

ABSTRACT
The DNA helicase HELQ is involved in homologous recombination repair, interstrand cross‐link repair, and replication stress response. Its functional defects are associated with infertility and abnormal gametogenesis. However, the specific mechanisms of HELQ in the development of germ cells remain to be elucidated. Here, we uncovered that HELQ deficiency led to proliferation defects of primordial germ cells (PGCs) in mouse embryos, thus compromising the establishment of reproductive reserve. Mechanistically, we found that HELQ interacted with the H3K9me3 demethylase KDM4B, and the absence of HELQ led to a marked increase in both total and chromatin‐bound protein levels of KDM4B, resulting in reduced H3K9me3 levels in the region of the retrotransposon LINE‐1, which triggered its high expression and subsequently caused DNA damage accumulation. Moreover, the developmental defects of HELQ‐deficient PGCs were alleviated by inhibition of retrotransposition. These results indicate that HELQ maintains the genome stability of PGCs by repressing LINE‐1 expression. Our study reveals a critical role of HELQ in PGC development and provides new insights into reproductive disorders caused by defects in DNA damage response factors.
Keywords: DNA damage response, HELQ, LINE‐1, primordial germ cells, retrotransposition
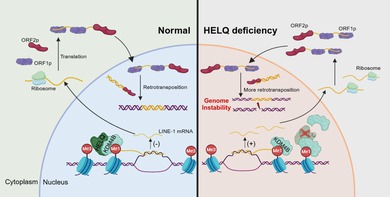
HELQ interacts with the histone demethylase KDM4B to maintain the level of H3K9me3 modification in the LINE‐1 region, thus inhibiting its expression and preserving genome stability. HELQ deficiency leads to elevated levels of KDM4B and reduces H3K9me3 modification, which increases LINE‐1 expression and subsequently genome instability in primordial germ cells.
1. Introduction
Germ cell development experiences intricate stages encompassing the specification of primordial germ cells (PGCs), migration, proliferation, sex determination, meiosis, and gamete maturation [1]. PGCs are precursors of the germ cells, which provide the basis for gametogenesis [2]. An insufficient number of PGCs diminishes the reproductive reserve and is an important cause of premature ovarian insufficiency (POI) in females and non‐obstructive azoospermia (NOA) in males [3, 4]. Therefore, ensuring the normal development of PGCs during the embryo stage is imperative for the maintenance of reproduction.
Germ cells maintain stringent genome stability to safeguard the faithful transmission of genetic information and species perpetuation [5, 6]. However, germ cells encounter diverse endogenous and exogenous DNA threats during their development, thus heavily relying on intricate DNA damage response (DDR) mechanisms to safeguard the integrity and stability of their genomes [7, 8]. Recent studies have uncovered the pivotal role of DDR pathways in governing germ cell development and maintaining fecundity [8, 9]. It has been well known that deficiencies in homologous recombination (HR) repair genes lead to aberrant repair of the programmed DNA double‐strand breaks (DSBs) arising during meiosis and are an important cause of reproductive defects in both mouse and humans [10, 11]. However, the functional loss of some DDR genes, including various Fanconi anemia (FA) genes, Rad54, and Mcm9, has resulted in profound PGC loss [12, 13, 14], highlighting the significance of the DDR mechanism in PGC development. Recently, we have found that a classical HR factor, MCM8, also protects the genome stability of PGCs from DNA damage caused by R‐loop accumulation [15], suggesting functional diversity of DDR factors in different stages of germ cell development.
HELQ (helicase POLQ‐like) is a DNA helicase involved in a variety of DDR processes. It maintains genome stability in mitotic cells by participating in inter‐strand crosslink (ICL) repair, replication stress response, and DSB repair [16]. Its deficiency leads to cellular sensitivity to the ICL inducer mitomycin C (MMC) independent of the FA pathway [17, 18]. When replication forks are stalled because of exogenous and endogenous replication stress, HELQ stabilizes the binding of RAD51 to nascent DNA on replication forks, thereby protecting stalled replication forks from nuclease degradation [19]. After DSB generation, HELQ promotes DSB end resection by enhancing EXO1 activity [19]; it also captures RPA‐bound ssDNA and strips RPA to promote annealing of complementary sequences and forms a complex with RAD51 to stimulate translocation during DNA unwinding to promote DSB repair [20, 21]. In addition, HELQ also assists the FA pathway in maintaining the stability of common fragile sites during mitosis [22].
HELQ is continuously and highly expressed during the development of germ cells from PGCs to spermatocytes and oocytes in males and females, respectively [23, 24]. A large‐scale population genetic study has found that HELQ is closely associated with early menopause [25], and variants of the HELQ gene have been identified in POI and NOA patients [26, 27, 28]. HELQ deficiency also results in atrophy of gonads and a decline in mouse fertility [17, 18]. Furthermore, the loss of function of HELQ homologs in Drosophila melanogaster (Mus301) and C aenorhabditis elegans (HELQ‐1) caused abnormal DSB repair during oocyte meiosis [29, 30], suggesting that HELQ deficiency leads to reproductive defects by impairing meiosis. Moreover, HELQ deficiency in male mice interrupted the transition of PGCs from mitosis to the mitotic arrest stage, resulting in an abnormal cell cycle and elevated apoptosis, suggesting that HELQ is also involved in regulating PGC development [23]. However, the specific role and molecular mechanism of HELQ in maintaining germ cell genome stability need further exploration.
The human genome contains a large amount of non‐coding regions, approximately 45% of which are transposable elements that are able to induce cellular apoptosis and senescence owing to their endonuclease activity and random transposition [31]. The long interspersed nuclear element 1 (LINE‐1) is the only retrotransposon with autonomous transposition activity in humans, and highly active LINE‐1 caused genome instability and even cell death [32]. Therefore, it is imperative for strict suppression of LINE‐1 expression and/or activity to maintain genome stability [33]. Yet, the genome‐wide demethylation process during the PGC development stage offers an opportunity for activation and transposition [1], indicating that additional regulatory mechanisms are required to defend against LINE‐1 activation and ensure normal development of germ cells.
In this study, we found a notable reduction of PGC number accompanied by proliferative defects and DNA damage accumulation in Helq knockout (KO) embryos. The absence of HELQ led to an upregulation of KDM4B, a demethylase of the repressive histone modification histone 3 lysine 9 trimethylation (H3K9me3), thus decreasing H3K9me3 levels in the LINE‐1 region and increasing its expression. The downregulation of LINE‐1 expression reduced the DNA damage and proliferative defects in KO cells, suggesting that elevated LINE‐1 expression contributed to DNA damage in HELQ‐deficient cells. This study revealed an essential role of HELQ in PGC development, offering a novel insight into the role of DDR genes in germ cell genome stability.
2. Materials and Methods
2.1. Mice
The Helq +/− mice were constructed by the Shanghai Model Organisms Center using the CRISPR‐Cas9 technology in the C57BL/6 genetic background. The gRNA sequences were as follows: gRNA‐1, TCA TTG GGA TAG TCC TTT GCT GG; gRNA‐2, CGA GAT CAG AGA CGG AAC CAG GG. Genomic DNA extracted from the tail or embryonic head was used for genotyping by PCR. The PCR primers were as follows: Helq‐F1, CAA AGA CTG GGC GGT TAG GT; Helq‐R1, CTG GGA ACT GAA CCC TGG TC; and Helq‐R2, CATC ACG GTC CCC ACA TTC A. All mice were kept under a specific pathogen‐free environment maintained at a temperature of 22°C–26°C and humidity of 40%–70%, with a consistent 12‐h light/dark cycle. According to the ethical animal guidelines, mice are euthanized before sampling to avoid unnecessary pain and discomfort. The Experimental Animal Ethics Review Committee of Shandong University approved all animal experiments in this study.
2.2. Generation of MEFs
Helq +/− female mice were mated with Helq +/− males. Noon of the day when copulation plugs were found was defined as E0.5. Embryos were removed from pregnant female mice at E13.5, and a small piece of tissue from the embryonic head was preserved for subsequent genotyping. The embryonic trunks were carefully collected and subjected to enzymatic digestion with 0.05% trypsin (Invitrogen) in a 37°C water bath. Following digestion, the cells were centrifuged, resuspended, and cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The obtained MEFs were then transduced with lentivirus SV40gp6 (Genechem) to facilitate immortalization. After 96 h, the MEFs were treated with 1 μg/mL puromycin for 4 days to select for successfully immortalized cells.
2.3. Construction of the HELQ Knockout Cell Line
The HELQ gene (NM_133636.5) in HEK293T cells was knocked out using the CRISPR/Cas9 technology. In brief, HELQ‐sgRNA sequences were inserted into the PsPca9(BB)‐2A‐Puro (PX459) plasmid to construct HELQ‐targeted plasmids. The sgRNA sequences were as follows: sgRNA‐F‐1, CAC CGT ATA TAC TTG GTA CAG CC CT; sgRNA‐R‐1, AAA CAG GGC TGT ACC AAG TAT ATA C; sgRNA‐F‐2, CAC CGT AGA GGA ATT ATG TCA GGT A; sgRNA‐R‐2, AAA CTA CCT GAC ATA ATT CCT CTAC. At 48 h after transfection with the targeted plasmids, the cells were subjected to selection using 1 μg/mL puromycin for another 4 days. Then, single HEK293T cells were isolated and cultured into colonies. DNA and protein were extracted from these single colonies for knockout validation by Sanger sequencing and western blot. The KO cell line with c.298‐312_1012 + 232del1259 deletion was selected for in vitro experiments.
