

Abstract
Heparanase (HPSE) is the sole mammalian endoglycosidase that degrades heparan sulfate (HS) proteoglycans, disrupting the extracellular matrix (ECM) and promoting cancer invasion and metastasis. Although HPSE overexpression is linked to tumor progression, no clinically approved HPSE inhibitors exist. We developed aminoglycoside-based HS mimetics with defined sulfation and hydrophobic modifications to target HPSE’s lipophilic pockets, a novel approach distinct from traditional HS glycans. Computational modeling showed that these mimetics engage HPSE through hydrophobic and π–π stacking interactions, enhancing affinity. The most potent compounds inhibited HPSE-driven ECM degradation, tumor cell proliferation, and invasion. In vivo, the lead candidate significantly reduced metastatic burden in B16 melanoma and MPC-11 myeloma models, showing tumor growth inhibition (TGI = 83.1%) versus SST0001 (TGI = 58.6%) and matching bortezomib. Importantly, the compound was well-tolerated with no notable toxicity. These results support HPSE as a cancer target and highlight aminoglycoside-based HS mimetics as promising therapeutics for metastatic cancer.
Graphical Abstract

INTRODUCTION
Cancer progression involves significant alterations in the tumor microenvironment that facilitate the growth and spread of cancerous cells.1 These changes involve restructuring the extracellular matrix (ECM) through various remodeling enzymes, which modify matrix macromolecules to promote the growth of primary tumors. Heparanase (HPSE), a mammalian endo-β-d-glucuronidase, is the sole enzyme responsible for the degradation of heparan sulfate proteoglycans (HSPGs),2 which are vital for ECM structure and function (Figure 1A). HPSE specifically cleaves the internal β-(1,4)-linkage between glucuronic acid (GlcA) and N-sulfated glucosamine (GlcNS) along the heparan sulfate (HS) side chains of HSPGs (Figure 1B).3 HS plays a critical role in various physiological and pathological processes due to its involvement in cell signaling, cell adhesion, and modulation of the ECM.4 Normal HPSE expression in the extracellular space helps maintain the ECM’s integrity.5 In contrast, overexpression of HPSE leads to ECM degradation, promoting cell migration and the release of HS-sequestered growth factors and cytokines, which ultimately stimulates tumor proliferation and metastasis.5,6 Clinical studies have shown a strong correlation between elevated HPSE levels and aggressive tumor behavior in solid tumors,7,8 associated with increased tumor growth, metastasis, and poorer patient prognosis.9
Figure 1.
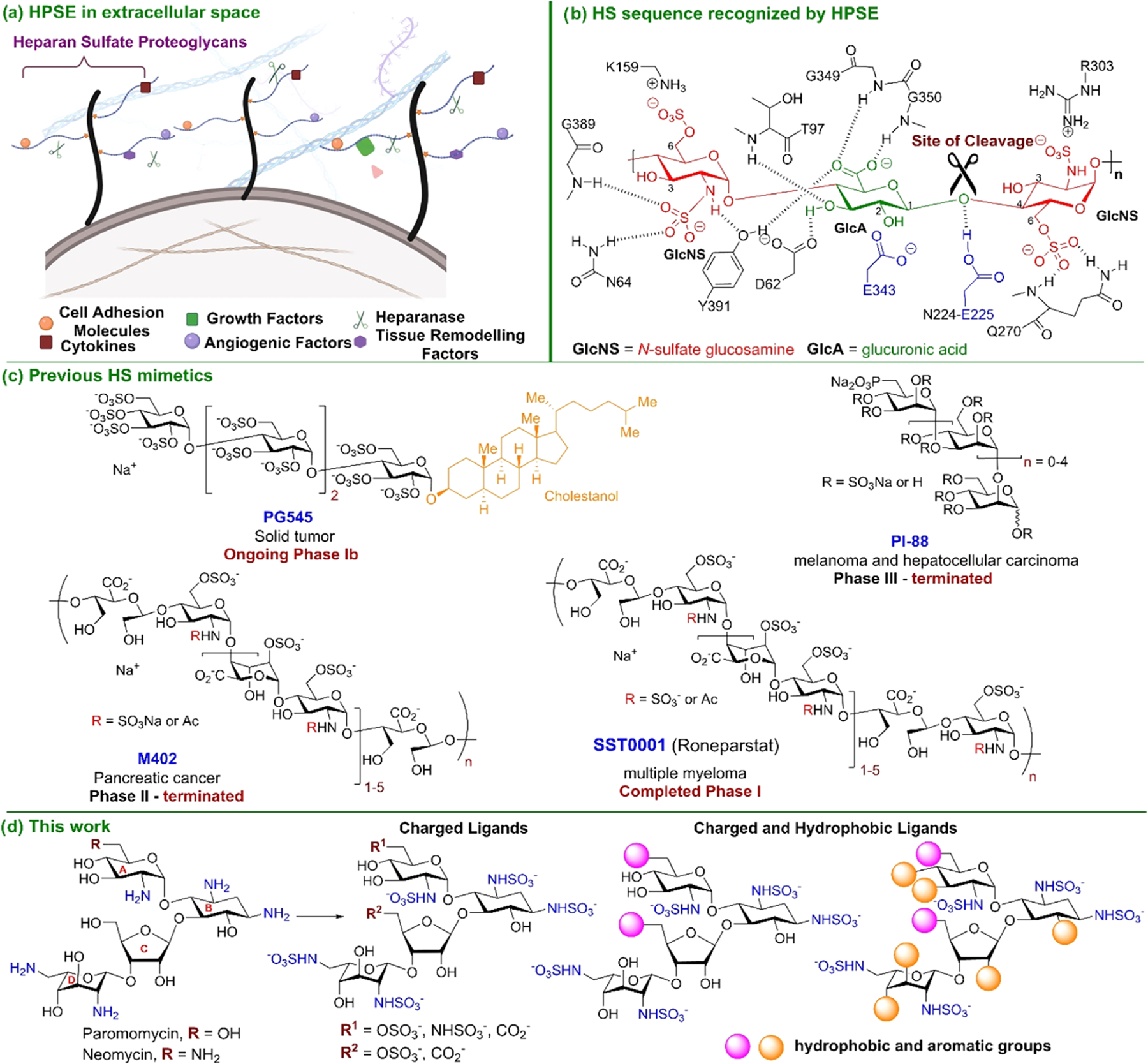
(A) Biological effects of overexpression of HPSE in the extracellular matrix. (B) Minimal preferred HSPE target sequence, HS trisaccharide that consists of a GlcA residue flanked by two GlcNS residues. (C) Previous HPSE-inhibiting ligands in clinical trials. (D) Design and development of aminoglycoside-HPSE inhibitors that consist of a pseudo-tetrasaccharide backbone with well-defined sulfation patterns and various hydrophobic groups.
Efforts to develop HPSE inhibitors have garnered significant attention. Many small molecules10 and antibodies11 have been developed as HPSE inhibitors. However, only four saccharide molecules (Figure 1C) have advanced to clinical trials. M40212 and SST000113 are chemically modified analogs of natural heparin with varying sulfation patterns, PI-8814 is a mixture of sulfated oligosaccharides, and PG54515 is a polysulfated tetrasaccharide incorporated with cholestanol. Clinical trials of compounds M402 and PI-88 have been terminated due to adverse side effects and poor efficacy after Phase II and III clinical trials, respectively. SST0001 has completed Phase I trials for the treatment of multiple myeloma, while PG545 is undergoing a Phase Ib clinical trial for the intervention of pancreatic cancer and other solid tumors.16 These challenges highlight the ongoing obstacles in developing effective HPSE-targeted therapies.
