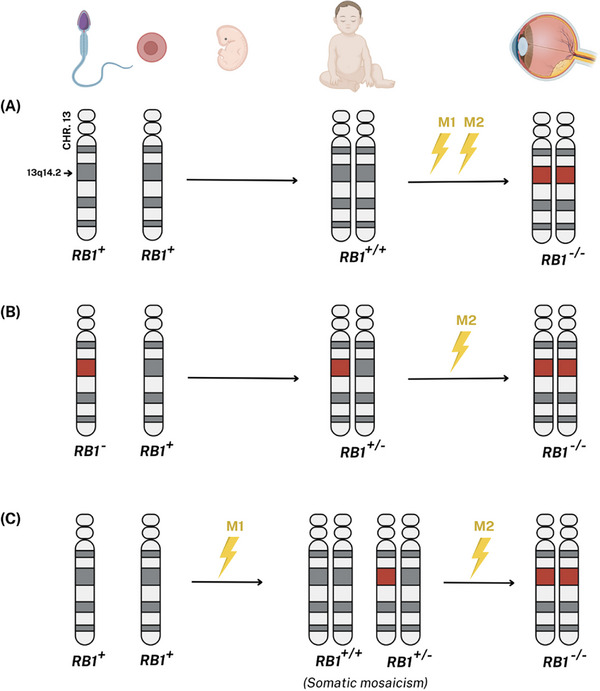
Genetics of Retinoblastoma. (A) The first two ‘hits’ to RB1 (M1 and M2) occur in somatic cells. (B) M1 is found in all germline cells, resulting from either de novo or inherited from a parent (in this case example, the father). Consequently, the genotype of the affected individual is heterozygous, carrying one pathogenic variant in germline cells (RB1 +/−). The M2 arises after a somatic mutational event in a retinal cell. Distinct second pathogenic variants in separate retinal cells give rise to independent tumor foci. (C) In some individuals, M1 occurs during embryonic development, resulting in somatic mosaicism. The M2 can then occur in retinal cells that harbor the M1, leading to tumor initiation. Heritable disease results from pathways depicted in pathways B and C. Both M1 and M2 are present in retinoma.
However, loss of both RB1 alleles does not inevitably result in a malignant phenotype; instead, it tends to induce genomic instability. A senescence response to this instability leads to non‐proliferative retinoma. 2 In other words, RB1 inactivation and the subsequent loss of pRB promote genomic instability, but senescence pathways can prevent transformation into retinoblastoma, leaving the tumor at the retinoma stage.
Senescence protein p16INK4a is found to be expressed in retinoma but not in either the normal retina or retinoblastoma. 2 Specifically, after loss of both RB1 alleles, CDKN2A gene (which encodes p16INK4a) in retinomas is upregulated, expressing high levels of p16INK4a. Interestingly, CDKN2A mRNA is also highly expressed in retinoblastoma but the absence of p16INK4a protein suggests the involvement of mechanisms for senescence evasion. 2 Rare cells might escape the induction of senescence either by inactivating p16INK4a or by not activating it at all. Normally, p16INK4a is involved in blocking the G1/S‐phase transition of the cell cycle, as described in Figure 2. Ιn retinomas, even in the absence of pRB, p16INK4a manages to block the cell cycle and mediate cellular senescence, likely through interaction with pRB family member, p130, which has partially overlapping functions with pRB and is highly expressed in retinomas but not in retinoblastomas. 2 The non‐proliferative nature of retinomas is also supported by the fact that immunostained tissues from the unaffected retina, retinoma, and retinoblastoma tested for proliferation markers Ki67 and proliferating cell nuclear antigen (PCNA), revealed identical staining patterns for retinomas and retinas (both negative for Ki67 and PCNA), in contrast to retinoblastomas (strong positivity for Ki67 and PCNA). 2 It should be highlighted that high expression of p130 and absence of Ki67 might indicate arrest in phase G0. Liu et al. 53 discovered that the “retinoma‐like” cells in their organoid study exhibited low levels of Ki67, which is consistent with ex vivo staining results. However, these cells showed elevated levels of PCNA, underscoring a notable difference from the findings observed in ocular histopathology.
