

Abstract
Introduction
Platelets play a crucial role, amongst others, in the host response to infection through the release of proinflammatory and procoagulant proteins. Pneumonia represents a major disease burden, which has further increased since the onset of the coronavirus disease 2019 (COVID-19) pandemic. We aimed to determine platelets’ protein release profiles in hospitalised patients with non-COVID community-acquired pneumonia (CAP) or COVID-19.
Methods
Platelets from patients with CAP or COVID-19 and from control subjects were incubated with platelet receptor agonists stimulating either glycoprotein (GP)VI or protease-activated receptor 1 (PAR1), or medium (spontaneous release); nine platelet products were measured in supernatants. Platelet activation was also assessed by measuring P-selectin and active GPIIbIIIa surface expression by flow cytometry.
Results
We enrolled 32 patients with CAP, 104 patients with COVID-19 and 47 control subjects. Platelets from CAP patients spontaneously released less chemokines as compared to controls, which sharply contrasted with the platelets’ release profile in COVID-19, which was characterised by enhanced release of chemokines, plasminogen activator inhibitor 1 (PAI-1) and CD40 ligand relative to both controls and CAP patients. These differences were partially maintained upon ex vivo stimulation of GPVI or PAR1. Enhanced GPVI-induced platelet release of P-selectin, CD40 ligand, PAI-1 and CCL5 was associated with an increased risk for the need of (non)invasive ventilation and/or mortality in COVID-19 patients. Platelet expression of P-selectin and GPIIbIIIa did not differ between groups.
Conclusion
Platelet release profiles differ strongly between CAP and COVID-19 patients, wherein platelets from COVID-19 patients released more bioactive products, linking with an adverse clinical outcome.
Shareable abstract
Platelets secrete inflammatory and procoagulant proteins. We report differential platelet mediator release profiles in community-acquired pneumonia and COVID-19. In COVID-19 enhanced platelet mediator release is associated with worse clinical outcomes. https://bit.ly/3TgUsSN
Introduction
Globally, lower respiratory tract infections are a leading cause of both morbidity and mortality [1]. Pneumonia alone is estimated to be responsible for over 1 million in-hospital deaths each year [1]. Until recently, community-acquired pneumonia (CAP), characterised by contraction of disease outside healthcare facilities, was mainly caused by a select subset of pathogens including Streptococcus pneumoniae and influenza [2]. However, with the rise of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections, the virus causing coronavirus disease 2019 (COVID-19), that has changed. The mortality associated with pneumonia strongly increased due to the emergence of COVID-19, with over 7 million deaths related to this infection reported since the start of the pandemic in 2019 [3].
During pneumonia, cellular and humoral defense mechanisms protect the host against the invading pathogen [4]. Platelets are a crucial, yet underappreciated, part of the host response to infection [5, 6] that have been shown to engage with, recruit and stimulate immune cells [6–9]. Notably, platelets are able to release a multitude of proinflammatory and procoagulant mediators [10–13], through which many of their immune functions are exerted, and in turn are key for the host response to infection.
Recent research from our group documented clear differences in the host response to CAP and COVID-19 [14, 15]. Given the importance of platelets and their released mediators in the host response to infection, we here sought to examine platelet function, particularly the release of immunologically active proteins, in patients with CAP. We concurrently analysed mediator release by platelets from patients with COVID-19, thereby aiming to obtain insight into potential differences and similarities between these conditions. Finally, we aimed to determine whether the release of platelet-derived mediators relate to the clinical course of patients.
Methods
A detailed description of the methods can be found in the supplementary methods.
Patient selection
Patients admitted to a general hospital ward with CAP or COVID-19 were enrolled in the ELDER-BIOME study (clinicaltrials.gov identifier NCT02928367) [14, 15]. The study was approved by the Amsterdam UMC ethics committee. Patients were eligible if they were over 18 years and met the following criteria upon hospital admission: clinical suspicion of an acute respiratory tract infection, one systemic symptom (fever/hypothermia or leukocytosis/leukopenia), and an infiltrate on chest radiograph or computed tomography scan. For detailed inclusion and exclusion criteria, see the supplementary methods. Controls were hospital employees or patients visiting the outpatient clinic without infectious disease symptoms or use of platelet aggregation inhibitors. Written informed consent was obtained from all subjects.
Preparation of platelet-rich plasma and washed platelets
Citrated blood was collected and prepared within 4 h of blood withdrawal. First, blood was centrifuged at 180 g for 10 min, platelet-rich plasma (PRP) was collected and centrifuged a second time at 200 g for 2 min to remove remaining erythrocytes and leukocytes. Subsequently platelets were centrifuged, washed and resuspended at physiological platelet levels (400×106 platelets·mL−1) in buffer. The platelet releasate was obtained by incubating washed platelets for 30 min at 37°C with buffer containing 0.1% bovine serum albumin (BSA) (spontaneous release), or 0.1% BSA containing buffer with cross-linked collagen-related peptide-XL (CRP-XL, CambCol Laboratory; 1 µg·mL−1) or thrombin receptor-activating peptide (TRAP)-6 (Bachem; 15 µM), after which supernatant was collected and snap frozen.