The progression of HPSE-inhibiting small molecules to clinical trials has encountered challenges due to a lack of specificity and off-target effects.8 Inspired by this, we aim to develop HPSE inhibitors with reduced off-target effects by repurposing low-cost and commercially available aminoglycosides, such as paromomycin and neomycin (Figure 1D). Their branched pseudo-tetrasaccharide structure enables them to fill the HPSE binding pocket effectively. While aminoglycosides target bacterial rRNA to inhibit protein synthesis,17 we hypothesize that modifying their hydroxyl and amine groups creates negatively charged glycans that interact with HPSE instead. This approach presents a promising opportunity for designing HS mimetics as HPSE inhibitors (Figure 1D).
In this study, we demonstrate that the incorporation of hydrophobic aromatic naphthalene groups and N-sulfate modifications on the free hydroxyl and amine groups, respectively, significantly enhances the potency of paromomycin- and neomycin-based HPSE inhibitors within the nanomolar range. Furthermore, these modifications markedly reduced cancer progression in vitro and in vivo. In addition, we investigate the incorporation of hydrophobic aromatic and aliphatic groups to probe the various hydrophobic regions on HPSE that have not been fully explored in ligand design. Although the inclusion of hydrophobic aromatic groups in our ligand design increases their molecular weight (MW), the final MW of our most potent compounds remains comparable to that of the clinically tested anti-HPSE ligand PG545. Consequently, we utilize PG545 as a benchmark for comparison when assessing the efficacy of our ligands.
RESULTS AND DISCUSSION
Design and Synthesis of Aminoglycoside-Based HS Mimetics.
The most effective HPSE inhibitors identified to date include a heterogeneous and polyanionic heparin-mimetic (SST0001) or a polysulfated tetrasaccharide combined with a lipophilic cholestanol moiety (PG545) (see Figure 1C). We aim to develop small-molecule glycans incorporating hydrophobic groups and well-defined sulfation patterns. We screened a diverse library of aminoglycoside-based ligands guided by computational insights.18 Consistent with previous studies, we identified essential residues within the binding pocket of HPSE that are critical for the recognition and binding of the endogenous HS substrate, including Arg272, Gly349, Thr97, and Lys159.2 These residues also play a crucial role in the recognition and binding of the newly designed HS mimetic ligands. Incorporating various hydrophobic moieties at the primary positions of the glucosamine ring A and the ribose ring C (Figure 1D) also significantly enhanced the interactions with hydrophobic residues located in the active site of HPSE (Figure S8). We incorporated aromatic hydrophobic groups, including benzyl, the smallest aromatic moiety, and biphenyl, to probe the hydrophobic pocket of heparanase. Cyclic aliphatic groups such as pyrrolidine and piperidine were also evaluated, given their widespread use in drug design. Given the enhanced binding affinity observed for incorporating aromatic groups at the primary positions of rings A and C of the sugar backbone, we explored the global integration of aromatic groups into our ligands. Our hypothesis was based on the understanding that the binding pocket of HPSE contained several hydrophobic regions that could be targeted by leveraging the hydrophobic effect.19,20 Our objective was to incorporate hydrophobic aromatic groups that could participate in hydrophobic and π interactions with the amino acid residues within the binding pocket of HPSE.
To achieve our goals, we developed aminoglycoside-based ligands, whose secondary hydroxyls are capped with O-benzyl, O-naphthyl, or O-biphenyl groups (Figure 2A). In the docking studies, we observed a substantial increase in hydrophobic interactions compared to the parent-free hydroxyl ligands (see Figure S15 for all interactions). The O-benzyl and the O-naphthyl ligands exhibited hydrophobic interactions with the glycine loop (Gly349 and Gly350). Furthermore, the O-naphthyl ligand maintained a hydrogen bonding interaction with Glu 225, which was not observed for the O-benzyl ligand. Based on the molecular docking results, we synthesized a library of 26 paromomycin- and neomycin-based analogs bearing the naphthyl (Nap), biphenyl, and benzyl (Bn) groups in 10–13 steps using a rapid and easy-to-scale-up synthetic route (see Scheme S1 for detailed synthesis procedures).
Figure 2.
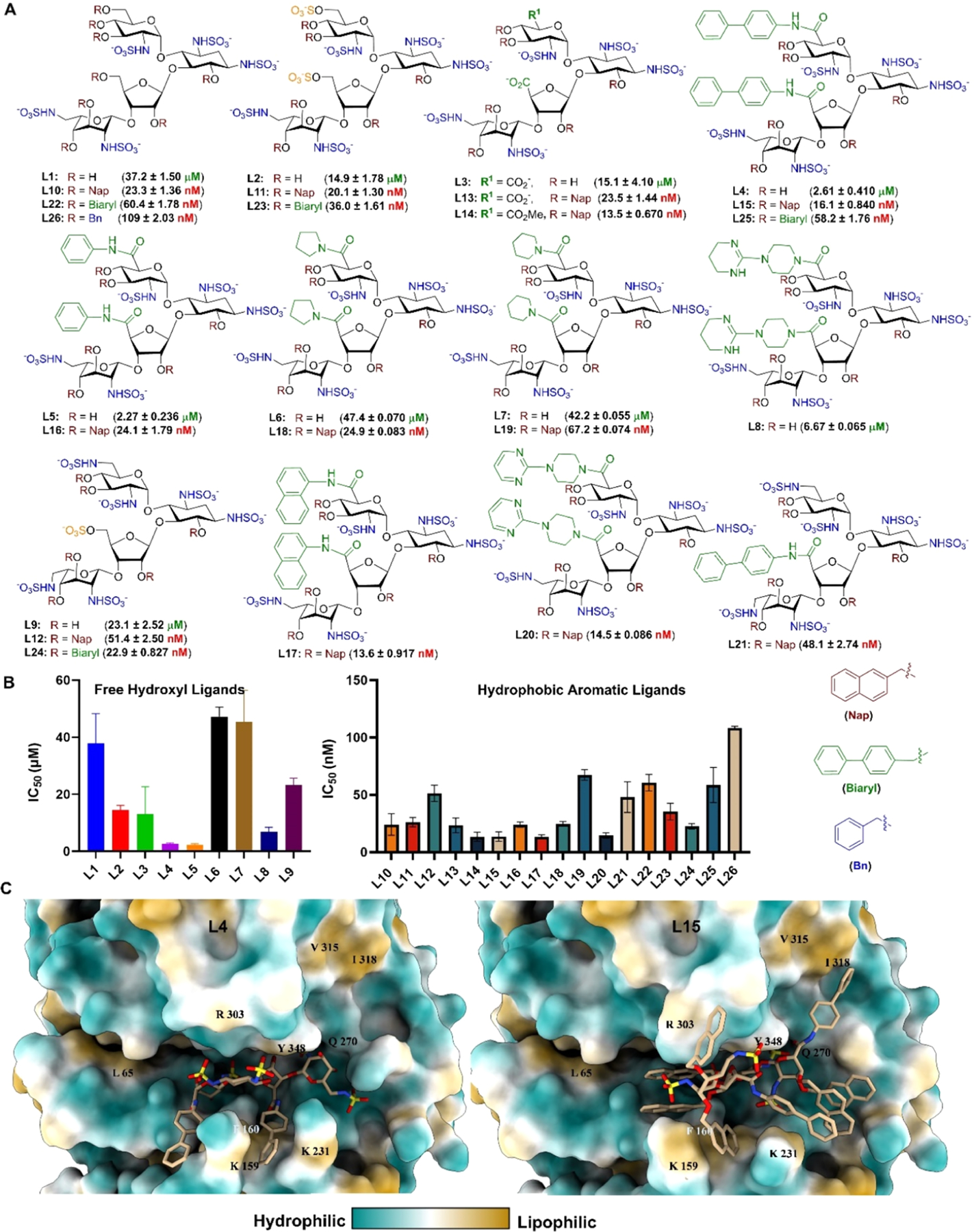
(A) Library of HPSE inhibitors from paromomycin and neomycin starting material. (B) Potency of ligands as determined by the TR-FRET assay. Data are shown as mean ± SD (n = 3). (C) Ligands L4 and L15 in the active site of HPSE with the molecular lipophilicity potential surface of HPSE as determined by USCF Chimera.