2.4. Cell Culture and Treatment
The HEK293T cell line, from Cell Bank, Chinese Academy of Sciences, and MEF cells were cultured in Dulbecco's modified eagle medium/high glucose medium (Gibco, 11965084) with 10% FBS (Gibco, 10091148) containing 1% penicillin–streptomycin (HyClone, SV30010) at 37°C with 5% CO2.
HEK293T cells were transduced with HELQ‐HA lentivirus (Genechem) and subsequently selected with 1 μg/mL puromycin for 3 days for further experiments. To downregulate LINE‐1 in MEF cells, the cells were infected with shLINE1‐adenovirus (VectorBuilder) containing the target sequence AGA ACA GAA TGC CAC CTT TAA. KDM4B siRNA (sense: 5′‐GGCAUAAGAUGACCCUCAUTT‐3′, antisense: 5′‐AUGAGGGUCAUCUUAUGCCTT‐3′) and negative control siRNA (sense: 5′‐UUCUCCGAACGUGUCACGUTT‐3′, antisense: 5′‐ACGUGACACGUUCGGAGA ATT‐3′) were designed and generated by Genepharma Inc. (Shanghai, China) and were transfected into WT and KO HEK293T cells using Lipofectamine 3000 (ThermoFisher Scientific, L3000015). Transfection was performed at a concentration of 50 nM when cells reached 80% confluence. After 48 h, the cells were digested into single cells and plated in 6‐well plates or on coverslips, and then fixed for immunostaining, or the proteins were extracted for Western blot. For cell proliferation assays, cells were planted in coverslips and then cultured with 20 μM EdU for 10 min. Then the cells were fixed with 4% paraformaldehyde (PFA) for 15 min and washed with PBS for the Click‐iT EdU assay following the manufacturer's instructions (Ribo Bioscience, C10371).
2.5. Histological Preparation and Immunostaining
For immunostaining, tissues were collected and fixed in 4% PFA. For the cell cycle analysis assays, pregnant mice were injected intraperitoneally with 100 mg/kg EdU reagent for 1 h before euthanasia. Tissues were washed using PBS and then freeze‐embedded to produce sections.
For immunostaining, the sections or the cells on coverslips were washed with PBS and then were permeabilized and blocked (0.3% Triton X‐100 + 25% donkey serum + PBS) for 1 h at room temperature. Samples were then incubated overnight at 4°C with primary antibody diluted with 1% bovine serum albumin containing 0.3% Triton X‐100 and then washed three times with PBS containing 0.1% Triton X‐100 (PBST). Subsequently, they were incubated with the corresponding secondary antibody for 1 h at room temperature, and washed with PBST three times. Finally, the nuclei were counterstained with Hoechst 33342 and mounted with a coverslip. Images were acquired using the confocal microscope (Andor Technology).
The primary antibodies used for immunostaining were anti‐DDX4 (1:50 dilution; Novus Biologicals, AF2030), anti‐FOXL2 (1:250 dilution; Abcam, ab246511), anti‐SOX9 (1:200 dilution; Millipore, AB5535), anti‐cleaved PARP1 (1:100 dilution; Cell Signaling Technology, 94885), anti‐Cyclin B1 (1:200 dilution; Cell Signaling Technology, 4138), anti‐53BP1 (1:500 dilution; Novus Biologicals, NB100‐304), anti‐γH2AX (1:50 dilution; Cell Signaling Technology, 9718), anti‐KDM4B (1:100 dilution; Affinity, DF13288), anti‐ORF1p (1:100 dilution, Abcam, ab216324), and anti‐ORF2p (1:200 dilution; Abcam, ab106004). Primary antibodies were detected with Alexa Fluor 488‐, 568‐ or 637‐conjugated secondary antibodies (1:800 dilution; Invitrogen).
2.6. Genital Ridge Section Preparation and PGC Counting
The genital ridges of the desired embryos were dissected and then embedded in OCT (Invitrogen) for cryosectioning at 10 μm thickness with a freezing microtome (ThermoFisher). For PGC counting, all sections from both gonadal ridges of each embryo were collected and subjected to immunofluorescence staining using an anti‐DDX4 antibody. PGCs indicated by DDX4‐positivity were counted in alternate sections on every other section, and the total number obtained from all sections was recorded as the absolute PGC number for that embryo.
2.7. Hematoxylin and Eosin Staining
Tissue sections were deparaffinized and hydrated, followed by staining of the nuclei and cytoplasm with hematoxylin and eosin, respectively. The sections were then dehydrated and transparentized using graded ethanol and xylene, before being mounted with coverslips. Histological images were obtained using an optical microscope (Olympus, BX53).
2.8. Alkaline Phosphatase Staining
Embryos were collected and fixed with 4% PFA and then washed with PBS, followed by washing with 25 mM Tris‐Maleate buffer (pH 9.0). The embryos were then immersed in a working buffer consisting of 25 mM Tris‐Maleate buffer (pH 9.0), 0.5 mM MgCl2, 0.4 mg/mL 1‐Naphthyl phosphate disodium salt, and 1 mg/mL Fast Red TR salt (Sigma) for 20 min at 37°C. An excess amount of PBS was added to halt the staining, and tissues were gently washed with PBS and then incubated in 40% glycerol for 1 h, followed by 80% glycerol overnight. Images were obtained using a stereoscope (Nikon, SMZ745T).
2.9. RNA Extraction and Quantitative PCR Analysis
Total RNA was extracted from HEK293T cells using the RNA‐Quick Purification Kit (ES Science, RN001) and subsequently reverse transcribed to cDNA using HiScript II Q RT SuperMix (Vazyme, R223‐01). The resulting cDNA products served as templates for real‐time PCR amplification, which was performed on the Roche LightCycler R480 PCR platform using TB Green Premix Ex Taq II (Takara Bio, RR820A). The primers were as follows: GAPDH‐F, GGA GCG AGA TCC CTCCAAAAT; GAPDH‐R, GGCTGTTGTCATACTTCTCATGG; ORF1‐F: TCA AAG GAA AGC CCA TCA GAC TA; ORF1‐R: TGG CCC CCA CTC TCT TCT; ORF2‐F: AAA TGG TGC TGG GAA AAC TG; ORF2‐R: GCC ATT GCT TTT GGT GTT TT.
2.10. Chromatin‐Binding Protein Extraction and Mass Spectrometry (MS) Analysis
An equal number of WT and KO HEK293T cells were taken for chromatin‐binding protein extraction according to the Subcellular Protein Fractionation Kit (ThermoFisher, 78840). The separated proteins were reduced and alkylated after enzymolysis. The peptides were dissolved in mobile phase A and separated using the NanoElute ultra‐high‐performance liquid chromatography system. After separation, they were injected into the capillary ion source for ionization and analyzed by the timsTOF Pro (Bruker) mass spectrometer.
For protein identification, the LC–MS/MS data files were processed using Proteome Discoverer 2.4, and the tandem mass spectra were matched against the Maxquant database (v1.6.15.0). The digestion enzyme used was trypsin/P, with a maximum of two missed cleavages allowed. Quality control of mass spectrometry data was implemented through five principal metrics: (1) peptide length distribution (7–20 amino acids) and charge state (2–3 charges), confirming proper tryptic digestion and fragmentation; (2) requirement of > 2 unique peptides per protein for reliable identification; (3) protein sequence coverage (< 30%) showing positive correlation with abundance; (4) uniform molecular weight distribution across detected proteins; and (5) peptides were permitted up to 5 modifications, with carbamidomethylation of cysteines set as a fixed modification and oxidation of methionines and N‐terminal acetylation of proteins considered as variable modifications. These parameters collectively ensured data reliability for subsequent proteomic analyses. The false discovery rate (FDR) for both protein identification and peptide spectrum match (PSM) identification was set to 1%. The mass error tolerances were set to 20 ppm for precursor ions and 0.02 Da for fragment matching. Gene ontology (GO) analysis was performed by ClusterProfiler R packages.
2.11. Immunoprecipitation (IP) Assay
The cells were washed twice with PBS, and then 500 μL of lysis buffer (Invitrogen) containing protease inhibitors was added to lyse the cells at 4°C for 30 min. The cell lysate was carefully transferred into a tube. After sonication, the samples were centrifuged to obtain the supernatant. The protein concentration was detected, and equal amounts of proteins were mixed with primary antibodies, followed by incubation at 4°C overnight. The mixture was then incubated with protein A/G beads and rotated at 4°C for 2 h. Non‐specific binding proteins were removed by washing with lysis buffer three times. Subsequently, 1× SDS loading buffer was added, and the mixture was denatured at 100°C for 10 min. The collected protein samples were then used for Western blot. The primary antibodies used for IP were anti‐HELQ (Novus Biologicals, NBP1‐91842), anti‐HA (Cell Signaling Technology, 3724), and rabbit IgG (Cell Signaling Technology, 2729).
2.12. Western Blot
Proteins were separated on SDS‐PAGE gels. After electrophoresis, separated proteins were transferred to polyvinylidene fluoride membranes (Millipore), which were blocked with 5% milk for 1 h at room temperature and then incubated with the corresponding primary antibodies at 4°C overnight. The membrane was incubated with horseradish peroxidase‐conjugated rabbit or mouse IgG secondary antibody (Proteintech) for 1 h at room temperature. Signal detection was performed using an enhanced chemiluminescence system (Millipore, WBKLS0500), and visualization was carried out on a Chemidoc MP System (Bio‐Rad). The primary antibodies used for Western blot were anti‐HELQ (1:100 dilution, Santa Cruz, sc‐81095), anti‐HA (1:1000 dilution, Cell Signaling Technology, 2367), anti‐β‐actin (1:5000 dilution, Proteintech, 66009‐1‐Ig), anti‐KDM4B (1:1000 dilution, ThermoFisher, A301‐478A), anti‐α‐Tubulin (1:5000 dilution, Proteintech, 66031‐1‐Ig), anti‐H3 (1:1000 dilution, Cell Signaling Technology, C4499), anti‐ORF1p (1:1000 dilution, Cell Signaling Technology, 88701 and 1:1000 dilution, Abcam, ab216324), and anti‐γH2AX (1:1000 dilution, Millipore, 05636).