Quantification of platelet factor release
The following platelet mediators were measured by Luminex multiplex assays (R&D Systems, Minneapolis, MN, USA; a custom-built 5-plex discovery assay (Luminex code: wPTpy9fm) and a custom-built 6-plex discovery assay (Luminex code: UTefe7K8), using the Bio-Plex 200 (Bio-Rad Laboratories Inc., Hercules, CA, USA) system: CD40 ligand (CD40L), a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), CD31, tissue factor pathway inhibitor (TFPI), tissue factor (TF), CCL5 (RANTES), CXCL4 (platelet factor 4), CXCL7 (NAP-2), plasminogen activator inhibitor type 1 (PAI-1) and P-selectin (CD62P). von Willebrand factor (VWF) antigen was measured by ELISA with a polyclonal rabbit anti-human VWF antibody (A0082) as capture antibody and a rabbit anti-human horseradish peroxidase-labelled VWF antibody (P0226) as detection antibody (Dako, Glostrup, Denmark). Supplementary table S1 depicts lower and upper limits of detection, and the proportion of samples within the detection range for each analyte.
Platelet flow cytometry
See supplementary methods for details. In a subset of patients, platelet activation was assessed through flow cytometry. For this, PRP was incubated with phosphate buffered saline with 0.1% BSA (medium control), or activated with CRP-XL (1 µg·mL−1) or TRAP-6 (15 µM) for 30 min at room temperature. Concomitantly, platelets were stained with anti-CD61-APC, and anti-CD62P-PE or anti-CD61-AlexaFluor700 and anti-CD62P-PerCP-Cy5.5 and anti-active GPIIbIIIa-AF467.
Statistical analysis
All tests were two-sided and a p-value <0.05 (corrected for multiple testing with the Benjamini–Hochberg method) was considered statistically significant. For effect size forest plots, we calculated the Hedges’ g [16] between groups. An effect size between −0.2 and 0.2 was considered negligible, an effect size >0.2 or < −0.2 small, >0.5 or < −0.5 moderate and >0.8 or < −0.8 large. Hierarchical clustering was performed using complete linkage.
Results
Patient characteristics and clinical outcomes
We enrolled 32 patients with CAP and 104 patients with COVID-19 within 48 h after admission to a general hospital ward (table 1). Most patients were sampled in the morning after admission (83.4% of CAP patients and 73.8% of COVID-19 patients); the remaining patients were sampled on the day of admission (CAP) or in the morning of day 2 after admission (COVID-19). 48 subjects without acute disease served as controls. Consistent with the literature [4], in most CAP patients the causative pathogen was not identified (20 out of 32 or 62.5%); in the other patients the most common causes were S. pneumoniae (n=4) and influenza A (n=3). Disease severity on admission was similar in COVID-19 patients and CAP patients, as indicated by the CURB-65 (confusion, blood urea nitrogen, respiratory rate, blood pressure, age 65 years or older) score [17] and the modified early warning score (MEWS) [18]. Platelet counts were lower in COVID-19 patients than in CAP patients, albeit in both groups within normal range (median 217×109/L and 262×109/L respectively, p=0.003). COVID-19 patients more often needed invasive mechanical ventilation during their hospital stay (14.6% versus 0% in CAP patients, p=0.054), while mortality rates were similar.
TABLE 1.
Baseline characteristics and outcome of community-acquired pneumonia (CAP) and COVID-19 patients
| Control | CAP | COVID-19 | Control versus CAP p-value | Control versus COVID-19 p-value | CAP versus COVID-19 p-value | |
|---|---|---|---|---|---|---|
| Patients, n | 48 | 32# | 104 | |||
| Baseline characteristics | ||||||
| Age | 48.5 (33.8–61.2) | 68.0 (53.0–74.5) | 60.0 (50.0–69.0) | 0.00 | <0.001 | 0.18 |
| Sex, male | 27 (56.2) | 19 (59.4) | 65 (63.1) | 0.96 | 0.53 | 0.86 |
| BMI | 24.7 (22.1–28.4) | 26.8 (23.5–31.7) | 28.6 (25.2–31.6) | 0.03 | <0.001 | 0.47 |
| Comorbidities | ||||||
| CCI | 1.0 (0.0–1.5) | 2.0 (1.0–4.0) | 2.0 (1.0–4.0) | 0.05 | 0.15 | 0.22 |
| COPD | 0 (0.0) | 4 (12.5) | 9 (8.7) | 0.05 | 0.08 | 0.76 |
| Hypertension | 7 (14.6) | 9 (28.1) | 34 (32.7) | 0.23 | 0.03 | 0.79 |
| Diabetes | 2 (4.2) | 4 (12.5) | 20 (19.2) | 0.34 | 0.03 | 0.54 |
| Cardiovascular disease | 16 (33.3) | 15 (46.9) | 45 (43.7) | 0.33 | 0.30 | 0.91 |
| Chronic kidney disease | 4 (8.3) | 2 (6.2) | 3 (2.9) | 1.00 | 0.28 | 0.73 |
| Prior malignancy | 1 (2.1) | 4 (12.5) | 9 (8.7) | 0.16 | 0.24 | 0.77 |
| Immunosuppression¶ | 5 (10.4) | 2 (6.2) | 2 (1.9) | 0.81 | 0.06 | 0.51 |
| Severity of disease | ||||||
| qSOFA score | 1 (0–1) | 1 (0–1) | 0.56 | |||
| MEWS | 5 (5–6) | 6 (5–6) | 0.89 | |||
| CURB-65 score | 2 (2–2) | 2 (1–2) | 0.03 | |||
| Admission laboratory results | ||||||
| C-reactive protein mg·L−1 | 167.0 (62.0–295.5) | 98.0 (50.0–141.3) | 0.01 | |||