Evaluation of Aminoglycoside-Based HS Mimetics as Inhibitors of Heparanase.
We evaluated the efficacy of our HS mimetic library against HPSE using a homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) assay with a fluorescent HS substrate (Figure 2A,B). The results indicate that N-sulfated paromomycin L1, containing free hydroxy groups in the primary positions of rings A and C, was less effective as an HPSE inhibitor than L2 and L3, which feature 6-O-sulfate and carboxylate, respectively. This finding supports previous reports, highlighting the significance of 6-O-sulfates in the design of HPSE inhibitors.21,22 Furthermore, incorporating hydrophobic aromatic moieties at the primary positions of rings A and C significantly improved potency, with L4 and L5 emerging as effective inhibitors with similar IC50 values in micromolar concentrations.
In the synthesis of our ligands, we capped their secondary hydroxyl groups with naphthalene (Nap) moieties and subsequently removed them to obtain free hydroxyl compounds. Our computational analysis suggested that the O-naphthyl groups could function as hydrophobic aromatic motifs to enhance ligand efficiency. Our results confirmed this hypothesis, showing significant potency increases in nanomolar concentrations for Nap-ligands L10 (IC50 = 23.40 ± 3.91 nM) and L11 (IC50 = 20.10 ± 1.30 nM) compared to the micromolar concentrations of free hydroxyl ligands L1 (IC50 = 37.24 ± 1.50 μM) and L2 (IC50 = 14.92 ± 1.79 μM) (Figure 2). Overall, Nap-ligands demonstrated superior potency against HPSE, indicating that larger conjugated aromatic groups enhance ligand efficacy.
Molecular Dynamics of HPSE and HPSE Inhibitors.
To further evaluate the impact of the hydrophobic groups on ligand efficacy, we performed molecular dynamics (MD) simulations on the complexes formed by L4 and L15 with HSPE (Figure 2C). Ligand L4 was chosen for our study because it is the most potent HPSE inhibitor among the free hydroxyl ligands we screened. In contrast, L15 was identified as one of the most effective Nap-capped hydroxyl ligands. Postsimulation analyses revealed significant differences in the stability of ligand interactions within the heparanase binding pocket. Ligand L15 maintained consistent hydrogen bonds (Figure S13) with the catalytic residues Glu 225 (82%), whereas L4 only interacted with Glu 225 in less than 10% of frames (Figure S13), indicating a comparatively weaker affinity. Furthermore, L15 demonstrated stabilizing π interactions with Ile318, Lys231, Phe160, Arg272, and Lys274 (Figure S10), which were largely absent in L4. Although L4 did show limited interactions with Lys 159 and Lys 231, it lacked critical contact with the lipophilic site formed by Ile 318 and Val 315, thereby reducing its binding stability.
Further structural analysis revealed that L15 induced a conformational shift in the binding pocket, resulting in a greater reduction in RMSD (Root Mean Square Deviation) than L4 (see Figure S14). This suggests enhanced stabilization of the binding pocket. The naphthalene groups in L15 facilitated deeper insertion of the sugar backbone into the binding pocket of heparanase, reducing solvent exposure of the sugar moiety. This conformation enabled the hydrophobic groups of L15 to align more effectively with the lipophilic surface of heparanase, maximizing contact with hydrophobic residues (Figure 2C). These additional hydrophobic interactions likely contribute to the overall stability and binding efficiency of the naphthalene-L15 within the active site of HPSE. The MD analysis revealed that the hydrophobic groups enhance ligand efficacy by promoting favorable hydrophobic interactions and conformational stability in the binding pocket, thereby potentially improving ligand efficiency and affinity (see Figures S11 and S12).
Evaluation of Potent Inhibitors to Prevent ECM Degradation.
The overexpression of heparanase in cancer cells is associated with enhanced metastasis, primarily attributed to the role of HPSE in degrading heparan sulfate proteoglycans (HSPGs) in the ECM, which enables primary tumor cells to enter the bloodstream and promote metastasis (Figure 3A).6,9 Building upon the findings derived from the Nap-ligands illustrated in Figure 2, we evaluated their efficacy in mitigating ECM degradation by inhibiting the enzymatic activity of HPSE.
Figure 3.

(A) Degradation of the ECM’s heparan sulfate by heparanase is a major contributing factor in disseminating malignant tumors to promote metastasis. (B) The % CPM (count per minute) of sulfate-labeled HS degradation fragments released into the incubation medium in the absence and presence of 0.5, or 5 μg/mL of the inhibitory ligands. HS degradation fragments are eluted from Sepharose 6B in fractions 15–35 (=peak II) (see the Biological Experimental Section).
We assessed the capacity of our potent ligands to inhibit the degradation of radiolabeled H35SPG by HPSE in an ECM environment.23 This evaluation was conducted by measuring the release of 35S-labeled HS degradation fragments from 35SO42−-labeled ECM, followed by quantification of radioactivity to determine the extent of HS degradation.5 The elution profile and biochemical characterization of labeled material eluted from a Sepharose 6B column (Figure S5) between fractions 15 and 35 (peak II) indicated that the released material consisted of HS degradation fragments, produced by heparanase enzymatic activity. In this study, L15, L17, and L18 were selected as they are the most effective HPSE inhibitors among the series of Nap-ligands tested. The glycopolymer GPM-2 was utilized as a reference compound due to its established efficacy as a heparanase inhibitor (IC50 = 0.10 ± 0.036 nM).24 Specifically, L17 achieved a nearly complete inhibition of ECM degradation at 5 μg/mL (Figure 3B), reducing the amount of released sulfate-labeled material to 1.32%, outperforming GPM-2, which resulted in a degradation reduction to 3.21% (Figure 3B). Furthermore, at a lower concentration of 0.5 μg/mL, L15 exhibited a substantial inhibitory effect, decreasing the amount of released radioactivity (CPM) to 36.38%, marking it as one of the top-performing paromomycin analogs.
Effect of Potent Inhibitors on U-87 Glioblastoma Cell Viability and Invasion.
Given the promising ability of L15 and L17 to prevent extracellular matrix degradation by inhibiting HPSE, we next evaluated their impact on the viability of U-87 glioblastoma cells, a highly aggressive cell line known to rely on HPSE for invasion.25 To assess cell viability, we first employed the WST-1 colorimetric assay, a highly sensitive method for detecting metabolic activity. Treatment with L15 and L17 resulted in a dose-dependent reduction in cell viability, with L17 showing the most pronounced effect, reducing viability to 39% at 120 μM, compared to PG545, which decreased viability to 45% at the same concentration (Figure 4A). Given the known resistance of U-87 cells, we attributed the need for relatively high concentrations to their aggressive phenotype rather than compound toxicity.26 To confirm this hypothesis, we used the CCK-8 assay, which offers improved accuracy for calculating half-maximal effective concentrations (EC50), to quantify the cytotoxic effects of each compound over a 48 h treatment period. Consistent with the WST-1 results, L15 (EC50 = 103.4 ± 1.95 μM), L17 (EC50 = 82.56 ± 2.38 μM), and PG545 (EC50 = 102.9 ± 2.95 μM) all exhibited relatively low cytotoxicity (Figure S6). While the WST-1 assay provided a snapshot of dose-dependent viability changes, the CCK-8 assay allowed us to quantitatively validate the low toxicity of the compounds.