2.13. CUT&Tag Assay
The CUT&Tag experiments were executed utilizing the Hyperactive Universal CUT&Tag Assay Kit (Vazyme Biotech, TD904) according to the user manual. Briefly, 1 × 105 cells were collected and washed with wash buffer once at room temperature, then were immobilized with 10 μL of activated concanavalin A‐coated magnetic beads for 10 min. Then 1 μg of primary antibody was added, followed by an overnight incubation at 4°C. The next day, the primary antibody buffer was discarded, and the cells were incubated with 0.5 μL the secondary antibody for 1 h at room temperature. After washing with DIG wash buffer, the cells were transferred to a solution containing pA/G‐Tnp for 1 h. Following three washes using DIG‐300 buffer, the cells were incubated in TTBL at 37°C for 1 h to release genomic DNA. The genomic DNA was then transferred to pre‐activated DNA Extract Beads Pro for 20 min. After washing with B&W buffer twice and air‐drying, the samples were dissolved in 20 μL nuclease‐free water. The amplified genomic DNA underwent further processing, with PCR library products being purified using VAHTS DNA Clean Beads (Vazyme Biotech, N411). Finally, the CUT&Tag libraries were sequenced on the advanced Illumina NovaSeq 6000 platform. The primary antibodies used for immunostaining were anti‐H3K9me3 (Active Motif, 39 161) and anti‐HA (Sigma‐Aldrich, H3663).
Quality control of raw data was performed using Trimmomatic, which included adapter trimming and removal of low‐quality bases. Initial assessment showed over 90% of bases had Q20 scores (99% base‐call accuracy), signifying high sequencing quality. Further improvements were seen after removing adapter sequences and low‐quality reads, etc., reflected by a higher proportion of Q20 bases. The filtering protocol retained over 90% of the original high‐quality bases, efficiently eliminating technical artifacts while preserving biologically relevant data. Bowtie2 was utilized to map the data to the reference genome. MACS2 was employed to conduct peak‐calling analysis on the genome (setting a p‐value cutoff of less than 0.05 to identify significant peaks) and compare differential peaks between groups. ChIPseeker was used for relevant annotation and visualization of peak distribution. The bwtool v1.0 was applied to visualize the signals of different regions for graphical analysis. Bedtools was employed to calculate peak intersections without length filtering, considering any overlap of 1 bp or more as intersecting peaks.
2.14. 3TC Treatment
Helq +/− female mice were mated with Helq +/− males. Lamivudine (Selleck, S1706) was administered daily by gavage to pregnant Helq +/− females at a dosage of 50 mg/kg daily from E7.5 to E11.5. The control pregnant females received the same dose of saline. Genital ridges were isolated from the desired embryos at E12.5 and prepared for STELLA, 53BP1, and γH2AX immunostaining.
2.15. Statistical Analysis
Statistical analyses were conducted using GraphPad Prism 9 and IBM SPSS Statistics 26. The data were presented as mean ± SD or median with interquartile range (IQR). When the numbers of data were more than 20, the data were tested whether they were normally distributed. Student's t‐test or Mann–Whitney U test was applied for comparisons between two groups; one‐way ANOVA was used for comparisons among multiple groups, followed by Dunnett's post hoc test to identify the differences between two groups. p < 0.05 was considered statistically significant.
3. Results
3.1. HELQ Is Essential for PGC Development
To investigate the role of HELQ in germ cell development, we constructed KO mice with the CRISPR/Cas9 technology. Genotyping was performed using polymerase chain reaction (PCR) and the HELQ protein was undetectable in the testes of KO mice (Figure S1A–C). Consistent with previous studies, adult KO mice had significantly reduced gonad size and few germ cells (Figure S1D,E). The follicles have depleted in the ovaries of three‐month‐old female KO mice; in adult males, some spermatogenic tubules in the testes showed no obvious abnormality, and some only have supporting cells. There were mature spermatozoa in the epididymis of KO male mice, but the number was reduced compared with that of the wild type (Figure S1E). In addition, no significant differences were found in the expression levels of the granulosa cell marker FOXL2 and the Sertoli cell marker SOX9 between WT and KO gonads, but three‐day‐old KO mice suffered a substantial loss of DDX4+ germ cells, suggesting abnormal germ cell development during the embryonic period (Figure 1A). Further analysis revealed that the number of PGCs slightly decreased at E11.5 (691.2 ± 69.5 vs. 589.0 ± 44.0) and significantly reduced at E12.5 in female KO embryos (1933.0 ± 443.2 vs. 708.6 ± 100.9) (Figure 1B–D). In contrast, in male KO embryos, the number of PGCs was not statistically different at E11.5 compared to WT embryos (762.8 ± 219.5 vs. 610.4 ± 52.0) and showed a remarkable decrease at E12.5 (2421.0 ± 375.1 vs. 915.6 ± 186.0) (Figure 1B–D). These results indicate that the reduction in germ cells in KO mice originates from the developmental defects of PGCs at the embryonic stage.
FIGURE 1.
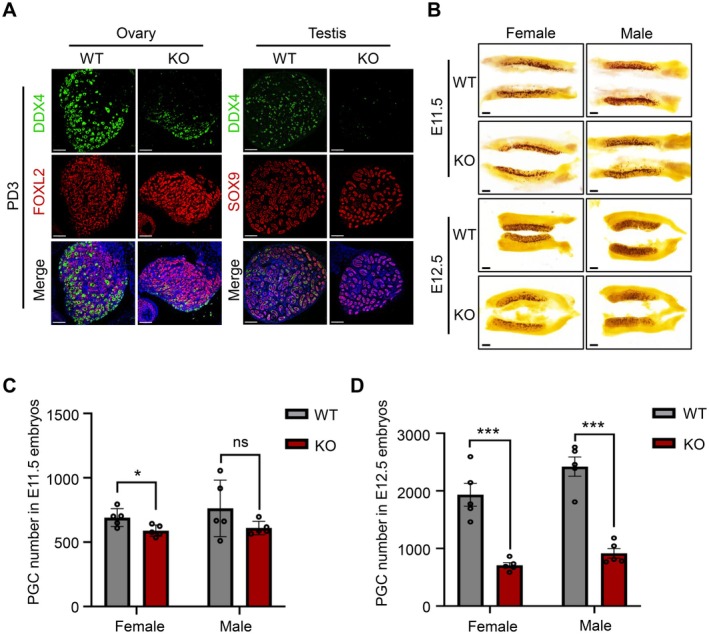
HELQ is essential for PGC development. (A) Immunofluorescence staining for DDX4 (a marker of germ cells), FOXL2 (a marker of granulosa cells), and SOX9 (a marker for Sertoli cells) in the gonads of three‐day‐old mice. (B) Representative images of alkaline phosphatase (a PGC marker) staining of genital ridges. In A and B, scale bars, 100 μm. (C, D) Absolute counts of PGCs from female and male embryos at E11.5 and E12.5, respectively. E11.5: female, 691.2 ± 69.5 versus 589.0 ± 44.0; male, 762.8 ± 219.5 versus 610.4 ± 52.0. E12.5: female, 1933.0 ± 443.2 versus 708.6 ± 100.9; male, 2421.0 ± 375.1 versus 915.6 ± 186.0. Data are presented as the mean ± SD, n = 5 embryos, *p < 0.05, ***p < 0.001, and ns, not significant by unpaired two‐tailed Student's t test.
3.2. HELQ Deficiency Impairs Proliferation and Increases DNA Damage in PGCs
Due to the noticeable decrease in PGCs in both male and female KO embryos at E12.5, we chose this time point to further investigate the cause of their reduction. The immunofluorescent staining of cleaved‐PARP1 revealed an increased level of apoptosis in PGCs of KO embryos compared to WT embryos (female: 1.30% ± 0.03% vs. 0.61% ± 0.41%; male: 1.78% ± 0.53% vs. 0.80% ± 0.26%) (Figure 2A,B). However, the overall number of apoptotic PGCs was less than 10 per KO embryo, indicating that apoptosis was not the leading cause for PGC loss. Then we examined the proliferation and cell cycle progress of PGCs by labeling S‐phase cells with 5‐ethynyl‐2′‐deoxyuridine (EdU) and co‐staining for the cell cycle protein Cyclin B1 (Figure 2C). The results showed a notable decrease in the proportion of S‐phase PGCs (female: 29.77% ± 6.80% vs. 51.57% ± 7.63%; male: 34.29% ± 5.58% vs. 43.10% ± 4.18%), accompanied by an increase in the proportion of G2‐phase PGCs in KO embryos compared to WT (female: 36.11% ± 2.74% vs. 22.97% ± 5.14%; male: 30.74% ± 5.20% vs. 22.55% ± 3.00%) (Figure 2D,E). In female KO embryos, there was no significant difference in M‐phase PGCs (7.75% ± 2.37% vs. 5.27% ± 1.91%), whereas a slight increase was observed in males (8.69% ± 0.86% vs. 6.44% ± 1.27%) (Figure 2D,E). Therefore, proliferation defects caused by HELQ deficiency were mainly responsible for PGC loss during embryonic development. PGCs are sensitive to DNA damage and rely significantly on DDR factors to maintain genome stability [34, 35]. Given the pivotal role of HELQ as a DDR factor in the maintenance of genome stability, we hypothesized that its deficiency might result in the accumulation of DNA damage, consequently impacting both cell proliferation and apoptosis. The 53BP1 and γH2AX foci were frequently utilized as markers for DSB formation, and the immunostaining revealed a notable increase in the proportion of nuclear 53BP1 and γH2AX foci in KO PGCs (female 53BP1: 24.06% ± 2.88% vs. 14.89% ± 2.63%; male 53BP1: 24.74% ± 2.34% vs. 19.82% ± 1.19%; female γH2AX: 18.94% ± 1.84% vs. 11.30% ± 1.78%; male γH2AX: 21.43% ± 3.25% vs. 14.91% ± 3.23%) (Figure 3A–D). These results suggest that HELQ preserves the genome stability of PGCs to facilitate their normal proliferation.