| WBC ×109/L | 14.2 (11.2–18.0) | 6.4 (4.8–8.0) | <0.001 | |||
| Neutrophils ×109/L | 10.9 (7.8–13.5) | 4.8 (3.3–6.8) | <0.001 | |||
| Lymphocytes ×109/L | 1.1 (0.5–1.8) | 0.9 (0.7–1.2) | 0.22 | |||
| Creatinine µmol·L−1 | 89 (73.2–122.5) | 79 (65.0–91.8) | 0.03 | |||
| Platelets ×109/L | 262 (193.5–392.5) | 217 (171.0–260.8) | 0.00 | |||
| Outcomes | ||||||
| ICU admission | 1 (3.2) | 16 (15.5) | 0.13 | |||
| Noninvasive ventilation | 1 (3.2) | 5 (4.9) | 1.00 | |||
| Invasive ventilation | 0 (0.0) | 15 (14.6) | 0.05 | |||
| Death in hospital | 2 (6.2) | 10 (9.6) | 0.82 | |||
| 30-day mortality | 2 (6.2) | 10 (9.6) | 0.82 | |||
| 90-day mortality | 2 (6.2) | 10 (9.6) | 0.82 | |||
Continuous data are presented as median (interquartile range), and compared using a two-sided ANOVA or two-sided Kruskal-Wallis test, respectively. Categorical data are presented as n (%) and compared using Fisher's exact test. BMI: body mass index; CCI: Charlson comorbidity index; qSOFA: quick sequential organ failure assessment score; MEWS: modified early warning score; CURB-65: confusion, blood urea nitrogen, respiratory rate, blood pressure, age 65 or older; WBC: white blood cells; ICU: intensive care unit. #: microbiological results in this group: no pathogen identified (n=20), Streptococcus pneumoniae (n=4), influenza A virus (n=3), rhinovirus (n=1), parainfluenza virus 1–4 (n=1), respiratory syncytial virus (n=1), Streptococcus intermedius (n=1), Aspergillus spp. (n=1), Streptococcus milleri (n=1), Legionella (n=1). The total number of causative pathogens does not match the total number of CAP patients, as some patients were infected with multiple pathogens. ¶: defined as a history of an organ transplant, immune deficiency or chronic use of immunosuppressants.
Platelets from CAP and COVID-19 patients show differential spontaneous mediator release
To obtain insight into the secretory activity of platelets in CAP and COVID-19 we quantified the levels of nine factors released by platelets through degranulation of α-granules (CCL5, CXCL4, CXCL7, PAI-1, VWF, P-selectin, CD40L), membrane shedding (P-selectin, CD40L, CD31) or other mechanisms (TFPI) in their supernatant. We first explored differences in spontaneous mediator release by platelets harvested from patients with CAP or COVID-19, or from control subjects. Platelets from CAP patients spontaneously released less CCL5, CXCL7 and CXCL4 as compared to platelets from control subjects, while the release of the other proteins measured was similar between these groups (figure 1). By contrast, platelets from COVID-19 patients released more CCL5, CXCL7, CXCL4, PAI-1 and CD40L when compared with platelets from controls or CAP patients, and more VWF relative to CAP patients. Figure 2 highlights the difference between platelets from CAP and COVID-19 patients by displaying the Hedges’ g effect size [16], demonstrating the overall reduced protein release by platelets from CAP relative to COVID-19 patients. The observed differences between CAP and COVID-19 patients persisted after adjusting for disease severity (supplementary table S2). Moreover, these differences could not be explained by the activation status of platelets, quantified by the expression of P-selectin and activated GPIIbIIIa on the platelet surface, which was similar in CAP and COVID-19 patients, and not divergent from control subjects (figure 3).
FIGURE 1.

Spontaneous mediator release of platelets in patients with community-acquired pneumonia (CAP) or COVID-19, and controls. Boxplots detailing platelet release of proteins: a) proteins released from platelet α-granules; b) proteins released/shed from other locations within or on platelets. For all proteins except von Willebrand factor (VWF): CAP n=31, COVID-19 n=101 and control n=48. For VFW: CAP n=30, COVID-19 n=100 and control n=47. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. PAI-1: plasminogen activator inhibitor 1; WHO std.: World Health Organization standard; CD40L: CD40 ligand; TFPI: tissue factor pathway inhibitor.
FIGURE 2.
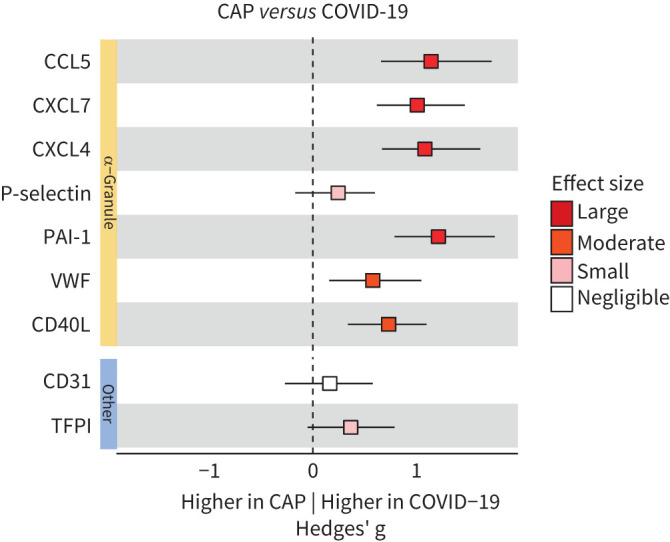
Effect size comparison of platelets’ spontaneous mediator release in patients with community-acquired pneumonia (CAP) or COVID-19. Forest plot depicting the magnitude of spontaneously released platelet-mediator differences (Hedges’ g) between CAP and COVID-19 patients. An effect size between −0.2 and 0.2 was considered negligible, an effect size >0.8 or <−0.8 large, >0.5 or <−0.5 moderate and >0.2 or <−0.2 small. Shades of red depict a positive effect size, with increasing intensity of colour for higher values. PAI-1: plasminogen activator inhibitor 1; VWF: von Willebrand factor; CD40L: CD40 ligand; TFPI: tissue factor pathway inhibitor.