Figure 4.

(A) L17 shows better efficacy in reducing the proliferation of U-87 malignant glioma cells than L15 and PG545. (B) Flow cytometry analysis using Annexin V stain to determine apoptosis post-treatment of U-87 cells with L15 and L17. (C) Gene expression level of HPSE post-treatment with L15 and L17 at 120 μM and 60 μM compared to the untreated control. (D) L15 and L17 reduce the invasiveness of U-87 cells as determined by the Matrigel invasion assay (positive: 10% FBS growth media, negative: serum-free media). (E) Selectivity profile of highly potent L15, L17, and the clinically tested PG545 against HPSE, PF4, and FXa. Data are shown as mean ± SEM (n = 3). ns >0.9999, *p < 0.01, **p < 0.001, ***p < 0.0001, ****p < 0.0001 for L15 or L17 vs PG545.
Finally, to determine whether reduced viability correlated with apoptotic cell death, we conducted flow cytometry analysis using Annexin V staining (Figure 4B). These results also revealed a dose-dependent decrease in viable U-87 cells, with L17 showing the strongest apoptotic response within 24 h post-treatment (Figure 4B).
Next, we evaluated the effect of L15 and L17 on the mRNA expression levels of HPSE in U-87 MG glioblastoma cells27 using quantitative real-time polymerase chain reaction (RT-qPCR) (Figure 4C). We hypothesized that the ligands could downregulate HPSE gene expression by interrupting the feedback regulation that normally sustains HPSE expression. HPSE gene expression is tightly controlled by cellular signaling and feedback loops.6,28 Under normal conditions, HPSE transcription is driven by pro-inflammatory and growth signals (e.g., TNF-α/IL-1β via NF-κB, HGF/PI3K/Akt) and repressed by tumor suppressors such as wild-type p53.29 Inhibiting HPSE disrupts these regulatory circuits. For example, HPSE normally cleaves heparan sulfate to release bound growth factors (HGF, VEGF, FGF, Hedgehog), which then feed back to induce HPSE expression via PI3K/Akt/NF-κB pathways.29,30 Inhibiting HPSE enzymatic activity could likely prevent this positive feedback, so those factors remain sequestered and fail to stimulate HPSE transcription. Previous work by Richardson et al. reported the ability of defibrotide, an HPSE inhibitor, to reduce the expression levels of HPSE in myeloma cells.28
In our studies, we specifically targeted a concentration of 60 μM for initial experiments, as preliminary cytotoxicity assays indicated that this dose had a minimal impact on cell viability in U-87 cells, maintaining over 70% cell viability (Figure 4A). At 60 μM, we observed a significant reduction in HPSE expression levels (Figure 4C), suggesting effective downregulation with low cytotoxic impact. To further assess the dose-dependent effects, we examined a higher ligand concentration. At 120 μM, HPSE expression was also reduced; however, the elevated cell death rates at these doses may indicate additional cellular stress responses that could influence gene expression patterns. Therefore, while a higher concentration continued to demonstrate HPSE downregulation, the increased cytotoxicity emphasizes the importance of balancing ligand efficacy with cellular viability to avoid stress-induced gene expression artifacts.
The invasive nature of U-87 cells has been linked to the aggressiveness of glioblastoma cancer;31 therefore, we sought to evaluate the potential of our ligands to inhibit their cellular invasion. We employed a Matrigel invasion assay, which measures the ability of U-87 cells to migrate through a basement membrane (BM) coated filter in a Boyden chamber. Invasive U-87 cells migrate through the Matrigel-coated filter into the lower chamber containing 10% fetal bovine serum (FBS).32 Our results demonstrated a significant reduction in U-87 cell invasion at nontoxic ligand concentrations of L15 and L17 (15–50 μM), indicating that these ligands effectively attenuate the invasive potential of these highly aggressive cells (Figure 4D). This inhibition of invasion was observed even under conditions where cell viability remained high, suggesting that L15 and L17 can modulate invasive behavior without inducing substantial cytotoxic effects.
Evaluation of Cross-Bioactivity of L15 and L17.
Most HS-based antiheparanase inhibitors were discontinued during clinical trials due to their off-target effects, which involved heparin-binding proteins, such as platelet factor 4 (PF4), a natural chemokine.33 PF4 binds to heparin and heparin/HS derivatives to form a complex that can trigger an autoimmune response, leading to heparin-induced thrombocytopenia (HIT).34 HIT is characterized by a reduction in platelet count and a hypercoagulable state, which can lead to bleeding complications, embolisms, and thrombosis.35 Consequently, we evaluated L15 and L17 against PF4 using a biolayer interferometry (BLI) assay, which has been established to test the ability of HS mimetics to compete with biotinylated heparin.36 We observed high selectivity for HPSE over PF4, with L17 showing over 100-fold selectivity, while L15 showed greater than 40-fold selectivity (Figure 4E). Both L15 and L17 exhibited better selectivity for HPSE over PF4 than the clinically tested ligand PG545. Although PF4 is a known heparin-binding protein, its binding site is relatively shallow, as confirmed by our computational studies and other researchers.37,38 In contrast, HPSE possesses a deeper active site that more effectively accommodates our ligands, resulting in significantly higher binding affinity. This structural distinction accounts for the minimal binding observed between our compounds and PF4, reinforcing their selectivity for HPSE.
Furthermore, given the increased risk of bleeding complications associated with many HS-based HPSE inhibitors,39 we assessed the anticoagulant properties of L15 and L17 (Figure 4E). Heparin exerts its anticoagulant effect by binding to antithrombin III (ATIII), inducing a conformational change that inhibits the binding of clotting factors such as factor Xa (FXa) and FIIa.40 This inhibition prevents the conversion of fibrinogen to fibrin, consequently impeding clot formation. As such, we evaluate the anticoagulant activity of L15 and L17 compared to PG545 against FXa using a chromogenic substrate assay.41 FXa was selected due to its compatibility with low-molecular-weight HS mimetics.42 Our results revealed that L15 and L17 exhibited no anticoagulant activity against FXa with IC50 values exceeding 500 μM, in contrast to PG545 (IC50 = 15.6 μM).
L17 Attenuates Metastasis of Melanoma into the Lungs.
Ligand L17 was selected for in vivo analysis due to its better efficacy in attenuating tumor cell proliferation while exhibiting minimal cytotoxicity in primary cells compared to the other tested ligands. Additionally, L17 demonstrated strong performance in the ECM degradation assay, an established indicator of HPSE inhibition. Compounds that effectively inhibit HPSE in this assay have consistently demonstrated promising efficacy in preclinical animal models.43 Based on these promising results, we prioritized L17 for further in vivo studies using the experimental metastasis assay. This well-established and widely used model is particularly valuable for assessing the efficacy of inhibitors to impede metastatic spread.44 Given its simplicity, it has become a preferred choice among numerous research groups and pharmaceutical companies for identifying and screening the potential antimetastatic properties of compounds that target known molecular mechanisms, primarily those involved in cell adhesion, migration, invasion, and degradation of the glycocalyx and extracellular matrix. The experimental metastasis assay played a crucial role in the development of heparanase-inhibiting compounds, SST0001 and PG545, which are currently being examined in clinical trials. This is the approach/rationale applied in our study. We first screened L17 in the experimental metastasis model using the B16 melanoma lung colonization model to assess its antimetastatic effects.