Cellular origins of retinoma
Precise identification of the cellular origin of retinoblastoma, and consequently retinoma, has garnered significant scientific interest and prompted extensive research efforts. Cone maturation begins as retinal progenitor cells leave the cell cycle to differentiate into cone precursors, which give rise to mature cone photoreceptors. Two oncoproteins, MYCN and MDM2, which are inherently significantly expressed in normal human cone precursors, are crucial for the proliferative dynamic and survival of pRB‐depleted cells; specifically, MDM2 acts as a negative regulator of the p53 tumor suppressor, while MYCN facilitates abnormal cell growth and proliferation. 54 Later, it was found that human cone precursors are uniquely sensitive to RB1 mutation. 55 pRB knockdown stimulates cone precursor proliferation, which relies on MYCN and MDM2 as well as other factors that have strong expression in maturing cone precursors and are essential in retinoblastoma cell proliferation. 55 Also, pRB‐depleted human cone precursors develop into tumors with features typical of human retinoblastoma in orthotopic xenografts. 55 These pieces of evidence suggested retinoblastoma's cone precursor origin. Further investigations, including the most recent studies on retinal organoids derived from genetically modified human embryonic cells with biallelic RB1 mutations, have indicated that retinoma, and by extension retinoblastoma, originates from maturing retinal cone arrestin‐3 positive (ARR3+) cone precursors. 53 , 56 Specifically, in human retinas, loss of pRB stimulates maturing (ARR3+) cone precursor proliferation via MYCN‐dependent cell‐cycle entry and MDM2‐mediated suppression of apoptosis. 55 , 56 In due course, most of these cells exit the cell cycle with highly expressed p16INK4a and p130, forming indolent premalignant lesions, in which rare cells that hold proliferative dynamics give rise to quiescent retinomas.56 Some cells escape this mechanism, forming highly proliferative masses that express cone cell markers characteristic of retinoblastoma (Figure 4). 56
FIGURE 4.
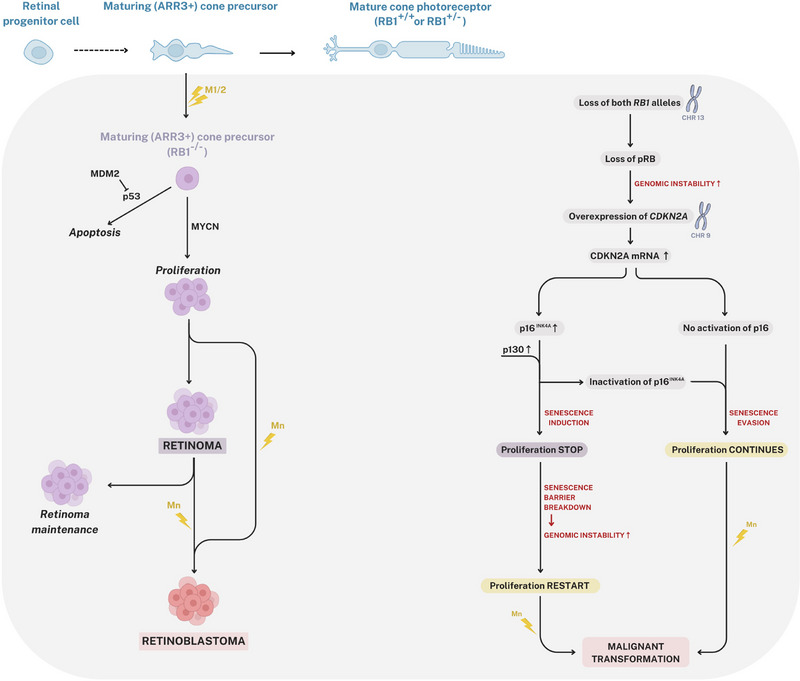
Retinoma pathogenesis. In human retinas, the loss of pRB promotes the proliferation of maturing (ARR3+) cone precursor cells (CPs) through MYCN‐dependent cell‐cycle activation and MDM2‐mediated inhibition of apoptosis. Most of these cells exit the cell cycle, exhibiting high levels of p16INK4a and p130, which leads to the formation of indolent premalignant lesions. Within these lesions, a small number of cells maintain a proliferative dynamic, ultimately giving rise to quiescent retinomas. Some cells evade this regulatory mechanism, leading to the development of highly proliferative masses that express cone cell markers typical of retinoblastoma. While the majority of retinomas remain benign throughout an individual's lifetime and do not progress to retinoblastoma, transformation into RB1−/− retinoblastoma may occur when senescence pathways fail and genetic instability reaches a critical threshold. M1, first hit; M2, second hit; Mn, nth hit.