FIGURE 2.
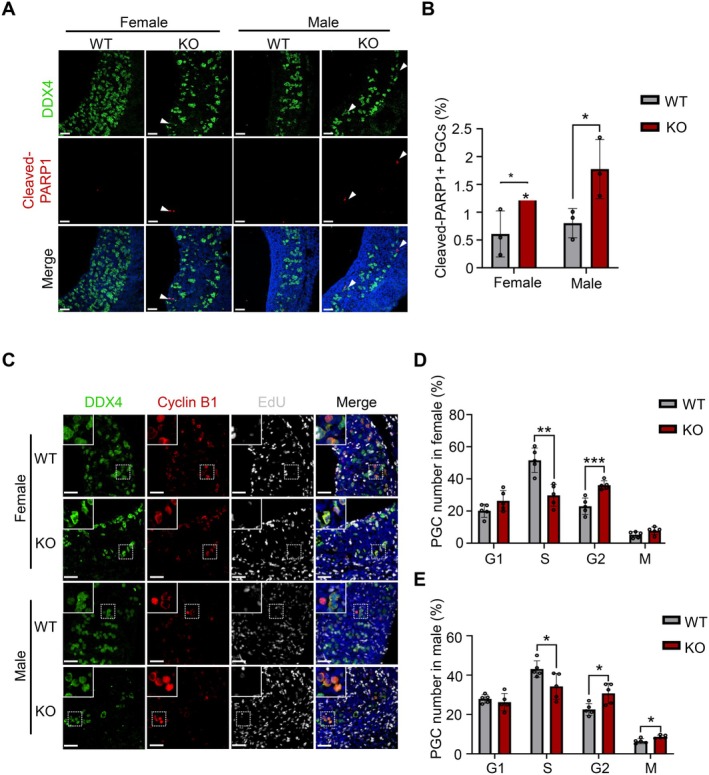
HELQ deficiency impairs proliferation of PGCs. (A) Immunofluorescence staining for cleaved‐PARP1 (a marker of apoptotic cells) in the genital ridges of E12.5 embryos. Scale bars, 50 μm. Arrowheads indicate representative cells. (B) The percentage of apoptotic PGCs in E12.5 embryos. n = 3 embryos. All PGCs were scored in each embryo. Female: 1.30% ± 0.03% versus 0.61% ± 0.41%; Male: 1.78% ± 0.53% versus 0.80% ± 0.26%. (C) Immunofluorescence staining for DDX4, Cycline B1, and EdU incorporation assay to analyze cell cycle distribution of PGCs in the genital ridges of E12.5 embryos. Scale bars, 50 μm, and 20 μm in the enlarged images. (D, E) The cell cycle distribution of PGCs in E12.5 embryos. n = 5 embryos. At least 1000 PGCs were scored in each WT embryo, and all PGCs were scored in each KO embryo. G1‐phase cells, cyclin B1 negative; S‐phase cells, EdU positive; G2‐phase cells, cyclin B1 expressed in the cytoplasm; M‐phase cells, cyclin B1 highly expressed in the nucleus. S‐phase PGCs: female, 29.77% ± 6.80% versus 51.57% ± 7.63%; male, 34.29% ± 5.58% versus 43.10% ± 4.18%. G2‐phase PGCs: female, 36.11% ± 2.74% versus 22.97% ± 5.14%; male, 30.74% ± 5.20% versus 22.55% ± 3.00%. M‐phase PGCs: female, 7.75% ± 2.37% versus 5.27% ± 1.91%; male, 8.69% ± 0.86% versus 6.44% ± 1.27%. In B, D and E, data are presented as the mean ± SD, *p < 0.05, **p < 0.01, and ***p < 0.001 by unpaired two‐tailed Student's t test.
FIGURE 3.

HELQ deficiency increases DNA damage in PGCs. (A) The immunofluorescence staining for DDX4 and 53BP1 (a marker of DNA double‐strand breaks (DSBs)) in the genital ridges of E12.5 embryos. Scale bars, 5 μm. (B) The percentage of 53BP1‐positive PGCs (cells containing at least one 53BP1 foci were considered positive) in E12.5 embryos. At least 200 PGCs were scored in each embryo. Female: 24.06% ± 2.88% versus 14.89% ± 2.63%; Male: 24.74% ± 2.34% versus 19.82% ± 1.19%. (C) The immunofluorescence staining for DDX4 and γH2AX (a marker of DNA DSBs) in the genital ridges of E12.5 embryos. Scale bars, 5 μm. (D) The percentage of γH2AX‐positive PGCs (cells containing at least one γH2AX foci were considered positive) in E12.5 embryos. At least 200 PGCs were scored in each embryo. Female: 11.30% ± 1.78% versus 18.94% ± 1.84%; Male: 14.91% ± 3.23% versus 21.43% ± 3.25%. In B and D, data are presented as the mean ± SD, n = 4 embryos, *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired two‐tailed Student's t test.
3.3. Deletion of HELQ Leads to an Elevation of the Histone Demethylase KDM4B
Since the PGC scarcity and the difficulty in isolating enough KO PGCs, we turned to constructing HELQ knockout HEK293T cells to delve into its role in maintaining genome stability. Given the low expression levels of HELQ in HEK293T cells and the limited efficacy of commercially available HELQ antibodies, IP experiments were conducted to enrich HELQ protein from both WT and KO HEK293T cells, and subsequent Western blotting assays confirmed the absence of HELQ protein in KO cells (Figure S2A). The KO HEK293T cells exhibited phenotypes similar to that of KO PGCs, characterized by the accumulation of DNA damage and impaired proliferation, suggesting the feasibility of HEK293T cells for further investigating the function of HELQ (Figure S2B‐G).
Previous investigations have indicated that the DNA helicase HELQ exerts its biological functions through binding to chromatin, and a discernible increase of nuclear foci could be observed under replication stress [36]. To further elucidate the role of HELQ in maintaining genome stability, we treated WT and KO HEK293T cells with aphidicolin (APH) to induce replication stress, then analyzed chromatin‐bound proteins by mass spectrometry. A total of 302 differentially expressed proteins were identified, including 284 upregulated proteins and 18 downregulated proteins in the KO cells compared to WT cells (Figure 4A and Table S1). We noted that these proteins were mainly involved in biological processes of gene expression regulation by Gene Ontology (GO) analysis, including various DNA‐ or RNA‐binding proteins, epigenetic modification factors, and post‐transcriptional regulators (Figure 4B). Notably, H3K9me3 demethylase KDM4B, a regulator of repressive epigenetic modification, was significantly increased in KO cells (Figure 4B and Table S1). Previous research has revealed that KDM4B exerts dual functions in DDR. Following the formation of DSBs, KDM4B is recruited to the site of damage, depending on PARP1, to promote H3K9me3 demethylation, thereby loosening heterochromatin structure to facilitate the recruitment of downstream factors for DNA damage repair [37]. However, overexpression of KDM4B reduces H3K9me3 levels in the retrotransposon element LINE‐1 region, resulting in the hyperactivation of LINE‐1 and the accumulation of DNA damage [38]. Therefore, the precise regulation of KDM4B levels is important for maintaining genome stability. We observed a significant elevation in both whole and chromatin‐bound protein levels of KDM4B in KO cells (Figure 4C–E) and also found the interaction between HELQ and KDM4B by the Co‐IP assay (Figure 4F). These results suggest that the accumulation of DNA damage in HELQ‐deficient cells may be associated with the elevated KDM4B levels.
FIGURE 4.

Deletion of HELQ leads to an elevation of the histone demethylase KDM4B. (A) Chromatin‐binding proteomics analysis of WT and KO HEK293T cells showed the up‐and down‐regulated proteins in KO cells compared to WT cells. (B) GO analysis of differentially expressed proteins obtained from proteomics. (C) Levels of KDM4B were detected by Western blot in WCE and chromatin‐binding proteins from WT and KO cells. (D) The relative expression level of KDM4B in WCE normalized to α‐Tubulin. (E) The relative expression level of chromatin‐binding KDM4B normalized to H3. (F) Co‐IP assay to detect the interaction between exogenously expressed HELQ‐HA with KDM4B in HEK293T cells. In D and E, data are presented as the mean ± SD, n = 3 independent experiments, *p < 0.05 by unpaired two‐tailed Student's t test. Chr‐binding, chromatin‐binding protein; IB, immunoblotting; IP, immunoprecipitation; WCE, whole cell extracts.