In the B16 melanoma study, C57BL/6J mice were intravenously inoculated with B16 melanoma cells (1.5 × 105 cells), resulting in extravasation and lung colonization. After postinjection of the tumor cells, mice received an intraperitoneal injection of either PBS (control), L17, or GPM-2 at a dosage of 200 μg per mouse. In this study, GPM-2 was used as the reference compound due to the limited availability of PG545. Additionally, we have previously demonstrated the substantial antimetastatic properties of GPM-2 in a 4T1 breast carcinoma experimental metastasis model.21 On day 17 postinjection, the mice were euthanized, and their lungs were harvested for analysis. The number of metastases was then quantified, and the results were presented graphically. As demonstrated in Figure 5A, C57BL/6J mice treated with L17 exhibited a significant reduction in lung metastasis compared to the PBS control. Ligand L17 demonstrated slightly diminished efficacy relative to GPM-2 (Figure 5A). The B16 experimental metastasis assay does not aim to assess whether heparanase inhibition slows down the growth of tumor cells that have colonized metastatic sites. Rather, it measures the ability of blood-borne tumor cells to extravasate and colonize in the lung tissue. We have demonstrated the capacity of ligand L17 to attenuate this process.
Figure 5.

(A) L17 shows reduced lung metastasis of B16 melanoma cells. (B) L17 shows reduced tumor growth of MPC-11 myeloma tumors compared to GPM-2 and PBS (control). Data are shown as mean ± SEM **p < 0.005 for L17 vs control and GPM-2 vs control.
L17 Attenuated the Tumor Growth of MPC-11 Myeloma.
The extravasation process occurs rapidly, with highly metastatic cells that enter the bloodstream being able to extravasate within minutes. Consequently, the drug must be administered shortly before or immediately after the intravenous inoculation of these cells. The experimental metastasis model primarily reflects the later stages of metastasis, which potentially overlooks earlier metastatic events, such as local invasion and intravasation. Therefore, we further investigate the ability of ligand L17 to inhibit the primary tumor growth. We utilized a murine plasma cell myeloma (MPC-11) model to assess the effect on tumor growth. The MPC-11 myeloma cell line has been extensively studied in vivo, particularly for experiments involving HPSE inhibition.45,46 Accordingly, MPC-11 myeloma cells (3 × 105 cells) were implanted into BALB/c mice subcutaneously. Following implantation, the mice were administered daily intraperitoneal injections of either PBS (control), L17, or GPM-2 (1 mg/mL) over 16 days. The mice were euthanized at the end of the treatment period, and the tumors were excised and weighed for subsequent analysis. The results showed that L17 significantly reduced tumor growth compared to the untreated control group and the GPM-2-treated group, highlighting its potential efficacy in suppressing tumor growth (Figure 5B).
Multiple myeloma (MM) is a blood cancer that depends on the bone marrow microenvironment for growth.47 HPSE plays a crucial role in modifying this environment. It contributes to the shedding of syndecan-1 from the myeloma cell surface and the associated increased angiogenesis, tumor growth, metastasis, and bone disease (Figure 6A). High levels of shed syndecan-1 in myeloma patients are correlated with poor prognosis and reduced overall survival.48–50 Furthermore, elevated HPSE levels in myeloma cells from patients who have undergone multiple chemotherapy rounds51 are associated with resistance against the leading antimyeloma drug, bortezomib (Figure 6A).52 This correlation highlights the role of HPSE in modulating the efficacy of antimyeloma treatments. Some patients who experience a relapse of myeloma, despite receiving all available therapeutic modalities, exhibit responsiveness and favorable tolerability to the HPSE inhibitor SST0001 in Phase 1 clinical trials (Figure 6A).13
Figure 6.

(A) Heparanase is involved in chemoresistance in myeloma patients. (B) L17 reduces the growth of MPC-11 myeloma tumors compared to the clinically tested HPSE inhibitor SST0001, which has completed a Phase I clinical trial for treating patients with relapsed myeloma. L17 is slightly more effective than the antimyeloma drug bortezomib in reducing MPC-11 tumor growth. Data are shown as mean ± SEM *p < 0.03 for SST0001 vs control, SST0001 vs L17, and SST0001 vs GPM-2 (see the Methods section for additional details).
In a subsequent study, we assessed the efficacy of SST0001, derived from the glycol-split of heparin polysaccharide, on tumor growth utilizing MPC-11 myeloma cells that were subcutaneously implanted into BALB/c mice. This treatment was compared to the HSPE inhibitor L17 (Figure 6B). Our results demonstrated that L17 displayed a significantly greater efficacy in inhibiting MPC-11 tumor growth, achieving a tumor growth inhibitor index (TGI) value of 83.1%. In contrast, SST0001 showed lower efficacy, with a TGI value of 58.6%. Bortezomib, a proteasome inhibitor, binds to the 26S proteasome and activates signaling pathways that disrupt the cell cycle, induce apoptosis, and alter the bone marrow microenvironment.53 Given that HPSE inhibitors and bortezomib work through different mechanisms (Figure 6A), we compared the efficacy of the proteasome inhibitor bortezomib with that of the HPSE inhibitor L17 (Figure 6B). Initial findings showed that bortezomib exhibited a comparable efficacy in attenuating MPC-11 tumor growth, achieving a TGI value of 81.7%. Overall, the HPSE inhibitor L17 and the antimyeloma drug bortezomib showed higher efficacy against MPC-11 cell growth compared to the clinically tested HPSE inhibitor SST0001. Lastly, we assessed the synergistic effect of combining L17 and bortezomib but observed no additional impact of the combined treatment, resulting in a TGI value of 81.8%.
CONCLUSIONS
Extensive research has established that heparanase (HPSE) is a key driver of tumor progression and metastasis through the degradation of the extracellular matrix (ECM) and the release of growth factors. Despite its therapeutic potential, no HPSE inhibitors have been clinically approved due to off-target effects and limited efficacy. To address this, we developed a streamlined strategy for synthesizing HPSE inhibitors from readily available aminoglycosides, allowing for the rapid screening of diverse compounds.
Our approach leverages hydrophobic interactions to enhance ligand binding within HPSE’s 609.6 Å2 binding pocket,54 using bulky pseudo-tetrasaccharide aminoglycosides. Incorporating hydrophobic aromatic groups enhanced ligand stability and potency in the low nanomolar range, while maintaining high selectivity over other heparin-binding proteins, such as PF4 and ATII. Compared to the clinically tested PG545, our inhibitors demonstrated superior HPSE selectivity and efficacy.
In vitro studies demonstrated that our ligands effectively inhibited ECM degradation, reduced the viability of U-87 glioblastoma cells, and blocked their invasion. In vivo, the lead compound L17 significantly suppressed tumor growth and metastasis in B16 and MPC-11 models, achieving a tumor growth inhibition (TGI) of 83.1%, surpassing the clinically tested SST0001 (TGI 58.6%) and the leading antimyeloma drug, bortezomib (TGI 81.7%). Given bortezomib resistance in relapsed myeloma patients, L17 offers a promising alternative. Their high efficacy, selectivity, and accessibility position aminoglycosides as a compelling strategy for developing targeted HPSE inhibitors for solid and hemato-logical malignancies.
BIOLOGICAL EXPERIMENTAL SECTION
Reagents.
All compound solutions were prepared in sterile ultrapure water (Thermo Scientific Smart2Pure 6 UV/UF). Dr. Alessandro Noseda (Leadiant Biosciences S.p.A, Rome, Italy) and Dr. Edward Hammond (Zucero Therapeutics, Brisbane, Queensland, Australia) kindly provided HSPE inhibitors SST0001 and PG545, respectively.
Heparanase Inhibition Assay.