From retinoma to retinoblastoma
Although both retinomas and (most) retinoblastomas share the same genetic status (RB1 −/−), retinomas exhibit lower levels of aneuploidy and higher levels of senescence‐associated proteins. 2 , 4 While M1 and M2 may constitute the initial rate‐limiting steps in tumorigenesis, additional genomic imbalances (M3‐Mn) involving specific oncogenes and tumor suppressors are necessary for malignant transformation. 57 In essence, when senescence pathways fail and genetic instability accumulates to a critical level, transformation into RB1−/− retinoblastoma can occur. Genomic instability can elevate the risk of further mutational events in critical cancer genes, potentially causing a breakdown of the senescence barrier (Figure 4). This progression is marked by the altered gene copy numbers, a progressive gain of certain oncogenes (MDM4 and KIF14 on chromosome 1q, MYCN on chromosome 2p, E2F3 and DEK on chromosome 6p), and loss of tumor suppressor genes (CDH11 on 16q). 2 Table 2 summarizes the molecular differences between retinoma and retinoblastoma.
TABLE 2.
Gene expression retinoma versus retinoblastoma
| Gene expression | Retinoma | Retinoblastoma |
|---|---|---|
| pRB | − | − |
| CDKN2A mRNA | + (high levels) | + (high levels) |
| p16INK4a | + (high levels) | − |
| p130 | + (high levels) | − |
| p107 | + (equal levels) | + (equal levels) |
| Ki67 | − | + (high levels) |
| PCNA† | − | + (high levels) |
| p53 | +/− | +/− |
| p75NTR | + (high levels) | − |
| CDH11 | + (high levels) | + (decreased levels) |
| DEK | + | + (high levels) |
| KIF14 | + | + (high levels) |
| E2F3 | − | + (high levels) |
The comparison is based on immunostaining for proteins and quantitative RT‐PCR for mRNA. Data are adapted from Reference 2.
†The retinoma‐like cell cluster in an organoid study by Liu et al. 53 strongly expressed PCNA.
+, detection; −, absence; +/−, detection in some but not all cells; pRB, retinoblastoma protein; PCNA, proliferating cell nuclear antigen.
UNANSWERED QUESTIONS AND FUTURE DIRECTIONS
Despite the challenges in understanding retinoma, ongoing research continues to yield valuable insights into this condition. However, retinoma research encounters various obstacles and limitations that hinder rapid progress toward a thorough understanding of the condition. One primary challenge is its rarity, leading to a limited patient population for clinical studies. Additionally, the subtle and often asymptomatic nature of retinomas means that they are frequently underdiagnosed, further constraining the available data pool. Furthermore, the specimens analyzed were predominantly those from enucleated eyes due to adjacent malignant retinoblastoma, while eyes with only retinoma were typically not enucleated. This results in an underrepresentation of benign retinomas in molecular studies. Ex vivo culture and retinal organoid models have proven valuable in advancing our understanding of retinal development and tumorigenesis, providing a controlled environment for investigating molecular pathways. However, these models have limitations, including an inability to fully replicate the in vivo retinal microenvironment, and they have not yet been optimized for studying the benign nature of retinomas. Moreover, the lack of well‐established mouse models for retinoma further constrains preclinical investigations. Consequently, despite existing research efforts, numerous critical questions about retinoma remain unanswered, underscoring the need for further investigations.