FIGURE 3.

Platelet surface protein expression. Expression of platelet surface proteins representative of platelet activation, measured using flow cytometry. a) Platelet surface P-selectin expression of community-acquired pneumonia (CAP) and COVID-19 patients normalised to the respective median level of control samples (dotted line) measured concomitantly to samples of interest. Controls n=45, CAP n=17, COVID-19 n=53. b) Platelet surface expression of GPIIbIIIa in active conformation in CAP and COVID-19 patients normalised to the respective median level of control samples (dotted line) measured concomitantly to samples of interest. Control n=40, CAP n=13, COVID-19 n=37. MFI: median fluorescence intensity; Unstim: unstimulated platelets.
In our study, COVID-19 patients were enrolled during the second and third “wave” of the pandemic in the Netherlands [19]; during both waves the SARS-CoV-2 α-strain was predominant and dexamethasone the standard of care, while during the third wave vaccinations and (where indicated) anti-interleukin-6 therapies were introduced [19]. Previous studies have suggested that the extent of platelet activation detected in COVID-19 patients at the start of the pandemic was mitigated later on [20–23]. In the current investigation, platelets from COVID-19 patients recruited during the second and third wave showed similar spontaneous protein release (supplementary figure S1).
A prevailing trend emerged, showing lower platelet mediator levels in CAP patients compared to controls, while levels appeared elevated in COVID-19 patients. To further validate this observation, we employed principal component analysis (PCA) on all samples evaluating the spontaneous release in an unsupervised manner (supplementary figure S2), revealing distinct mediator release patterns. The first principal component (PC1) accounted for 68.0% of the dataset variance (figure 4a), with CAP patients below and COVID-19 patients above control levels (figure 4b). The Wilcoxon's-rank test confirmed significant differences in PC1 across all groups, with the largest difference between CAP and COVID-19 patients, suggesting a disease-specific effect on platelet functionality (mediator release) independent of activation status as measured by surface expression. An exploratory analysis hampered by low sample size suggested that the mediator release profile of platelets from patients with a pure (non-SARS-CoV-2) viral etiology (n=4) was dissimilar to that of platelets from COVID-19 patients (supplementary figure S3).
FIGURE 4.

Principal component analysis and hierarchical clustering based on platelet mediator release. a) Variance of the data explained by different principal components (PC), showing that PC1 is able to capture the majority of variance. b) Boxplots comparing PC1 coordinates per group. p-values were derived from a Wilcox-test comparing the PCs between groups. c) Unsupervised hierarchical clustering. Subjects are clustered based on the value of the different mediators. d) Correlation between spontaneous platelet mediator release and modified early warning score (MEWS) in community-acquired pneumonia (CAP) patients; shaded areas indicate 95% confidence interval. *p<0.05; ***p<0.001; ****p<0.0001. PAI-1: plasminogen activator inhibitor 1; VWF: von Willebrand factor; CD40L: CD40 ligand; TFPI: tissue factor pathway inhibitor.
The most notable differences in platelet release were observed between CAP and COVID-19 patients, even more so than when each was compared to the control group (figures 1 and 4b). To investigate whether CAP and COVID-19 patients can be distinguished based solely on the profiles of released platelet factors – without considering the original group assignments – we applied hierarchical clustering. Figure 4c illustrates that, utilising the data from the spontaneously released platelet products, CAP and COVID-19 patients clustered to opposite sides of the heatmap. Two CAP patients clustered to the left, exhibiting platelet release profiles similar to those of COVID-19 patients. This similarity might be attributed to their exceptionally high levels of systemic inflammation (C-reactive protein 597 and 375 mg·L−1; supplementary table S3). Figure 4c further confirms that severity of disease, portrayed by MEWS, or platelet counts were not related to the diverging platelet release profiles, providing additional support for an overall reduced release of inflammatory mediators by platelets from CAP patients relative to platelets from COVID-19 patients. Next, we assessed the relationship between spontaneous platelet mediator release and disease severity across the groups. Correlation analyses between MEWS and platelet releasate concentrations in both CAP and COVID-19 patients showed no significant correlations in COVID-19 patients. However, in CAP patients, several significant inverse correlations were observed, suggesting decreased α degranulation with increased disease severity, except for VWF and CD40 L (supplementary table S4, figure 4d).
Diverging mediator release by platelets of CAP and COVID-19 patients partially persists upon ex vivo stimulation
During acute inflammation platelets are exposed to several agonists, resulting in platelet activation through specific agonist–receptor interactions. Glycoprotein (GP)VI and protease-activated receptor-1 (PAR1) are platelet receptors triggered by collagen and thrombin respectively that are considered to play an important role in platelet activation during acute infection [6, 9]. Thus, we exposed platelets of patients (and controls) to the GPVI agonist CRP-XL or the PAR1 agonist TRAP-6. Both stimuli strongly increased the release of all nine platelet products measured (supplementary figure S4). Some differences in spontaneous platelet mediator release between groups (figure 1) were also observed upon ex vivo stimulation of platelets with CRP-XL or TRAP-6: in CAP platelet release of CCL5 and CXCL7 (induced by CRP-XL or TRAP-6) was lower than in controls, while in COVID-19 platelet release of PAI-1 (induced by CRP-XL or TRAP-6) was higher than in controls (figure 5). In contrast, the platelet release of P-selectin (induced by CRP-XL or TRAP-6) was lower in COVID-19 than controls. This same effect on P-selectin release was seen in platelets of CAP patients, albeit to a lesser extent. Direct comparison of mediator release by stimulated platelets disclosed significantly increased release by platelets from COVID-19 patients (relative to platelets from CAP patients) of CCL5 and PAI-1 upon exposure to CRP-XL or TRAP-6, and of CXCL7 and CD40L upon exposure to CRP-XL. Figure 6a highlights the differences in agonist-stimulated protein release by platelets from CAP versus COVID-19 patients expressed as Hedges’ g.