HPSE inhibition was evaluated using a fluorescence resonance energy transfer (FRET)-based assay. In 0.6 mL Eppendorf tubes, 42 μL of paromomycin or neomycin analog solutions (dissolved in Milli-Q water) or 42 μL of Milli-Q water (control) was added to 42 μL of HPSE solution (5.3 nM; R&D Systems, Cat #: 7570-GH) prepared in 20 mM Tris–HCl buffer, pH 7.5, containing 0.15 M NaCl and 0.1% CHAPS. For blank controls, the buffer alone was used instead of HPSE. The reaction mixtures were incubated at 37 °C for 10 min to allow pre-equilibration of the enzyme and ligands. Following preincubation, 84 μL of biotin-heparan sulfate labeled with europium cryptate (Cisbio, Cat #: 61BHSKAA), containing 58.6 ng of reagent in 0.2 M sodium acetate buffer (pH 5.5), was added to each tube to initiate the enzymatic reaction. The reactions were then incubated at 37 °C for 60 min. To terminate the reaction, 168 μL of Streptavidin-XLent! reagent (Cisbio, Cat #: 611SAXLA), prepared at a concentration of 1.0 μg/mL in a dilution buffer (0.1 M NaH2PO4, 0.8 M KF, 0.1% BSA, pH 7.5), was added. For signal measurement, 100 μL of each reaction mixture was transferred to a white 96-well half-area microplate (Corning, Cat #: 3693), prepared in triplicate. HTRF emissions were recorded at 665 and 616 nm with an excitation wavelength of 340 nm using a Tecan Spark plate reader.
Platelet Factor 4 Binding Assay.
Biolayer interferometry (BLI) assays were conducted on an Octet Red instrument (fortéBIO) at 25 °C. A solution-phase affinity assay, adapted from SPR analysis, was used by BLI to determine ligand binding affinities.55 Human recombinant CXCL4/platelet factor 4 (PF4) protein (R&D Systems, Cat. 795-P4) was incubated with varying concentrations of ligands or heparin. The remaining free protein in this equilibrium mixture was tested for binding to immobilized heparin. All proteins (R&D Systems) were carrier-free. Heparin-biotin (Creative PEGworks, 18 kDa, with one biotin per polymer) at 5 μg/mL was immobilized onto Octet Streptavidin (SA) biosensors (Sartorius, Cat. 18–5019) for 5 min. The immobilization and binding studies were performed at a shaking speed of 1000 rpm in 1× HBS-EP buffer (Cytiva Life Sciences, Cat. BR100669). Binding experiments were analyzed using GraphPad Prism, and the percentage of PF4 binding was calculated based on the signal response relative to control conditions.
Factor Xa Binding Assay.
Factor Xa (FXa) activity was assessed using BIOPHEN Heparin Anti-Xa (2 stages) USP/EP kits (Cat #: A221005-USP), following the manufacturer’s instructions. All reagents were reconstituted, prepared per protocol, and preincubated at 37 °C for 15 min. To evaluate FXa inhibition, 40 μL of varying concentrations of heparin (0.002–500 nM), PG545, Ligand 17, or Ligand 15 (0.00097–1000 μM) were mixed with 40 μL of antithrombin III (ATIII; 0.04 IU) in a Nunc 96 DeepWell block (1.0 mL/well, clear). The mixtures were incubated at 37 °C for 2 min. 40 μL of FXa (0.32 μg) was then added using a multichannel pipet and incubated at 37 °C for another 2 min (stage 1). Following this, 40 μL of FXa-specific chromogenic substrate (0.048 mmol) was added to initiate the reaction. The reaction was terminated after 2 min by adding 240 μL of citric acid (20 g/L). A 100 μL aliquot from each reaction was transferred to a clear 96-well microplate in triplicate, and absorbance at 405 nm was measured using a TECAN Spark plate reader. For blank measurements, reagents were mixed in reverse order (citric acid added first, followed by FXa substrate, FXa, ATIII, and test samples). Absorbance values from these blanks were subtracted from corresponding assay values to correct for the background signal.
Extracellular Matrix Degradation Assay.
A sulfate [35S] labeled ECM coating the surface of 35 mm tissue culture dishes is deposited by cultured corneal endothelial cells and prepared as described.23 For measurements of heparanase enzymatic activity, the ECM is incubated (3 h, 37 °C, pH 6.0, 1 mL final volume) with recombinant human heparanase (100 ng/mL) in the absence and presence of paromomycin ligands at 5 and 0.5 μg/mL. The reaction mixture contains 50 mM NaCl, 1 mM DTT, 1 mM CaCl2, and 10 mM buffer Phosphate-Citrate, pH 6.0. After incubation, the medium was collected to assess proteoglycan degradation by gel filtration on Sepharose 6B columns (0.9 × 30 cm2). Fractions of 0.2 mL were eluted with PBS and measured for radioactivity using a liquid scintillation analyzer. Blue dextran marks the excluded volume (Vo), while phenol red indicates the total included volume (Vt). Degradation fragments of heparan sulfate (HS) side chains, previously characterized,9,12 are eluted from Sepharose 6B at 0.5 < Kav < 0.8 (fractions 15–35).
Cell Culture.
Human glioblastoma U-87 MG was obtained from the American Type Culture Collection (ATCC) and cultured in Eagle’s Minimum Essential Medium (EMEM, ATCC) supplemented with 10% Fetal bovine serum (FBS, ATCC) and 1% penicillin-streptomycin (P.S, ATCC). A culture of Human epithelial cells (MCF-10A) was gifted from Dr. Hasan Korkaya (Wayne State University) and cultured in a mammary epithelial cell growth medium bullet kit (Lonza, CC-3150) supplemented with 100 ng cholera toxin (Sigma-Aldrich, C8052). Cells were cultured in a humidified incubator at 37 °C and 5% CO2. Ligand solutions were prepared in sterile ultrapure water (Thermo Scientific Smart2Pure 6 UV/UF).
CCK-8 Assay.
Cytotoxicity was assessed using the Cell Counting Kit-8 (CCK-8) assay. 1 × 104 cells were seeded in 96-well plates to a final volume of 100 μL culture medium. 24 h after seeding, cells were treated with varying concentrations of ligands. All dilutions were made with culture media to a final volume of 100 μL. Cells were incubated 24 or 48 h post-treatment before adding 10 μL of CCK-8 reagent to each well. Blank control wells contained 100 μL culture media +10 μL of CCK-8 reagent. The plate was then incubated for 2 h in standard culture conditions. After incubation, the plates were shaken for 1 min, and then the absorbance was read at 440 nm using the TECAN Spark microplate reader.
Matrigel Invasion Assay.
The inhibition of U-87 MG cell invasion was analyzed using the cell invasion assay (Matrigel-coated filters, pore size 8 μm) kit from Abcam (cat# ab235697). A culture of U-87 cells at 80% confluency was starved in serum-free EMEM for 24 h. After starvation, the cells were harvested, resuspended in Wash Buffer II, and counted. They were then resuspended at a concentration of 1 × 106 cells/mL in serum-free media. In the bottom chamber, 200 μL of EMEM containing FBS was added, and for positive control, 10% control migration inducer was added to the media. EMEM containing no FBS was used as a negative control. In the top chamber, 5 × 104 cells were added to a final volume of 50 μL, and an equal volume of treatment was added to each well, resulting in a final volume of 100 μL. The plate was incubated for 24 h, during which time a standard curve was generated as recommended by the manufacturer’s protocol. After incubation, cells were collected, and the migrated cells in the bottom chamber were stained using a cell invasion dye and quantified by reading the plate at EM/Ex = 530/590 nm using a TECAN Spark microplate reader.
Annexin V Apoptosis Flow Assay.