Some of the most pressing queries include identifying which retinomas never progress to retinoblastoma and remain benign for life, and understanding the exact pathophysiological mechanisms behind this stability. On the other hand, in the case of malignant transformation; the full range of genetic mutations involved, and the intracellular or extracellular factors that might influence this process are largely unknown. Unraveling these mechanisms will not only help prevent the occurrence of retinoblastoma in patients with retinoma but also provide crucial insights into cancer prevention in general and open new avenues for therapeutic interventions.
CONCLUSION
In conclusion, retinoma, though a rare and often underdiagnosed condition, provides significant insights into retinoblastoma genesis. This overview has highlighted the critical aspects of retinoma, including its genetic foundations, clinical manifestations, diagnostic challenges, and potential implications for patient management. By understanding the unique characteristics of retinoma and its relationship with its malignant counterparts, healthcare professionals can better identify and monitor patients at risk, ensuring timely intervention and improved outcomes. Future research should focus on elucidating the molecular mechanisms underlying retinoma progression and exploring innovative diagnostic and therapeutic approaches, ultimately enhancing our ability to manage this enigmatic retinal pathology.
CONFLICT OF INTEREST
None.
ACKNOWLEDGMENTS
We thank Yasmeen Khokhar, Christina Lam, and Cynthia VandenHoven for the acquisition of clinical images. Figures 2, 3, 4 were created using BioRender.com.
Toumasis PN, Mallipatna A, Corson TW, Dimaras H. Retinoma: An overview. Pediatr Investig. 2025;9:139–149. 10.1002/ped4.12472
REFERENCES
- 1. Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, et al. Retinoblastoma. Nat Rev Dis Primers. 2015;1:15021. DOI: 10.1038/nrdp.2015.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, et al. Loss of RB1 induces non‐proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363‐1372. DOI: 10.1093/hmg/ddn024 [DOI] [PubMed] [Google Scholar]
- 3. Eagle RC Jr. High‐risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study. Arch Pathol Lab Med. 2009;133:1203‐1209. DOI: 10.5858/133.8.1203 [DOI] [PubMed] [Google Scholar]
- 4. Sampieri K, Amenduni M, Papa FT, Katzaki E, Mencarelli MA, Marozza A, et al. Array comparative genomic hybridization in retinoma and retinoblastoma tissues. Cancer Sci. 2009;100:465‐471. DOI: 10.1111/j.1349-7006.2008.01070.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gallie BL, Dupont B, Whitsett C, Kitchen FD, Ellsworth RM, Good RA. Histocompatibility typing in spontaneous regression of retinoblastoma. Prog Clin Biol Res. 1977;16:229‐237. [PubMed] [Google Scholar]
- 6. Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J Cancer. 1982;45:513‐521. DOI: 10.1038/bjc.1982.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Margo C, Hidayat A, Kopelman J, Zimmerman LE. Retinocytoma. A benign variant of retinoblastoma. Arch Ophthalmol. 1983;101:1519‐1531. DOI: 10.1001/archopht.1983.01040020521003 [DOI] [PubMed] [Google Scholar]
- 8. Aaby AA, Price RL, Zakov ZN. Spontaneously regressing retinoblastomas, retinoma, or retinoblastoma group 0. Am J Ophthalmol. 1983;96:315‐320. DOI: 10.1016/s0002-9394(14)77821-3 [DOI] [PubMed] [Google Scholar]