FIGURE 5.

Release of platelet proteins following GPVI or PAR1 stimulation in patients with community-acquired pneumonia (CAP) or COVID-19, and controls. Boxplots illustrate protein release from platelets following ex vivo stimulation of GPVI (by CRP-XL) or PAR1 (by TRAP-6) treatment for 30 min at 37°C. a) Proteins from platelet α-granules; b) proteins released or shed from other platelet compartments. Sample sizes for each panel, excluding von Willebrand factor (VWF), are as follows: for CRP-XL stimulated samples, CAP n=32, COVID-19 n=97, control n=48; for TRAP-6 stimulated samples, CAP n=28, COVID-19 n=78, control n=41; for VWF, CRP-XL stimulated samples, CAP n=30, COVID-19 n=96, control n=47; and for TRAP-6 stimulated samples, CAP n=28, COVID-19 n=76, control n=41. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. GPVI: glycoprotein VI; PAR1: protease-activated receptor 1; PC: principal component; CRP-XL: synthetic cross-linked collagen-related peptide; TRAP-6: thrombin receptor-activating peptide-6; PAI-1: plasminogen activator inhibitor 1; CD40L: CD40 ligand; TFPI: tissue factor pathway inhibitor; WHO std.: World Health Organization standard.
FIGURE 6.

Effect size comparison of platelet release and correlations with surface expression following activation in CAP and COVID-19 patients. a) Forest plot depicting the magnitude of platelet–mediator differences (Hedges’ g) between CAP and COVID-19 patients. An effect size between −0.2 and 0.2 was considered negligible, an effect size >0.8 or <−0.8 large, >0.5 or <−0.5 moderate and >0.2 or <−0.2 small. Shades of red depict a positive effect size, with increasing intensity of colour for higher values. b) The upper panel displays the correlation between the release of platelet mediators and P-selectin surface expression following CRP-XL treatment in CAP and COVID-19 patients. The lower panel presents a similar analysis for TRAP-6-treated platelets. c) Correlation between platelet mediator release and GPIIbIIIa expression. For both b and c the vertical coloured bar at the right indicates the Spearman correlation coefficient: darker red signifies a higher rho, while darker blue indicates a lower rho. Correlations significant at p<0.05 are marked with an asterisk.
Like in unstimulated platelets (figure 3), differential mediator release by stimulated platelets was not accompanied by consistent changes in the platelet surface activation marker P-selectin when comparing between groups (supplementary figure S5). However, for CAP patients, the reduction in mediator release corresponded with a decrease in surface expression of activated GPIIbIIIa. In contrast, for COVID-19 patients, the elevation in mediator release compared to controls was associated with a reduction in activated GPIIbIIIa surface expression, suggesting a distinct and independent mechanism at play. Through Spearman correlation analysis, we further explored the relationship between the inducibility of platelet mediator release and the surface expression of P-selectin and GPIIbIIIa in CAP and COVID-19 patients (figure 6b,c). This analysis revealed a moderate positive correlation between mediator release and P-selectin expression in CAP patients, although this was generally not significant. In COVID-19 patients, however, mediator release showed an inverse relationship with P-selectin expression, albeit with low statistical significance. Interestingly, while mediator release in CAP patients appeared to be correlated with activated GPIIbIIIa expression, this association was not present in COVID-19 patients, again suggestive of diverging mechanisms.
Platelets’ capacity to release mediators is predictive of need for ventilation or mortality
We next investigated how the ability of platelets to release mediators after stimulation relates to clinical outcomes of patients. Utilising logistic regression adjusted for age and sex, this analysis was only conducted for COVID-19 patients since the very low event rate in CAP patients precluded this. The analysis focused on markers that were differentially abundant between the platelets of COVID-19 patients and those of the controls/CAP. We used the need for noninvasive or invasive ventilation and mortality at 30 days as a clinically relevant composite end-point. Platelet release of P-selectin (p=0.036), CD40L (p=0.040), CCL5 (p=0.040) and PAI-1 (p=0.040) following GPVI stimulation could forecast the occurrence of the composite end-point, with p-values Benjamini–Hochberg adjusted for multiple comparisons. Furthermore, we determined that a 10% increase in the levels of P-selectin, CD40L, PAI-1 or CCL5 corresponded to an increase in the odds ratio (OR) for requiring ventilation or mortality (see figure 7 for OR for composite end-point of mediators). Platelet protein release following PAR1 stimulation did not associate with the need for invasive ventilation or 30-day mortality in COVID-19 patients.
FIGURE 7.