Apoptosis analysis was performed using the Annexin V Apoptosis Detection Kit (Cat. 88–8005–72). U-87MG cells (1 × 106) were treated with our ligands at varying concentrations. Post-treatment incubation was done for 24 and 48 h, after which the cells were harvested and washed with PBS (ATCC, cat. 30–2200) and 1× binding buffer. Cells were resuspended in binding buffer, and 5 μL of FITC Annexin V was added to the cell suspension and incubated for 15 min at room temperature. Cells were washed and resuspended in 1× binding buffer, and 5 μL of Propidium iodide solution was added. The mixture was then incubated for 15 min before subsequent analysis by flow cytometry. Analysis was carried out using the Attune CytPix Flow Cytometer.
Real-Time qPCR Assay.
Total RNA was isolated and purified from U-87MG cells using the QIAwave RNA Mini Kit (Qiagen, Cat. 74534) after treatment with ligands for 24 h. Using the iScript Advanced cDNA Synthesis Kit (Bio-Rad, Cat. No. 1725037), the purified total RNA was retrotranscribed according to the manufacturer’s procedure. RT-qPCR was performed using the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Cat. 1725270). Expression of HPSE was quantified using the PrimePCR SYBR Green Assay qHsaCID0015510 (Heparanase). Various house-keeping genes were screened for U-87 glioblastoma, and TATA-binding protein (TBP) gene was the most stable reference for gene expression analysis.56 TBP forward 5′ GAG CTG TGA TGT GAA GTT TCC 3′, reverse 5′ TCT GGG TTT GAT CAT TCT GTA G 3′. Relative expression of target genes was quantified using the QuantStudio 5 PCR system.
In Vivo Mouse Studies.
Research involving vertebrate animals will take place at Technion’s Faculty of Medicine, Haifa, Israel. All procedures involving experimental animals, including their transportation, routine care, and use in experiments, are conducted in accordance with the Standards for the Humane Care and Use of Laboratory Animals. The Technion is certified by the NIH to run experiments using live animals. All experiments were performed in accordance with the Technion’s Institutional Animal Care and Use Committee (IL-078–05–2021; OPRR-A5027–01).
Euthanasia was performed using light anesthesia of mice, followed by cervical dislocation, following the recommendations of the panel on euthanasia of the American Veterinary Medical Association. We routinely use this method because it is painless and efficient for the mice.
B16 Melanoma Experimental Metastasis.
C57BL/6J mice (n = 6 per group) were intravenously injected with B16 melanoma cells (1.5 × 105 cells), followed by intraperitoneal injections of PBS (control), GMP-2 (200 μg/mouse), or ligand L17 (200 μg/mouse) after cell inoculation. Lungs were harvested 17 days after cell inoculation, fixed with Bouin solution, photographed, and the number of metastatic foci counted. The experiment was repeated twice, and the variation did not exceed ± 15% of the mean.
MPC-11 Mouse Myeloma Tumor Growth.
Mouse MPC-11 myeloma/plasmacytoma cells are detached with trypsin/EDTA, washed with PBS, brought to a concentration of 3 × 105 cells/0.2 mL, and inoculated subcutaneously at the right flank of 6- to 8-week-old Balb/c mice. On day 1, mice are randomly divided into groups (n = 5–7) receiving daily intraperitoneal injections of PBS alone (control), ligand L17 (0.1 mg/mouse), GPM-2 (0.1 mg/mouse), or SST0001 (1.2 mg/mouse). Xenograft size is determined twice a week by externally measuring the tumors in 2 dimensions using a caliper. At termination (day 17), mice are sacrificed, and xenografts are resected, weighed, and fixed in 4% PFA. Each experiment was repeated at least twice, and the variation did not exceed ±15% of the mean. The percent tumor growth inhibition (%TGI) was calculated as follows:
Combination Experiment.
MPC-11 myeloma/plasmacytoma cells are detached with trypsin/EDTA, washed with PBS, brought to a concentration of 3 × 105 cells/0.2 mL, and inoculated subcutaneously at the right flank of 6–8-weeks-old Balb/c mice. On day 1, mice are randomly divided into groups (n = 7) receiving daily intraperitoneal injections of PBS alone (control), ligand L17 (0.1 mg/mouse/day), bortezomib (0.02 mg/mouse, twice a week), or L17 plus bortezomib. At termination (day 17), mice are sacrificed, and the tumors are weighed and fixed in 4% PFA. Each experiment was repeated at least twice, and the variation did not exceed ±15% of the mean.
Statistical Analysis.
All assays were conducted independently at least three times unless stated otherwise. Statistical analyses were performed using GraphPad Prism (version 10.2.3). Data are presented as mean ± SEM, with significance defined as *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, unless otherwise specified.
SYNTHETIC EXPERIMENTAL SECTION
Reagents and Method.
All reactions were performed with an oven-dried flask fitted with a rubber septum under inert argon conditions unless otherwise stated. All organic solvents used were either anhydrous, as purchased from the vendor (Sigma-Aldrich, Alfa Aesar, TCI, and Combi-Blocks), or dried using a solvent purification system. Paromomycin and neomycin B were purchased from the vendors Biosynth and Chem-Impex, respectively. Organic solvents from aqueous workup were concentrated with a Buchi rotary evaporator at 35 °C or less and 25 Torr. Reaction progress was monitored with analytical thin layer chromatography (TLC) plates (TLC silica gel 60 F254) with 230–400 mesh silica gel. Products and intermediates were visualized with a UV light and stained with 5% H2SO4 for sugar compounds and ceric ammonium molybdate stain for nonsugars. Column chromatography was performed using 40–63 μm silica gel (SiliaFlash F60 from Silicycle) or Sephadex LH-20 from Cytiva. The enzymes for the TR-FRET recombinant human heparanase (HPSE) and platelet factor 4 (recombinant human CXCL4/PF4 protein) assays were procured from R&D Systems, heparin-biotin (18 kDa, 1 biotin per HP polymer) from Creative PEGWorks, streptavidin biosensors from Sartorius, HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3.0 mM EDTA, and 0.005% (v/v) surfactant Tween 20) from Cytiva, and biotin-heparan sulfate-Eu cryptate from Cisbio. Details of buffer and enzyme concentrations, as well as additional information, can be found in the protocols.
Instrumentation.
Newly synthesized compounds were analyzed by NMR spectroscopy and high-resolution mass spectrometry. 1H NMR spectra were recorded on either an Agilent 400 or 600 MHz or a Bruker 500 MHz spectrometer, while 13C NMR spectra were recorded with an Agilent 151 MHz or a Bruker 126 MHz spectrometer. The NMR chemical shifts are referenced to the solvent residual peak and are expressed in parts per million (ppm, δ scale). Deuterated solvents used include CDCl3 (δ 7.27 ppm, δ 77.16 ppm), D2O (δ 4.79 ppm), and CD3OD (d-4) (δ 3.31 ppm, δ 49.00 ppm). The automatic phasing and polynomial baseline correction features of the MestReNova software were utilized to process the acquired NMR data. Data presented as follows: chemical shift (s: singlet, d: doublet, t: triplet, q: quartet, m: multiplet), integration, and coupling constant in hertz (Hz). Low-resolution mass spectrometry was recorded using negative-mode ESI-MS with a Shimadzu LCMS-2020 instrument. High-resolution mass spectrometry was performed using a Thermo Orbitrap Exploris 120 in the Lumigen Instrument Center Mass Spectrometry and Analytical Instrument Core at Wayne State University.
GENERAL SYNTHETIC PROCEDURES
Azide Protection.