- 9. Shields CL, Srinivasan A, Lucio‐Alvarez JA, Shields JA. Retinocytoma/retinoma: comparative analysis of clinical features in 78 tumors and rate of transformation into retinoblastoma over 20 years. J AAPOS. 2021;25:147.e1‐147.e8. DOI: 10.1016/j.jaapos.2020.11.024 [DOI] [PubMed] [Google Scholar]
- 10. Yi XQ, Qian J, Guo J, Xue K. Clinical features of patients with retinocytoma (in Chinese). Chin J Ophthalmol. 2021;57:526‐530. DOI: 10.3760/cma.j.cn112142-20200831-00564 [DOI] [PubMed] [Google Scholar]
- 11. Santos C, Ramalho M, Coutinho I, Teixeira S. Photopsia revealing a retinocytoma. BMJ Case Rep. 2015;2015:bcr2015211018. DOI: 10.1136/bcr-2015-211018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Balmer A, Munier F, Gailloud C. Retinoma. Case studies. Ophthalmic Paediatr Genet. 1991;12:131‐137. DOI: 10.3109/13816819109029394 [DOI] [PubMed] [Google Scholar]
- 13. Singh AD, Santos CM, Shields CL, Shields JA, Jr Eagle RC. Observations on 17 patients with retinocytoma. Arch Ophthalmol. 2000;118:199‐205. DOI: 10.1001/archopht.118.2.199 [DOI] [PubMed] [Google Scholar]
- 14. Abouzeid H, Balmer A, Moulin AP, Mataftsi A, Zografos L, Munier FL. Phenotypic variability of retinocytomas: preregression and postregression growth patterns. Br J Ophthalmol. 2012;96:884‐889. DOI: 10.1136/bjophthalmol-2011-300896 [DOI] [PubMed] [Google Scholar]
- 15. Lueder GT, Héon E, Gallie BL. Retinoma associated with vitreous seeding. Am J Ophthalmol. 1995;119:522‐523. DOI: 10.1016/s0002-9394(14)71246-2 [DOI] [PubMed] [Google Scholar]
- 16. Hadjistilianou T, De Francesco S, Martone G, Malandrini A. Retinocytoma associated with calcified vitreous deposits. Eur J Ophthalmol. 2006;16:349‐351. DOI: 10.1177/112067210601600227 [DOI] [PubMed] [Google Scholar]
- 17. Shah PK, Narendran V, Manayath GJ, Chowdhary S. Atypical retinocytoma with diffuse vitreous seeds: An insight. Oman J Ophthalmol. 2011;4:81‐83. DOI: 10.4103/0974-620X.83659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garoon RB, Medina CA, Scelfo C, Harbour JW. Retinocytoma with vitreous seeding: new insights from enhanced depth imaging optical coherence tomography and high‐resolution posterior segment ultrasonography. Retin Cases Brief Rep. 2021;15:68‐70. DOI: 10.1097/ICB.0000000000000732 [DOI] [PubMed] [Google Scholar]
- 19. Mashayekhi A, Shields CL, Eagle RC Jr, Shields JA. Cavitary changes in retinoblastoma: relationship to chemoresistance. Ophthalmology. 2005;112:1145‐1150. DOI: 10.1016/j.ophtha.2005.01.041 [DOI] [PubMed] [Google Scholar]
- 20. Gallie BL, Phillips RA, Ellsworth RM, Abramson DH. Significance of retinoma and phthisis bulbi for retinoblastoma. Ophthalmology. 1982;89:1393‐1399. DOI: 10.1016/s0161-6420(82)34622-9 [DOI] [PubMed] [Google Scholar]
- 21. Sampieri K, Mencarelli MA, Epistolato MC, Toti P, Lazzi S, Bruttini M, et al. Genomic differences between retinoma and retinoblastoma. Acta Oncol. 2008;47:1483‐1492. DOI: 10.1080/02841860802342382 [DOI] [PubMed] [Google Scholar]
- 22. Lam A, Shields CL, Manquez ME, Shields JA. Progressive resorption of a presumed spontaneously regressed retinoblastoma over 20 years. Retina. 2005;25:230‐231. DOI: 10.1097/00006982-200502000-00025 [DOI] [PubMed] [Google Scholar]
- 23. Nork TM, Poulsen GL, Millecchia LL, Jantz RG, Nickells RW. p53 regulates apoptosis in human retinoblastoma. Arch Ophthalmol. 1997;115:213‐219. DOI: 10.1001/archopht.1997.01100150215011 [DOI] [PubMed] [Google Scholar]
- 24. Papac RJ. Spontaneous regression of cancer. Cancer Treat Rev. 1996;22:395‐423. DOI: 10.1016/s0305-7372(96)90023-7 [DOI] [PubMed] [Google Scholar]