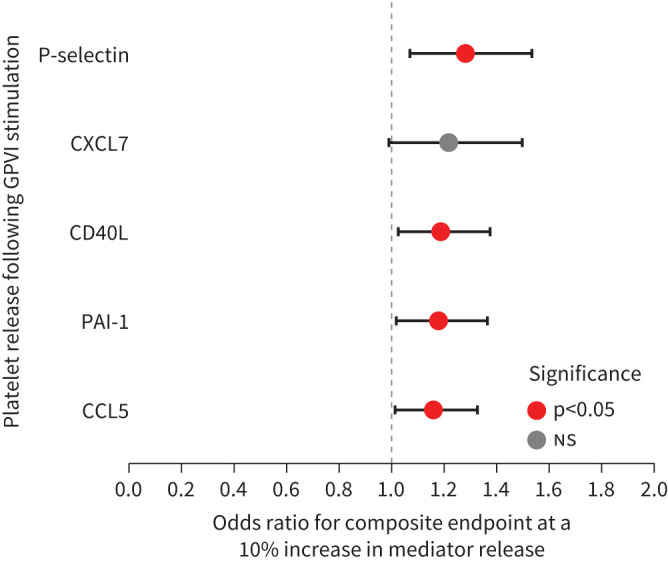
Odds ratio for (non)invasive ventilation and/or 30-day mortality. Forest plot illustrating the odds ratio of a composite end-point of (non)invasive ventilation and/or 30-day mortality for a 10% increase in the release of platelet proteins, derived from a logistic regression model. The horizontal lines depict the 95% confidence interval, the colour of the dot represents significance. PAI-1: plasminogen activator inhibitor 1; CD40L: CD40 ligand; GPV1: glycoprotein VI.
Discussion
Platelets are able to synthesise and release a multitude of mediators with distinct functions in the host response to infection. Platelet products like CCL5, P-selectin, CXCL7 and CXCL4 contribute to the initiation of defense mechanisms against invading pathogens by stimulating leukocyte recruitment to and activation at sites of infection [24–26]. Platelet-released mediators such as VWF and PAI-1, but also CXCL7 and CXCL4, are important for the initiation of platelet aggregation and coagulation in response to endothelial damage or spreading of pathogens [26–28]. In addition, platelet products like TFPI and CD31 may assist in maintaining homeostasis by inhibiting coagulation and thrombosis, or inhibiting trans-endothelial migration of leukocytes respectively [29–31]. While many previous investigations studied platelet activation by determining surface expression of activation markers and/or platelet aggregation [20, 21, 23], this study is the first to evaluate the release of inflammatory and procoagulant mediators by platelets of patients presenting with CAP.
Platelets from CAP patients exhibited reduced release of the chemokines CCL5 (all conditions), CXCL7 (spontaneous and GPVI stimulated) and CXCL4 (spontaneous) compared to control platelets. We observed that with increasing severity of disease, platelets of CAP patients were less able to release inflammatory mediators. This is in line with a recent study showing that P2Y1-dependent inflammatory function in platelets is lost in patients with sepsis resulting from CAP, while preserving aggregation abilities [32]. While the proteins that are less released by platelets of CAP patients are all derived from α-granules, our results argue against a general defect in α-granule release in CAP, considering that other α-granule constituents (PAI-1, VWF) were not different in platelet supernatants of CAP patients and controls. Possibly, reduced mediator release by platelets from CAP patients results from diminished incorporation of specific mediators into α-granules during the megakaryocyte stage or early platelet development, or from the selective release of α-granule subpopulations favouring the release of certain proteins over the other [33].
In contrast, platelets from COVID-19 spontaneously released more bioactive proteins than platelets from either controls or CAP patients, involving both α-granule products and mediators from other platelet compartments. Platelet-released proteins in COVID-19 have both proinflammatory (CCL5, CXCL4, CXCL7) and procoagulant properties (VWF, PAI-1), which could in part explain why COVID-19 is associated with an increased risk of thromboembolic disease [34–37]. In this respect, the strongly enhanced release of PAI-1, a main inhibitor of fibrinolysis and a prothrombotic factor [34, 35], by GPVI or PAR1 stimulated platelets of COVID-19 patients is also of relevance. A possible explanation for why COVID-19 patients’ platelets are more proinflammatory could stem from endothelial inflammation and injury, which are key pathogenic mechanisms in COVID-19 [38, 39] and may prime platelets [40], making them hyperreactive to agonist stimulation. Of note, platelet numbers were normalised in all ex vivo experiments, while admission platelet counts were slightly lower in blood of COVID-19 patients relative to CAP patients. Although in both groups platelet counts were within normal range, it is uncertain whether this difference could impact the overall in vivo inflammatory effect of platelet mediator release.
Prior research has thoroughly investigated the use of flow cytometry to measure platelet activation in patients with pneumonia [21, 23, 41, 42]. We here demonstrate that flow cytometry measurements of platelet activation may not always align with the release of key platelet mediators involved in inflammation and coagulation, even when both originate from the same sub-compartment. Indeed, in COVID-19 platelet surface expression of activation markers and release of platelet mediators were not, or even inversely, related. The induced surface expression of P-selectin, traditionally seen as a marker of platelet activation indicating α-degranulation [43], had no correlation or an inverse relationship with the release of several α-granule proteins in the COVID-19 patients’ platelet supernatant. This finding indicates the need for caution in interpreting platelet P-selectin surface expression levels as a proxy for the release of α-granule proteins. In contrast, in CAP patients these two responses displayed consistent positive correlations, although not always statistically significant, likely due to sample size limitations. Particularly, the induced surface expression of active GPIIbIIIa and the release of CCL5, P-selectin, PAI-1 and TFPI upon stimulation with CRP-XL were positively correlated. These results suggest that platelet activation measured by flow cytometry or protein release reflect distinct mechanisms that vary between COVID-19 and CAP.