3.4. HELQ Maintains Genome Stability by Inhibiting KDM4B Expression
The full‐length LINE‐1 contains two open reading frames (ORFs), ORF1 and ORF2, which encode the ORF1p and ORF2p proteins, respectively [32]. The mRNA levels of ORF1 and ORF2 were significantly increased in KO HEK293T cells compared to WT cells (Figure 5A,B), and LINE‐1 protein levels were similarly increased (Figure 5C–F), which were also observed in KO PGCs and MEF cells (Figures 5G,H and S3A,B). Due to the transposition process of LINE‐1 and the nucleic acid endonuclease activity possessed by ORF2p, elevated expression of LINE‐1 threatens genome stability [39]. To determine whether the high expression of LINE‐1 and the accumulation of DNA damage in HELQ‐deficient cells were associated with the elevated KDM4B levels, we performed small interfering RNA (siRNA)‐mediated knockdown of KDM4B in WT and KO HEK293T cells. The reduction of KDM4B protein levels after siRNA transfection was confirmed (Figure 6A,B). Notably, KDM4B knockdown significantly suppressed the aberrantly increased expression of LINE‐1 retrotransposons in HELQ KO cells (Figure 6A,C), which is consistent with the reported role of KDM4B in promoting LINE‐1 activation via H3K9me3 demethylation [38]. Moreover, depletion of KDM4B markedly reduced DNA damage accumulation (Figure 6A,D) and restored the proliferative capacity of KO HEK293T cells (Figure 6E,F). Collectively, these results demonstrate that HELQ maintains genome stability, at least in part, by restraining KDM4B expression.
FIGURE 5.
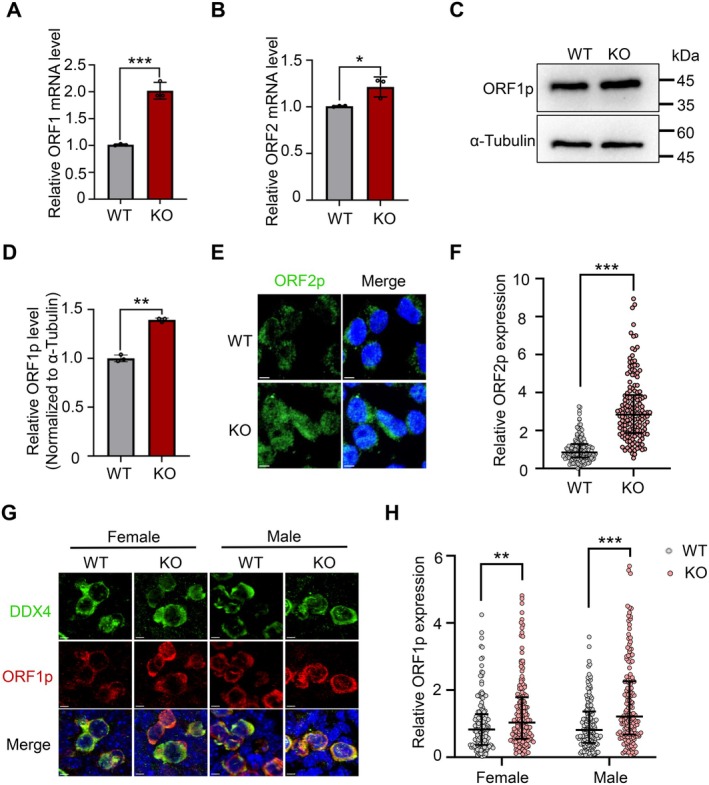
HELQ deficiency results in increased LINE‐1 expression. (A, B) The ORF1 and ORF2 mRNA levels were determined by RT‐qPCR in WT and KO HEK293T cells. n = 3 independent experiments. (C, D) The expression level of LINE‐1 ORF1p protein in WT and KO HEK293T cells were detected by Western blot, and the α‐tubulin was used as the loading control. n = 3 independent experiments. (E, F) Representative immunostaining images and the relative expression level of ORF2p in WT and KO HEK293T cells. Scale bars, 5 μm. At least 150 cells were scored in each embryo. (G, H) Immunofluorescence staining for DDX4 and ORF1p in E12.5 genital ridges and the relative expression level of ORF1p in PGCs. Scale bars, 5 μm. At least 150 cells were scored in each embryo. In A, B, and D, data are presented as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 by unpaired two‐tailed Student's t test. In F and H, data are presented as median with IQR, **p < 0.01, ***p < 0.001 by two‐tailed Mann–Whitney U test.
FIGURE 6.

HELQ maintains genome stability by inhibiting KDM4B expression. (A) The expression levels of ORF1p, γH2AX and KDM4B in WT and KO HEK293T cells after knockdown of KDM4B were detected by Western blot. Ctrl, control;siKDM4B, KDM4B‐targeting siRNA. (B–D) The relative expression levels of KDM4B, ORF1p, and γH2AX in WT and KO MEFs after knockdown of KDM4B. (E) Representative images of EdU assay to evaluate the proliferation of WT and KO HEK293T after knockdown of KDM4B. Scale bars, 10 μm. (F) The percentage of EdU‐positive cells in WT and KO HEK293T cells after knockdown of KDM4B. At least 200 cells were scored per group. WT‐Ctrl: 43.32% ± 3.26%, KO‐Ctrl: 34.02% ± 3.84%, WT‐siKDM4B: 44.32% ± 2.48%, KO‐siKDM4B: 42.78% ± 1.70%. In B, C, D and F, data are presented as mean ± SD, n = 3 independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001 by one‐way ANOVA, followed by Dunnett's post hoc test.
3.5. HELQ Regulates the Level of H3K9me3 in the LINE‐1 Region
H3K9me3 plays a key role in transcriptional silencing and heterochromatin formation, which are involved in maintaining genome stability [40, 41]. To further explore the regulatory function of HELQ in H3K9me3, we performed the CUT&Tag assay in both wild‐type and HELQ‐HA stably expressing HEK293T cells to detect the distribution features of H3K9me3 and HELQ in the genome. We found that H3K9me3 exhibited a preferential distribution in non‐coding regions, specifically in intergenic (39.10%) and intronic (38.00%) regions, but was less abundant in open chromatin regions like promoters and exons (Figure 7A). Meanwhile, HELQ exhibited a similar distribution pattern, with enrichment in intergenic (33.50%) and intron (40.30%) regions (Figure 7A).
FIGURE 7.

HELQ regulates the level of H3K9me3 in the LINE‐1 region. (A) Distribution ratio of H3K9me3 and HELQ‐HA on genomic elements of HEK293T cells. (B) Proportion of distribution of H3K9me3 and HELQ‐HA on transposable elements of HEK293T cells. (C, D) Overlap of HELQ‐HA binding sites with H3K9me3 modification sites and the proportion of distribution of these shared regions on transposable elements of HEK293T cells. (E) Heatmap of the overall signal levels of H3K9me3 in WT and KO HEK293T cells. (F) The signal level of H3K9me3 in the LINE‐1 region of WT and KO HEK293T cells.
Given the regulatory significance of H3K9me3 in transposable elements, we further analyzed the distribution of H3K9me3 and HELQ‐HA on transposable elements and observed a pronounced enrichment of peaks in the LINE‐1 region, constituting 25.78% (H3K9me3) and 23.29% (HELQ‐HA) of the total transposable element peaks, respectively (Figure 7B). Subsequently, the overlap analysis of the CUT&Tag data for HELQ‐HA and H3K9me3 identified 232,917 overlapping peaks, accounting for about 23% of all peaks (Figure 7C). Furthermore, LINE‐1 elements constituted the highest proportion, accounting for 16.28% of the total overlapping peaks, followed closely by Alu elements at 9.65% (Figure 7D). Considering that H3K9me3 under the regulation of KDM4B was primarily localized in the LINE‐1 region [38], we hypothesized that HELQ maintained H3K9me3 levels in the LINE‐1 region by downregulating KDM4B.
To elucidate the impact of HELQ deletion on H3K9me3, we compared the H3K9me3 levels in WT and KO HEK293T cells and found that there was no significant alteration in the overall genome‐wide H3K9me3 levels following HELQ deletion, but the level of H3K9me3 in the LINE‐1 region was decreased (Figure 7E,F), suggesting that the transcriptional activity in the LINE‐1 region may be increased upon HELQ deletion.
3.6. HELQ Reduces DNA Damage by Inhibiting LINE‐1 Expression
To further determine whether the accumulation of DNA damage in HELQ‐deficient cells originates from the high expression of LINE‐1, we conducted the rescue experiments by inhibiting the expression or activity of LINE‐1. Knockdown of LINE‐1 in MEFs by adenovirus infection reduced DNA damage levels (Figure S3A,C) and restored the proliferative capacity (Figure S3D,E). Moreover, inhibiting retrotransposition of LINE‐1 with Lamivudine (3TC) in pregnant mice partially rescued PGC number and decreased DNA damage levels in KO PGCs (Figures 8 and S4). These results demonstrated that HELQ deficiency caused high expression of LINE‐1, leading to DNA damage accumulation and proliferation impairment in cells (Figure 9).
FIGURE 8.

Inhibiting retrotransposition protects PGC development in KO embryos. (A) Presentative images of DDX4 and 53BP1 immunostaining in E12.5 genital ridges of embryos after pregnant mice were administered with saline or 3TC. Scale bars, 5 μm. Arrowheads indicate representative cells. Ctrl, control; 3TC, 2′‐3′ deoxy‐3′‐thiocytidine. (B) Absolute counts of PGCs in E12.5 embryos after pregnant mice were administered with saline or 3TC. (Female: WT‐Ctrl: 2223 ± 311.70, KO‐Ctrl: 744.30 ± 23.12, WT‐3TC: 3554 ± 409.10, KO‐3TC: 1646 ± 235.10. Male: WT‐Ctrl: 2298 ± 348.40, KO‐Ctrl: 699.70 ± 176.20, WT‐3TC: 2412 ± 406, KO‐3TC: 1699 ± 501.80) (C) The proportion of 53BP1‐positive PGCs (with at least one foci) in E12.5 embryos after pregnant mice were administered with saline or 3TC. At least 200 PGCs were scored in each embryo. (Female: WT‐Ctrl: 8.67% ± 2.08%, KO‐Ctrl: 21.07% ± 1.91%, WT‐3TC: 10.83% ± 2.02%, KO‐3TC: 13.17% ± 2.26%. Male: WT‐Ctrl:12.17% ± 2.93%, KO‐Ctrl: 21.17% ± 2.26%, WT‐3TC: 10.67% ± 2.75%, KO‐3TC: 11.83% ± 1.04%) In B and C, data are presented as mean ± SD, n = 3 embryos, *p < 0.05, **p < 0.01, ***p < 0.001 by one‐way ANOVA, followed by Dunnett's post hoc test.