- 25. Eagle RC Jr, JA Shields, Donoso L, Milner RS. Malignant transformation of spontaneously regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology. 1989;96:1389‐1395. DOI: 10.1016/s0161-6420(89)32714-x [DOI] [PubMed] [Google Scholar]
- 26. Uysal Y, Shields CL, Shields JA, Eagle RC Jr. Malignant transformation of retinocytoma into retinoblastoma. Retin Cases Brief Rep. 2008;2:256‐258. DOI: 10.1097/ICB.0b013e318154b70b [DOI] [PubMed] [Google Scholar]
- 27. Kiratli H, Koç I. Malignant transformation of retinocytoma treated with intra‐arterial chemotherapy. Can J Ophthalmol. 2016;51:e105‐107. DOI: 10.1016/j.jcjo.2015.12.023 [DOI] [PubMed] [Google Scholar]
- 28. Navaratnam J, Faber R, Eide N, Lund‐Iversen M, Garred Ø, Munier FL. Retinocytoma undergoing retinoblastoma transformation in an adult patient. Case Rep Ophthalmol Med. 2023;2023:8127245. DOI: 10.1155/2023/8127245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dimaras H, Khetan V, Halliday W, Héon E, Chan HS, Gallie BL. Retinoma underlying retinoblastoma revealed after tumor response to 1 cycle of chemotherapy. Arch Ophthalmol. 2009;127:1066‐1068. DOI: 10.1001/archophthalmol.2009.178 [DOI] [PubMed] [Google Scholar]
- 30. Karcioglu ZA. Fine needle aspiration biopsy (FNAB) for retinoblastoma. Retina. 2002;22:707‐710. DOI: 10.1097/00006982-200212000-00004 [DOI] [PubMed] [Google Scholar]
- 31. Sabir M, Jha P, Chawla R, Shaikh N. Multimodal imaging in sporadic retinocytoma. BMJ Case Rep. 2023;16:e252260. DOI: 10.1136/bcr-2022-252260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rootman DB, Gonzalez E, Mallipatna A, Vandenhoven C, Hampton L, Dimaras H, et al. Hand‐held high‐resolution spectral domain optical coherence tomography in retinoblastoma: clinical and morphologic considerations. Br J Ophthalmol. 2013;97:59‐65. DOI: 10.1136/bjophthalmol-2012-302133 [DOI] [PubMed] [Google Scholar]
- 33. Kamihara J, Bourdeaut F, Foulkes WD, Molenaar JJ, Mossé YP, Nakagawara A, et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res. 2017;23:e98‐e106. DOI: 10.1158/1078-0432.CCR-17-0652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mirzayev I, Gündüz AK. Hamartomas of the retina and optic disc. Turk J Ophthalmol. 2022;52:421‐431. DOI: 10.4274/tjo.galenos.2022.25979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shields CL, Say E, Fuller T, Arora S, Samara WA, Shields JA. Retinal astrocytic hamartoma arises in nerve fiber layer and shows “Moth‐Eaten” optically empty spaces on optical coherence tomography. Ophthalmology. 2016;123:1809‐1816. DOI: 10.1016/j.ophtha.2016.04.011 [DOI] [PubMed] [Google Scholar]
- 36. Soliman SE, VandenHoven C, MacKeen LD, Héon E, Gallie BL. Optical coherence tomography‐guided decisions in retinoblastoma management. Ophthalmology. 2017;124:859‐872. DOI: 10.1016/j.ophtha.2017.01.052 [DOI] [PubMed] [Google Scholar]
- 37. Yang B, Li D, Xiao J. Spontaneous regression of an isolated retinal astrocytic hamartoma in a newborn: a case report. BMC Ophthalmology. 2023;23:395. DOI: 10.1186/s12886-023-03135-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chung J, Turaka K, Shields CL. Retinocytoma shows lack of response to chemoreduction. J Pediatr Ophthalmol Strabismus. 2010;47 Online:e1‐3. DOI: 10.3928/01913913-20101217-03 [DOI] [PubMed] [Google Scholar]