The GPVI-induced release of platelet-derived inflammatory mediators was associated with a composite end-point of (non)invasive ventilation and 30-day mortality in COVID-19; this association was not observed following PAR1 stimulation. Previous studies have identified strongly elevated levels of GPVI ligands in the lungs during pneumonia [44]. These findings lead us to hypothesise that in infected patients with elevated GPVI levels, those who exhibit the strongest response to this heightened stimulus, resulting in enhanced mediator release, may be more prone to develop lung injury in COVID-19.
Our study has limitations. We enrolled a relatively low number of CAP patients, attributable to the decreased incidence of non-COVID CAP during the pandemic, which may limit the generalisability of our findings. While most patients were sampled in the morning after admission, in the remaining CAP patients samples were obtained on the day of hospital presentation versus on day 2 in the remaining COVID-19 patients, which may have introduced bias. Another limitation was the necessity to use different antibodies for the flow cytometry analysis of certain surface proteins, which was a result of logistical challenges during the pandemic.
Nonetheless, we provide comprehensive insight into the unique release patterns of platelet proteins implicated in inflammation and coagulation in pneumonia across different etiologies. This study documents associations between platelet–mediator release and an adverse clinical outcome in COVID-19. Notably, our results reveal a distinct divergence in platelet release profiles between CAP and COVID-19 patients, with CAP patients’ platelets releasing lower mediator quantities relative to the other groups, and COVID-19 patients' platelets releasing higher mediator levels, despite similar surface activation markers between CAP and COVID-19 patients.
Footnotes
Provenance: Submitted article, peer reviewed.
Ethics statement: The study was approved by the Amsterdam UMC ethics committee. Written informed consent was obtained from all subjects.
Conflict of interest: None declared.
Support statement: O. Chouchane was supported by the Landsteiner Foundation (LSBR # 1901). V. Léopold was funded by Fondation Bettencourt-Schueller and Institut Servier, EHAM. J. de Brabander and C.C.A. van Linge received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 847786 (FAIR). Funding information for this article has been deposited with the Crossref Funder Registry.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material
00863-2024.SUPPLEMENT
References
- 1.Troeger C, Blacker B, Khalil IA, et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis 2018; 18: 1191–1210. doi: 10.1016/S1473-3099(18)30310-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoar S, Musher DM. Etiology of community-acquired pneumonia in adults: a systematic review. Pneumonia (Nathan) 2020; 12: 11. doi: 10.1186/s41479-020-00074-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.W orld Health Organization . WHO COVID-19 dashboard. Date last accessed: 25 March 2025. Date last updated: 9 March 2025. https://data.who.int/dashboards/covid19/deaths
- 4.Torres A, Cilloniz C, Niederman MS, et al. Pneumonia. Nat Rev Dis Primers 2021; 7: 25. doi: 10.1038/s41572-021-00259-0 [DOI] [PubMed] [Google Scholar]
- 5.Stocker TJ, Ishikawa-Ankerhold H, Massberg S, et al. Small but mighty: platelets as central effectors of host defense. Thromb Haemost 2017; 117: 651–661. doi: 10.1160/th16-12-0921 [DOI] [PubMed] [Google Scholar]
- 6.Deppermann C, Kubes P. Start a fire, kill the bug: the role of platelets in inflammation and infection. Innate Immun 2018; 24: 335–348. doi: 10.1177/1753425918789255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of innate immune responses by platelets. Front Immunol 2019; 10: 1320. doi: 10.3389/fimmu.2019.01320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost 2015; 114: 449–458. doi: 10.1160/th14-12-1067 [DOI] [PubMed] [Google Scholar]
- 9.de Stoppelaar SF, van't Veer C, van der Poll T. The role of platelets in sepsis. Thromb Haemost 2014; 112: 666–677. doi: 10.1160/TH14-02-0126 [DOI] [PubMed] [Google Scholar]
- 10.Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357: 2482–2494. doi: 10.1056/NEJMra071014 [DOI] [PubMed] [Google Scholar]
- 11.von Hundelshausen P, Weber KS, Huo Y, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 2001; 103: 1772–1777. doi: 10.1161/01.cir.103.13.1772 [DOI] [PubMed] [Google Scholar]
- 12.Henn V, Slupsky JR, Gräfe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998; 391: 591–594. doi: 10.1038/35393 [DOI] [PubMed] [Google Scholar]
- 13.Whiteheart SW. Platelet granules: surprise packages. Blood 2011; 118: 1190–1191. doi: 10.1182/blood-2011-06-359836 [DOI] [PubMed] [Google Scholar]
- 14.Schuurman AR, Reijnders TDY, Saris A, et al. Integrated single-cell analysis unveils diverging immune features of COVID-19, influenza, and other community-acquired pneumonia. eLife 2021; 10: e69661. doi: 10.7554/eLife.69661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuurman AR, Reijnders TDY, van Engelen TSR, et al. The host response in different aetiologies of community-acquired pneumonia. EBioMedicine 2022; 81: 104082. doi: 10.1016/j.ebiom.2022.104082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hedges LV. Distribution theory for glass's estimator of effect size and related estimators. J Educ Stat 1981; 6: 107–128. doi: 10.2307/1164588 [DOI] [Google Scholar]
- 17.Lim WS, van der Eerden MM, Laing R, et al. Defining community acquired pneumonia severity on presentation to hospital: an international derivation and validation study. Thorax 2003; 58: 377–382. doi: 10.1136/thorax.58.5.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subbe CP, Kruger M, Rutherford P, et al. Validation of a modified Early Warning Score in medical admissions. QJM 2001; 94: 521–526. doi: 10.1093/qjmed/94.10.521 [DOI] [PubMed] [Google Scholar]