FIGURE 9.

HELQ inhibits LINE‐1 expression to maintain genome stability. HELQ interacts with the histone demethylase KDM4B to maintain the level of H3K9me3 modification in the LINE‐1 region, thus inhibiting its expression and preserving genome stability. HELQ deficiency leads to elevated levels of KDM4B and reduces H3K9me3 modification, which increases LINE‐1 expression and subsequently genome instability in primordial germ cells.
4. Discussion
In this study, we found that HELQ maintained the H3K9me3 modification in the LINE‐1 region by down‐regulating the KDM4B level to inhibit the expression of LINE‐1, thus maintaining genome stability, which may be involved in PGC rapid proliferation and reproductive reserve establishment. We have uncovered a novel function of the DDR factor HELQ in inhibiting the activation of the retrotransposon LINE‐1, providing new insights into the mechanisms by which DDR genes maintain the genome stability of germ cells.
HELQ is a versatile DDR factor and functions in multiple stages of gametogenesis. HELQ homologs functioned in DSB repair during meiosis in D. melanogaster and C. elegans [29, 30], whereas Helq knockout decreased spermatogonia before the initiation of meiosis in PD5 male mice [17, 18]. Furthermore, HELQ deficiency led to aberrant mitotic arrest transition of PGCs in male mouse embryos and impaired the consequent formation of the spermatogonial stem cell pool [23]. In contrast, our study showed a massive loss of PGCs at E12.5 with proliferation defects and DNA damage in both male and female KO embryos, thus affecting the germline reserve establishment. Although HELQ is ubiquitously expressed in various cell types, the abnormality was particularly severe in KO PGCs compared to somatic cells. These results suggest that germline progenitor cells may harbor a heightened dependency on the function of HELQ to protect the genome stability and integrity from specific source of endogenous DNA damage in PGCs. In this study, we first found that HELQ deficiency led to DNA damage accumulation induced by high LINE‐1 expression in PGCs, suggesting that activation of LINE‐1 is a crucial endogenous threat to the genome stability of PGCs. The phenotypic discrepancy between PGCs and somatic cells could potentially be explained by certain biological underpinnings. In fact, the specific loss of germ cells was observed in knockout mouse models of other ubiquitously expressed DNA repair genes, including genes in the Fanconi Anemia pathway and HR repair [12, 13, 14]. These findings suggest that PGCs could be considered as a group of special cells that are more dependent on DNA repair factors to respond to endogenous genome threats. Especially, extensive epigenetic reprogramming including global DNA demethylation and elevated expression level of LINE‐1 during PGC development [1], creates a vulnerability to retrotransposons, which are an endogenous source of genome threat. Therefore, it is necessary to suppress transposon activity in PGCs to maintain a high level of genome stability. While previous studies have revealed that HELQ maintains genome stability by participating in ICL repair, replication stress response, and DSB repair, we find a novel function of HELQ to inhibit LINE‐1 expression to maintain genomic stability during PGC development in this study.
LINE‐1 threatens genome stability through transposition and nuclease activity. The main mechanisms against its genotoxicity include epigenetic modifications and RNA‐mediated regulation [42, 43]. Given the helicase activity of HELQ in DNA repair and its localization to damaged replication forks [20, 36], the HELQ‐KDM4B‐H3K9me3 axis may represent a more generalized damage‐control mechanism to suppress transposon activation during DNA replication, compared with the canonical piRNA‐PIWI pathway that depends on small RNA‐guided sequence‐specific silencing [43]. Furthermore, emerging evidence indicated that DNMT1‐mediated DNA methylation is functionally coupled to H3K9me3‐marked heterochromatin [44]. Therefore, HELQ deletion may also compromise DNA methylation, which needs to be verified.
As epigenetic reprogramming leads to loss of transcriptional silencing and activation of the LINE‐1 expression, the transposable elements are significantly more active in germ cells compared to somatic cells [45]. The strict regulation of LINE‐1 expression and/or activity at different stages of germ cell development has been revealed [46, 47]. Knockdown of DNA methylase Dnmt3l decreases DNA de novo methylation that can cause high expression of transposable elements, including LINE‐1 and intracisternal A‐particles (IAP), leading to aberrant meiosis and impaired germ cell development in male mice [42]. Meanwhile, the PIWI/piRNA pathway also inhibits the LINE‐1 expression by promoting DNA methylation and H3K9me3 modifications in LINE‐1 regions in spermatogenesis [43]. On the contrary, the depletion of PIWI‐associated proteins results in the excessive activation of transposable elements, ultimately impeding spermatogenesis and causing infertility in male mice [43]. Interestingly, infertility is also present in female golden hamsters without PIWI family proteins [48, 49]. In contrast, reverse transcriptase inhibitors, which limit the genomic toxicity of ORF2p, can mitigate fetal oocyte attrition (FOA) and increase the ovarian reserve in mice with no significant negative effects on fertility [50]. Additionally, deficiency of the histone methyltransferase PRMT5 results in the downregulation of the suppressive modification H2A/H4R3me2s in PGCs, leading to increased LINE‐1 expression, accumulated DNA damage, and decreased PGCs [51]. In this study, we found that PGCs lacking HELQ showed high expression of LINE‐1, leading to genome instability, which could be rescued by the reverse transcription inhibitor, further emphasizing the importance of the proper regulation of LINE‐1 in germ cells, especially during PGC development. The critical link between HELQ deficiency and diminished reproductive reserve provides a new insight into genetic counseling, early diagnosis, and therapeutic strategies for patients with POI or NOA and further evidence supporting the fertility‐preserving potential of LINE‐1 inhibitors. Targeted pharmacologic interventions against LINE‐1 retrotransposition may become a promising approach to improve fertility, especially in cases associated with HELQ dysfunction or other related genetic defects.
Mechanistically, we found that HELQ deletion resulted in significantly higher levels of H3K9me3 demethylase KDM4B in both total and chromatin‐binding proteins, and KDM4B was also identified to interact with HELQ. It has been reported that KDM4B could recognize and catalyze the demethylation of H3K9me3 in the LINE‐1 region [38]. Consistently, we discovered a substantial overlap of the DNA binding sites of HELQ and the H3K9me3 modification sites in the LINE‐1 region. Notably, while the overall genomic level of H3K9me3 remained unaffected by HELQ deletion, a discernible decrease in H3K9me3 signals was detected within the LINE‐1 region specifically. This pattern resembled the altered H3K9me3 modification observed upon KDM4B overexpression [38], suggesting a potential correlation between H3K9me3 modulation and increased KDM4B expression in HELQ‐deficient cells. However, the mechanism underlying the upregulation of KDM4B expression and its enhanced chromatin binding after HELQ deletion needs future exploration. In addition, the increased levels of LINE‐1 expression were also found in KO PGCs, suggesting a novel function of HELQ in restricting LINE‐1 expression in PGC development. As a conserved epigenetic marker, H3K9me3 exhibits preferential enrichment at younger transposable elements, which usually are known as active elements [52]. H3K9me3 broadly represses a wide range of transposons by collaborating with various methyltransferases, each of which is specific to different class retrotransposons. For instance, H3K9 methyltransferases TRIM28 and SETDB1 predominantly regulate long terminal repeat elements [53, 54, 55], whereas methyltransferase SUV39H1/2 predominantly regulates the LINEs [56]. The regulation of transposons by H3K9me3 also exhibits developmental stage specificity, forming a multidimensional precise regulatory network [52, 57], suggesting that H3K9me3‐mediated transposon repression is both global and functionally specialized. Our study found that HELQ occupied several H3K9me3‐marked transposon elements, with LINE‐1 being the most highly represented. However, it is possible that other transposable elements may also be modulated by a similar mechanism, which requires further experimental validation.
Recently, many studies have also demonstrated the regulatory effects of DDR factors on LINE‐1 retrotransposition, and knockdown of FA and HR pathway‐related genes increases LINE‐1 transposition activity [58, 59, 60]. While the SLX4‐XPF‐ERCC1 incision complex in the FA DNA crosslink repair pathway cleaves nucleic acid intermediates of LINE‐1 retrotransposition to prevent reverse transcription [61], BRCA1 restricts LINE‐1 retrotransposition by promoting DNA‐end resection at DSBs in the nucleus and by binding to LINE‐1 mRNA and suppressing the translation of ORF2p in the cytoplasm [60]. Besides, DNA damage checkpoint proteins have emerged as critical regulators of LINE‐1 transposition. ATM deficiency in mouse neuronal cells leads to an increased frequency of LINE‐1 transposition [62]. In addition to the direct suppression of LINE‐1 transposition, p53 also downregulates LINE‐1 mRNA transcription by binding to its 5′‐UTR region [63]. Our finding emphasizes that HELQ could recruit histone methyltransferase KDM4B to LINE‐1 genomic loci, directly enforcing their epigenetic silencing through chromatin modification. This transcriptional‐level suppression, combined with downstream transposition blockade mediated by other DDR factors, potentially constitutes an evolutionarily conserved two‐tiered defense strategy to restrict LINE‐1 mobilization. Nevertheless, the coordination between these mechanisms and the specificity of HELQ in recognizing LINE‐1 transposons requires further study.