- 39. Berry JL, Xu L, Polski A, Jubran R, Kuhn P, Kim JW, et al. Aqueous humor is superior to blood as a liquid biopsy for retinoblastoma. Ophthalmology. 2020;127:552‐554. DOI: 10.1016/j.ophtha.2019.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gerrish A, Mashayamombe‐Wolfgarten C, Stone E, Román‐Montañana C, Abbott J, Jenkinson H, et al. Genetic diagnosis of retinoblastoma using aqueous humour‐findings from an extended cohort. Cancers. 2024;16:1565. DOI: 10.3390/cancers16081565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Polski A, Xu L, Prabakar RK, Kim JW, Shah R, Jubran R, et al. Cell‐free DNA tumor fraction in the aqueous humor is associated with therapeutic response in retinoblastoma patients. Transl Vis Sci Technol. 2020;9:30. DOI: 10.1167/tvst.9.10.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fabius AWM, van Hoefen Wijsard M, van Leeuwen FE, Moll AC. Subsequent malignant neoplasms in retinoblastoma survivors. Cancers. 2021;13:1200. DOI: 10.3390/cancers13061200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korswagen LA, Moll AC, Imhof SM, Schouten‐van Meeteren AY. A second primary tumor in a patient with retinoma. Ophthalmic Genet. 2004;25:45‐48. DOI: 10.1076/opge.25.1.45.29006 [DOI] [PubMed] [Google Scholar]
- 44. Mendoza PR, Specht CS, Hubbard GB, Wells JR, Lynn MJ, Zhang Q, et al. Histopathologic grading of anaplasia in retinoblastoma. Am J Ophthalmol. 2015;159:764‐776. DOI: 10.1016/j.ajo.2014.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh L, Kashyap S. Update on pathology of retinoblastoma. Int J Ophthalmol. 2018;11:2011‐2016. DOI: 10.18240/ijo.2018.12.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820‐823. DOI: 10.1073/pnas.68.4.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643‐646. DOI: 10.1038/323643a0 [DOI] [PubMed] [Google Scholar]
- 48. Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327‐334. DOI: 10.1016/S1470-2045(13)70045-7 [DOI] [PubMed] [Google Scholar]
- 49. Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297‐306. DOI: 10.1038/nrm3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, et al. Retinoblastoma‐protein‐dependent cell‐cycle inhibition by the tumour suppressor p16. Nature. 1995;375:503‐506. DOI: 10.1038/375503a0 [DOI] [PubMed] [Google Scholar]
- 51. Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715‐1725. DOI: 10.1002/ijc.27316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abouzeid H, Schorderet DF, Balmer A, Munier FL. Germline mutations in retinoma patients: relevance to low‐penetrance and low‐expressivity molecular basis. Mol Vis. 2009;15:771‐777. [PMC free article] [PubMed] [Google Scholar]
- 53. Liu H, Zhang Y, Zhang YY, Li YP, Hua ZQ, Zhang CJ, et al. Human embryonic stem cell‐derived organoid retinoblastoma reveals a cancerous origin. Proc Natl Acad Sci U S A. 2020;117:33628‐33638. DOI: 10.1073/pnas.2011780117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu XL, Fang Y, Lee TC, Forrest D, Gregory‐Evans C, Almeida D, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone‐specific MDM2 signaling. Cell. 2009;137:1018‐1031. DOI: 10.1016/j.cell.2009.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu XL, Singh HP, Wang L, Qi DL, Poulos BK, Abramson DH, et al. Rb suppresses human cone‐precursor‐derived retinoblastoma tumours. Nature. 2014;514:385‐388. DOI: 10.1038/nature13813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Singh HP, Wang S, Stachelek K, Lee S, Reid MW, Thornton ME, et al. Developmental stage‐specific proliferation and retinoblastoma genesis in RB‐deficient human but not mouse cone precursors. Proc Natl Acad Sci U S A. 2018;115:E9391‐E9400. DOI: 10.1073/pnas.1808903115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46:617‐634. DOI: 10.1002/gcc.20457 [DOI] [PubMed] [Google Scholar]