- 19.Slim MA, Appelman B, Peters-Sengers H, et al. Real-world evidence of the effects of novel treatments for COVID-19 on mortality: a nationwide comparative cohort study of hospitalized patients in the first, second, third, and fourth waves in the Netherlands. Open Forum Infect Dis 2022; 9: ofac632. doi: 10.1093/ofid/ofac632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID-19. Blood 2020; 136: 1317–1329. doi: 10.1182/blood.2020007214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Léopold V, Chouchane O, Butler JM, et al. Platelets of COVID-19 patients display mitochondrial dysfunction, oxidative stress and energy metabolism failure compatible with cell death. Res Pract Thromb Haemost 2023; 7: 102213. doi: 10.1016/j.rpth.2023.102213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schrottmaier WC, Pirabe A, Pereyra D, et al. Adverse outcome in COVID-19 is associated with an aggravating hypo-responsive platelet phenotype. Front Cardiovasc Med 2021; 8: 795624. doi: 10.3389/fcvm.2021.795624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Léopold V, Pereverzeva L, Schuurman AR, et al. Platelets are hyperactivated but show reduced glycoprotein VI reactivity in COVID-19 patients. Thromb Haemost 2021; 121: 1258–1262. doi: 10.1055/a-1347-5555 [DOI] [PubMed] [Google Scholar]
- 24.Lorant DE, Topham MK, Whatley RE, et al. Inflammatory roles of P-selectin. J Clin Invest 1993; 92: 559–570. doi: 10.1172/jci116623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakogiannis C, Sachse M, Stamatelopoulos K, et al. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 2019; 122: 154157. doi: 10.1016/j.cyto.2017.09.013 [DOI] [PubMed] [Google Scholar]
- 26.Brandt E, Ludwig A, Petersen F, et al. Platelet-derived CXC chemokines: old players in new games. Immunol Rev 2000; 177: 204–216. doi: 10.1034/j.1600-065x.2000.17705.x [DOI] [PubMed] [Google Scholar]
- 27.Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res 2007; 120: Suppl 1, S5–S9. doi: 10.1016/j.thromres.2007.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brogren H, Karlsson L, Andersson M, et al. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood 2004; 104: 3943–3948. doi: 10.1182/blood-2004-04-1439 [DOI] [PubMed] [Google Scholar]
- 29.van der Poll T. Tissue factor as an initiator of coagulation and inflammation in the lung. Crit Care 2008; 12: Suppl 6, S3. doi: 10.1186/cc7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu XW, Lian EC. Binding properties and inhibition of platelet aggregation by a monoclonal antibody to CD31 (PECAM-1). Arterioscler Thromb Vasc Biol 1997; 17: 3154–3158. doi: 10.1161/01.atv.17.11.3154 [DOI] [PubMed] [Google Scholar]
- 31.Liao F, Ali J, Greene T, et al. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med 1997; 185: 1349–1357. doi: 10.1084/jem.185.7.1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arkless KL, Fish M, Jennings A, et al. Investigation into p2y receptor function in platelets from patients with sepsis. Shock 2023; 60: 172–180. doi: 10.1097/SHK.0000000000002158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Italiano JE Jr, Battinelli EM. Selective sorting of alpha-granule proteins. J Thromb Haemost 2009; 7: Suppl 1, 173–176. doi: 10.1111/j.1538-7836.2009.03387.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frischmuth T, Hindberg K, Aukrust P, et al. Elevated plasma levels of plasminogen activator inhibitor-1 are associated with risk of future incident venous thromboembolism. J Thromb Haemost 2022; 20: 1618–1626. doi: 10.1111/jth.15701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meltzer ME, Lisman T, de Groot PG, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood 2010; 116: 113–121. doi: 10.1182/blood-2010-02-267740 [DOI] [PubMed] [Google Scholar]
- 36.Michels A, Lillicrap D, Yacob M. Role of von Willebrand factor in venous thromboembolic disease. JVS Vasc Sci 2022; 3: 17–29. doi: 10.1016/j.jvssci.2021.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whiteley W, Wood A. Risk of arterial and venous thromboses after COVID-19. Lancet Infect Dis 2022; 22: 1093–1094. doi: 10.1016/S1473-3099(22)00314-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu S-W, Ilyas I, Weng J-P. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol Sin 2023; 44: 695–709. doi: 10.1038/s41401-022-00998-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonaventura A, Vecchié A, Dagna L, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 2021; 21: 319–329. doi: 10.1038/s41577-021-00536-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gresele P, Falcinelli E, Momi S. Potentiation and priming of platelet activation: a potential target for antiplatelet therapy. Trends Pharmacol Sci 2008; 29: 352–360. doi: 10.1016/j.tips.2008.05.002 [DOI] [PubMed] [Google Scholar]
- 41.Gawaz M, Dickfeld T, Bogner C, et al. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med 1997; 23: 379–385. doi: 10.1007/s001340050344 [DOI] [PubMed] [Google Scholar]
- 42.Rondina MT, Brewster B, Grissom CK, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest 2012; 141: 1490–1495. doi: 10.1378/chest.11-2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tyagi T, Jain K, Gu SX, et al. A guide to molecular and functional investigations of platelets to bridge basic and clinical sciences. Nat Cardiovasc Res 2022; 1: 223–237. doi: 10.1038/s44161-022-00021-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claushuis TAM, de Vos AF, Nieswandt B, et al. Platelet glycoprotein VI aids in local immunity during pneumonia-derived sepsis caused by gram-negative bacteria. Blood 2018; 131: 864–876. doi: 10.1182/blood-2017-06-788067 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material
00863-2024.SUPPLEMENT