In summary, we revealed that the absence of HELQ elevated KDM4B levels and led to abnormal H3K9me3 levels in the LINE‐1 region, resulting in excessive expression of LINE‐1 and subsequently DNA damage accumulation, which may impair PGC proliferation and reproductive reserve establishment. This study uncovered a novel function of HELQ in suppressing transposable elements through the regulation of epigenetic modifications and also provided new insights into the insufficient establishment of reproductive reserve caused by DDR gene deficiency.
Author Contributions
Methodology, validation, data curation, visualization, and writing the original draft: Lili Cao and Jiayi Ren. Methodology and validation: Zhaojie Kong, Mengchun Hu, and Yaxuan Zhang. Supervision: Zi‐Jiang Chen and Yingying Qin. Investigation, resources, conceptualization, and revision of the manuscript: Yajuan Yang and Shidou Zhao.
Disclosure
The authors have nothing to report.
Ethics Statement
All animal experiments conducted in this study were approved by the Experimental Animal Ethics Review Committee at Shandong University.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. Generation and verification of Helq knockout mice.
Figure S2. HELQ deficiency impairs proliferation and increases DNA damage in HEK293T cells.
Figure S3. The high expression of LINE‐1 leads to DNA damage accumulation and proliferation defects in KO MEFs.
Figure S4. The reverse transcription inhibitor reduces DNA damage of PGC in KO embryos.
Table S1.
Acknowledgments
This study was supported by grants from the National Key Research and Development Program of China (2022YFC2703800), the Natural Science Foundation of Shandong Province for Distinguished Young Scholars (ZR2023JQ031), the Fundamental Research Funds of Shandong University (2023QNTD004), the National Natural Science Foundation of China (32170867, 32370906, 82371645, and 32200697), the Natural Science Foundation of Shandong Province (ZR2024QC195), the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐5‐001), the Shandong Provincial Key Research and Development Program (2020ZLYS02), and the Taishan Scholars Program for Young Experts of Shandong Province (tsqn202211370 and tsqn202312386). We would like to express our gratitude to all members of the project team for their dedication and hard work.
Cao L., Ren J., Kong Z., et al., “ HELQ Maintains Genome Stability of Primordial Germ Cells by Inhibiting LINE‐1 Expression,” The FASEB Journal 39, no. 12 (2025): e70720, 10.1096/fj.202403260R.
Funding: This study was supported by grants from the National Key Research and Development Program of China (2022YFC2703800), the Natural Science Foundation of Shandong Province for Distinguished Young Scholars (ZR2023JQ031), the Fundamental Research Funds of Shandong University (2023QNTD004), the National Natural Science Foundation of China (32170867, 32370906, 82371645, and 32200697), the Natural Science Foundation of Shandong Province (ZR2024QC195), the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐5‐001), the Shandong Provincial Key Research and Development Program (2020ZLYS02), and the Taishan Scholars Program for Young Experts of Shandong Province (tsqn202211370 and tsqn202312386).
Lili Cao and Jiayi Ren contributed equally to this work.
Contributor Information
Yajuan Yang, Email: yj0204@sdu.edu.cn.
Shidou Zhao, Email: shidouzhao@sdu.edu.cn.
Data Availability Statement
The datasets employed and analyzed during this research can be accessed from the corresponding author upon reasonable request.
References
- 1. Saitou M., Kagiwada S., and Kurimoto K., “Epigenetic Reprogramming in Mouse Pre‐Implantation Development and Primordial Germ Cells,” Development 139, no. 1 (2012): 15–31. [DOI] [PubMed] [Google Scholar]
- 2. Saitou M. and Yamaji M., “Primordial Germ Cells in Mice,” Cold Spring Harbor Perspectives in Biology 4, no. 11 (2012): a008375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. European Society for Human R , Embryology Guideline Group on P O I , Webber L., et al., “ESHRE Guideline: Management of Women With Premature Ovarian Insufficiency,” Human Reproduction 31, no. 5 (2016): 926–937. [DOI] [PubMed] [Google Scholar]
- 4. Bolcun‐Filas E. and Handel M. A., “Meiosis: The Chromosomal Foundation of Reproduction,” Biology of Reproduction 99, no. 1 (2018): 112–126. [DOI] [PubMed] [Google Scholar]
- 5. Murphey P., McLean D. J., McMahan C. A., Walter C. A., and McCarrey J. R., “Enhanced Genetic Integrity in Mouse Germ Cells,” Biology of Reproduction 88, no. 1 (2013): 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milholland B., Dong X., Zhang L., Hao X., Suh Y., and Vijg J., “Differences Between Germline and Somatic Mutation Rates in Humans and Mice,” Nature Communications 8 (2017): 15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winship A. L., Stringer J. M., Liew S. H., and Hutt K. J., “The Importance of DNA Repair for Maintaining Oocyte Quality in Response to Anti‐Cancer Treatments, Environmental Toxins and Maternal Ageing,” Human Reproduction Update 24, no. 2 (2018): 119–134. [DOI] [PubMed] [Google Scholar]
- 8. Ruth K. S., Day F. R., Hussain J., et al., “Genetic Insights Into Biological Mechanisms Governing Human Ovarian Ageing,” Nature 596, no. 7872 (2021): 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ke H., Tang S., Guo T., et al., “Landscape of Pathogenic Mutations in Premature Ovarian Insufficiency,” Nature Medicine 29, no. 2 (2023): 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie C., Wang W., Tu C., et al., “Meiotic Recombination: Insights Into Its Mechanisms and Its Role in Human Reproduction With a Special Focus on Non‐Obstructive Azoospermia,” Human Reproduction Update 28, no. 6 (2022): 763–797. [DOI] [PubMed] [Google Scholar]
- 11. Huang C., Guo T., and Qin Y., “Meiotic Recombination Defects and Premature Ovarian Insufficiency,” Frontiers in Cell and Development Biology 9 (2021): 652407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsui V. and Crismani W., “The Fanconi Anemia Pathway and Fertility,” Trends in Genetics 35, no. 3 (2019): 199–214. [DOI] [PubMed] [Google Scholar]
- 13. Messiaen S., Le Bras A., Duquenne C., et al., “Rad54 Is Required for the Normal Development of Male and Female Germ Cells and Contributes to the Maintainance of Their Genome Integrity After Genotoxic Stress,” Cell Death & Disease 4, no. 8 (2013): e774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo Y. and Schimenti J. C., “MCM9 Deficiency Delays Primordial Germ Cell Proliferation Independent of the ATM Pathway,” Genesis 53, no. 11 (2015): 678–684. [DOI] [PubMed] [Google Scholar]
- 15. Wen C., Cao L., Wang S., et al., “MCM8 Interacts With DDX5 to Promote R‐Loop Resolution,” EMBO Journal 43, no. 14 (2024): 3044–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang N., Wen W., Liu Z., Xiong X., and Wu Y., “HELQ as a DNA Helicase: Its Novel Role in Normal Cell Function and Tumorigenesis (Review),” Oncology Reports 50, no. 6 (2023): 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adelman C. A., Lolo R. L., Birkbak N. J., et al., “HELQ Promotes RAD51 Paralogue‐Dependent Repair to Avert Germ Cell Loss and Tumorigenesis,” Nature 502, no. 7471 (2013): 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luebben S. W., Kawabata T., Akre M. K., et al., “Helq Acts in Parallel to Fancc to Suppress Replication‐Associated Genome Instability,” Nucleic Acids Research 41, no. 22 (2013): 10283–10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao Y., Hou K., Li Y., et al., “Human HELQ Regulates DNA End Resection at DNA Double‐Strand Breaks and Stalled Replication Forks,” Nucleic Acids Research 51, no. 22 (2023): 12207–12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anand R., Buechelmaier E., Belan O., et al., “HELQ Is a Dual‐Function DSB Repair Enzyme Modulated by RPA and RAD51,” Nature 601, no. 7892 (2022): 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamp J. A., Lemmens B., Romeijn R. J., Changoer S. C., van Schendel R., and Tijsterman M., “Helicase Q Promotes Homology‐Driven DNA Double‐Strand Break Repair and Prevents Tandem Duplications,” Nature Communications 12, no. 1 (2021): 7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Traband E. L., Hammerlund S. R., Shameem M., et al., “Mitotic DNA Synthesis in Untransformed Human Cells Preserves Common Fragile Site Stability via a FANCD2‐Driven Mechanism That Requires HELQ,” Journal of Molecular Biology 435, no. 22 (2023): 168294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J., Lu P., Wan C., et al., “Cell‐Fate Transition and Determination Analysis of Mouse Male Germ Cells Throughout Development,” Nature Communications 12, no. 1 (2021): 6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo F., Yan L., Guo H., et al., “The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells,” Cell 161, no. 6 (2015): 1437–1452. [DOI] [PubMed] [Google Scholar]
- 25. Stolk L., Perry J. R., Chasman D. I., et al., “Meta‐Analyses Identify 13 Loci Associated With Age at Menopause and Highlight DNA Repair and Immune Pathways,” Nature Genetics 44, no. 3 (2012): 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heddar A., Ogur C., Da Costa S., et al., “Genetic Landscape of a Large Cohort of Primary Ovarian Insufficiency: New Genes and Pathways and Implications for Personalized Medicine,” eBioMedicine 84 (2022): 104246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murtaza G., Yang L., Khan I., et al., “Identification and Functional Investigation of Novel Heterozygous HELQ Mutations in Patients With Sertoli Cell‐Only Syndrome,” Genetic Testing and Molecular Biomarkers 25, no. 10 (2021): 654–659. [DOI] [PubMed] [Google Scholar]
- 28. Gorsi B., Hernandez E., Moore M. B., et al., “Causal and Candidate Gene Variants in a Large Cohort of Women With Primary Ovarian Insufficiency,” Journal of Clinical Endocrinology and Metabolism 107, no. 3 (2022): 685–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCaffrey R., St Johnston D., and González‐Reyes A., “Drosophila mus301/Spindle‐C Encodes a Helicase With an Essential Role in Double‐Strand DNA Break Repair and Meiotic Progression,” Genetics 174, no. 3 (2006): 1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ward J. D., Muzzini D. M., Petalcorin M. I., et al., “Overlapping Mechanisms Promote Postsynaptic RAD‐51 Filament Disassembly During Meiotic Double‐Strand Break Repair,” Molecular Cell 37, no. 2 (2010): 259–272. [DOI] [PubMed] [Google Scholar]
- 31. Lander E. S., Linton L. M., Birren B., et al., “Initial Sequencing and Analysis of the Human Genome,” Nature 409, no. 6822 (2001): 860–921. [DOI] [PubMed] [Google Scholar]
- 32. D'Ordine A. M., Jogl G., and Sedivy J. M., “Identification and Characterization of Small Molecule Inhibitors of the LINE‐1 Retrotransposon Endonuclease,” Nature Communications 15, no. 1 (2024): 3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jansz N., “DNA Methylation Dynamics at Transposable Elements in Mammals,” Essays in Biochemistry 63, no. 6 (2019): 677–689. [DOI] [PubMed] [Google Scholar]
- 34. Hill R. J. and Crossan G. P., “DNA Cross‐Link Repair Safeguards Genomic Stability During Premeiotic Germ Cell Development,” Nature Genetics 51, no. 8 (2019): 1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo Y., Hartford S. A., Zeng R., Southard T. L., Shima N., and Schimenti J. C., “Hypersensitivity of Primordial Germ Cells to Compromised Replication‐Associated DNA Repair Involves ATM‐p53‐p21 Signaling,” PLoS Genetics 10, no. 7 (2014): e1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tafel A. A., Wu L., and McHugh P. J., “Human HEL308 Localizes to Damaged Replication Forks and Unwinds Lagging Strand Structures,” Journal of Biological Chemistry 286, no. 18 (2011): 15832–15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young L. C., McDonald D. W., and Hendzel M. J., “Kdm4b Histone Demethylase Is a DNA Damage Response Protein and Confers a Survival Advantage Following γ‐Irradiation,” Journal of Biological Chemistry 288, no. 29 (2013): 21376–21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiang Y., Yan K., Zheng Q., et al., “Histone Demethylase KDM4B Promotes DNA Damage by Activating Long Interspersed Nuclear Element‐1,” Cancer Research 79, no. 1 (2019): 86–98. [DOI] [PubMed] [Google Scholar]
- 39. Gasior S. L., Wakeman T. P., Xu B., and Deininger P. L., “The Human LINE‐1 Retrotransposon Creates DNA Double‐Strand Breaks,” Journal of Molecular Biology 357, no. 5 (2006): 1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peters A. H., O'Carroll D., Scherthan H., et al., “Loss of the Suv39h Histone Methyltransferases Impairs Mammalian Heterochromatin and Genome Stability,” Cell 107, no. 3 (2001): 323–337. [DOI] [PubMed] [Google Scholar]
- 41. Nicetto D., Donahue G., Jain T., et al., “H3K9me3‐Heterochromatin Loss at Protein‐Coding Genes Enables Developmental Lineage Specification,” Science 363, no. 6424 (2019): 294–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bourc'his D. and Bestor T. H., “Meiotic Catastrophe and Retrotransposon Reactivation in Male Germ Cells Lacking Dnmt3L,” Nature 431, no. 7004 (2004): 96–99. [DOI] [PubMed] [Google Scholar]
- 43. Wang X., Ramat A., Simonelig M., and Liu M. F., “Emerging Roles and Functional Mechanisms of PIWI‐Interacting RNAs,” Nature Reviews. Molecular Cell Biology 24, no. 2 (2023): 123–141. [DOI] [PubMed] [Google Scholar]
- 44. Ren W., Fan H., Grimm S. A., et al., “Direct Readout of Heterochromatic H3K9me3 Regulates DNMT1‐Mediated Maintenance DNA Methylation,” Proceedings of the National Academy of Sciences of the United States of America 117, no. 31 (2020): 18439–18447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jachowicz J. W. and Torres‐Padilla M. E., “LINEs in Mice: Features, Families, and Potential Roles in Early Development,” Chromosoma 125, no. 1 (2016): 29–39. [DOI] [PubMed] [Google Scholar]
- 46. Percharde M., Sultana T., and Ramalho‐Santos M., “What Doesn't Kill You Makes You Stronger: Transposons as Dual Players in Chromatin Regulation and Genomic Variation,” BioEssays 42, no. 4 (2020): e1900232. [DOI] [PubMed] [Google Scholar]
- 47. Kohlrausch F. B., Berteli T. S., Wang F., Navarro P. A., and Keefe D. L., “Control of LINE‐1 Expression Maintains Genome Integrity in Germline and Early Embryo Development,” Reproductive Sciences 29, no. 2 (2022): 328–340. [DOI] [PubMed] [Google Scholar]
- 48. Lv X., Xiao W., Lai Y., et al., “The Non‐Redundant Functions of PIWI Family Proteins in Gametogenesis in Golden Hamsters,” Nature Communications 14, no. 1 (2023): 5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H., Zhang F., Chen Q., et al., “The piRNA Pathway Is Essential for Generating Functional Oocytes in Golden Hamsters,” Nature Cell Biology 23, no. 9 (2021): 1013–1022. [DOI] [PubMed] [Google Scholar]
- 50. Tharp M. E., Malki S., and Bortvin A., “Maximizing the Ovarian Reserve in Mice by Evading LINE‐1 Genotoxicity,” Nature Communications 11, no. 1 (2020): 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim S., Günesdogan U., Zylicz J. J., et al., “PRMT5 Protects Genomic Integrity During Global DNA Demethylation in Primordial Germ Cells and Preimplantation Embryos,” Molecular Cell 56, no. 4 (2014): 564–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gruhn W. H., Tang W. W. C., Dietmann S., et al., “Epigenetic Resetting in the Human Germ Line Entails Histone Modification Remodeling,” Science Advances 9, no. 3 (2023): eade1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Groh S. and Schotta G., “Silencing of Endogenous Retroviruses by Heterochromatin,” Cellular and Molecular Life Sciences 74, no. 11 (2017): 2055–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao S., Lu J., Pan B., et al., “TNRC18 Engages H3K9me3 to Mediate Silencing of Endogenous Retrotransposons,” Nature 623, no. 7987 (2023): 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matsui T., Leung D., Miyashita H., et al., “Proviral Silencing in Embryonic Stem Cells Requires the Histone Methyltransferase ESET,” Nature 464, no. 7290 (2010): 927–931. [DOI] [PubMed] [Google Scholar]
- 56. Bulut‐Karslioglu A., De La Rosa‐Velázquez I. A., Ramirez F., et al., “Suv39h‐Dependent H3K9me3 Marks Intact Retrotransposons and Silences LINE Elements in Mouse Embryonic Stem Cells,” Molecular Cell 55, no. 2 (2014): 277–290. [DOI] [PubMed] [Google Scholar]
- 57. Xu R., Li S., Wu Q., et al., “Stage‐Specific H3K9me3 Occupancy Ensures Retrotransposon Silencing in Human Pre‐Implantation Embryos,” Cell Stem Cell 29, no. 7 (2022): 1051–1066.e8. [DOI] [PubMed] [Google Scholar]
- 58. Liu N., Lee C. H., Swigut T., et al., “Selective Silencing of Euchromatic L1s Revealed by Genome‐Wide Screens for L1 Regulators,” Nature 553, no. 7687 (2018): 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ardeljan D., Steranka J. P., Liu C., et al., “Cell Fitness Screens Reveal a Conflict Between LINE‐1 Retrotransposition and DNA Replication,” Nature Structural & Molecular Biology 27, no. 2 (2020): 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mita P., Sun X., Fenyö D., et al., “BRCA1 and S Phase DNA Repair Pathways Restrict LINE‐1 Retrotransposition in Human Cells,” Nature Structural & Molecular Biology 27, no. 2 (2020): 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bona N. and Crossan G. P., “Fanconi Anemia DNA Crosslink Repair Factors Protect Against LINE‐1 Retrotransposition During Mouse Development,” Nature Structural & Molecular Biology 30, no. 10 (2023): 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Coufal N. G., Garcia‐Perez J. L., Peng G. E., et al., “Ataxia Telangiectasia Mutated (ATM) Modulates Long Interspersed Element‐1 (L1) Retrotransposition in Human Neural Stem Cells,” Proceedings of the National Academy of Sciences of the United States of America 108, no. 51 (2011): 20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tiwari B., Jones A. E., Caillet C. J., Das S., Royer S. K., and Abrams J. M., “p53 Directly Represses Human LINE1 Transposons,” Genes & Development 34, no. 21–22 (2020): 1439–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Generation and verification of Helq knockout mice.
Figure S2. HELQ deficiency impairs proliferation and increases DNA damage in HEK293T cells.
Figure S3. The high expression of LINE‐1 leads to DNA damage accumulation and proliferation defects in KO MEFs.
Figure S4. The reverse transcription inhibitor reduces DNA damage of PGC in KO embryos.
Table S1.
Data Availability Statement
The datasets employed and analyzed during this research can be accessed from the corresponding author upon reasonable request.