A stirring solution containing free amine starting material (5.00 g, 10.7 mmol) in 54 mL of H2O at room temperature was supplemented with K2CO3 (14.8 g, 107 mmol) and a catalytic amount of CuSO4·5H2O (0.214 g, 0.856 mmol). After cooling to 0 °C, 1H-imidazole-1-sulfonyl azide hydrochloride (14.8 g, 85.6 mmol) was incrementally added to the solution over 10 portions at 10 min intervals. The resulting mixture was stirred at room temperature for 12 h while monitoring the reaction progress via TLC. Co-evaporation with MeOH under vacuum at 25 °C yielded a dark residue, which was then dry-loaded onto silica gel and subjected to purification by column chromatography (using a 1:1 mixture of EtOAc and hexanes) to furnish the Azide-protected tetrasaccharide.
Precautions when working with azides: (a) Azide formation step should be handled with caution as organic azides are potentially explosive. (b) Organic azides can and will decompose in the presence of external energy (heat, light, or pressure) and should be stored at −20 °C in the dark. (c) Organic azide waste should be placed in a separate container designated only for azide waste. Ensure that any organic azide waste is kept separate from acid to avoid any potential contact between the two.
Amidation Reaction.
Oxalyl chloride (2 equiv per carboxylic acid) was added to a solution of the carboxylic acid crude substrate (100–250 mg) in dichloromethane (DCM) (0.200 M) under argon at room temperature. The resulting reaction mixture was stirred at room temperature for 12 h. The reaction was concentrated in vacuo upon completion. The crude acetyl chloride intermediate was then dissolved in tetrahydrofuran (THF) (0.200 M) and triethylamine (Et3N) (5 equiv per acyl chloride), and amine (4 equiv per acyl chloride) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 3 h. The reaction progress was monitored by TLC and stopped upon completion. The reaction mixture was diluted with EtOAc, washed with saturated aqueous sodium bicarbonate (NaHCO3) solution, and then with saturated aqueous sodium chloride (NaCl) solution, and dried over sodium sulfate (Na2SO4). The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography.
Azide Reduction.
To a stirred solution of an azide substrate in DMF (0.200 M) at room temperature, 1,3-propane dithiol (5 equiv per azide) and triethylamine (Et3N) (5 equiv per azide) were added. Azide reduction reactions were performed with a microwave synthesizer (Manufacturer: Biotage, model: Initiator+, Serial #: 013596–27J) at 100 °C and 1 atm for 3 h. The reaction progress was monitored by mass spectrometry and concentrated in vacuo upon completion. The crude amine residue (pale yellow solid) was subjected to a subsequent sulfation reaction without further purification.
Sulfation.
The crude free amine was dissolved in anhydrous pyridine (3 mL). Sulfur trioxide trimethylamine complex (SO3.Et3N) (5 equiv per NH2 or OH) and triethylamine (5 equiv per NH2 or OH) were then added. All sulfation reactions were performed with a microwave synthesizer (Manufacturer: Biotage, model: Initiator+, Serial #: 013596–27J) at 100 °C and 1 atm for 30 min. The reaction progress was monitored by ESI mass spectrometry (negative mode) and loaded onto a Sephadex LH-20 column for purification.
Ion Exchange.
To a solution of the sulfated substrate in MeOH (3 mL), Amberlite IR120 Na+ resin (five times the weight of substrate) was added. The mixture was stirred at room temperature for 48 h, filtered, and concentrated in vacuo to afford the sodium salt product confirmed by 1H and 13C NMR as well as HRMS.
Deprotection of 2-Naphthylmethyl Group.
To a stirred solution of 2-naphthylmethyl-protected substrate in 1:1 MeOH/H2O (0.200 M), 20% Pd/C (3× weight of substrate) and hydrogen gas (balloon) were added and stirred for 12 h. The reaction progress was monitored by ESI mass spectrometry (negative mode). The reaction mixture was filtered with Celite and concentrated in vacuo to give the desired crude product as a white powder. To ensure excellent purity of the synthesized sulfated HS mimetic library, each compound was purified with 5 feet Sephadex LH-20 column and confirmed by 1H and 13C NMR as well as HRMS.
HPLC Analysis.
10 uL of sample was injected using a Shimadzu Sil-30AC MP with LC-30AD pumps onto a Phenomenex Luna CN 100 m × 3.00 mm × 3 μm column eluting at 1 mL/min of acetonitrile and water containing 0.1% formic acid from 5% acetonitrile from 0 to 2 min with linear gradient to 100% at 12 min followed by rinse and equilibration. Analysis was done using a Shimadzu 8040 LCQQQ in Q3 scan mode with SPD-M30A PDA for UV detection. All compounds used for in vitro and in vivo studies are >95% pure by HPLC analysis.
1,2′,2‴,3,6‴-Pentaazido-1,2′,2‴,3,6‴-pentadeamino-paromomycin (1).
To a stirring solution of paromomycin sulfate (5.00 g, 5 mmol) in H2O (60 mL) at room temperature were added K2CO3 (14.8 g, 107 mmol) and a catalytic amount of CuSO4·5H2O (0.162 g, 0.684 mmol). The solution was cooled to 0 °C, and 1H-imidazole-1-sulfonyl azide hydrochloride (10.547 g, 50 mmol) was added to the solution in 10 portions at 10 min interval. The reaction mixture was stirred at room temperature for 12 h. The reaction progress was monitored by TLC. The mixture was co-evaporated with MeOH in vacuo at 25 °C. The resulting dark residue was dried and loaded onto silica gel, and then purified by column chromatography (1:9 MeOH/EtOAc) to afford 1 as white crystals. Isolated yield: 70.7%. 1H NMR (500 MHz, CD3OD) δ 5.81 (t, J = 4.3 Hz, 1H), 5.41 (dd, J = 14.8, 1.7 Hz, 1H), 5.12 (dd, J = 12.8, 2.7 Hz, 1H), 4.45 (dd, J = 6.7, 4.6 Hz, 1H), 4.34–4.27 (m, 2H), 4.13 (dddd, J = 18.1, 6.2, 4.9, 3.1 Hz, 1H), 4.02 (ddd, J = 8.4, 4.7, 2.0 Hz, 1H), 3.94 (ddt, J = 6.7, 4.4, 2.5 Hz, 2H), 3.85 (dt, J = 4.8, 2.5 Hz, 1H), 3.82 (q, J = 1.7 Hz, 1H), 3.80–3.66 (m, 6H), 3.66–3.63 (m, 1H), 3.59–3.33 (m, 7H), 3.08 (dd, J = 10.5, 3.7 Hz, 1H), 2.18 (dt, J = 12.8, 4.4 Hz, 1H), 1.39 (q, J = 12.5 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 109.10, 109.08, 99.92, 98.97, 98.15, 98.08, 85.44, 83.63, 83.18, 77.26, 77.16, 77.13, 77.02, 76.45, 75.68, 75.47, 75.35, 74.26, 73.71, 72.89, 72.77, 72.38, 72.35, 72.07, 72.02, 71.30, 69.74, 65.02, 64.83, 64.80, 63.41, 62.62, 62.57, 62.01, 61.97, 61.66, 61.65, 52.77, 52.57, 33.18, 30.84.
1,2′,2‴,3,6′,6‴-Hexaazido-1,2′,2‴,3, 6′,6‴-hexadeaminoneomycin (2).
To a stirring solution of neomycin B sulfate (5.00 g, 5.5 mmol) in H2O (60 mL) at room temperature were added K2CO3 (9.12 g, 66 mmol) and a catalytic amount of CuSO4·5H2O (0.206 g, 0.825 mmol). The solution was cooled to 0 °C, and 1H-imidazole-1-sulfonyl azide hydrochloride (12.51 g, 66 mmol) was added to the solution in 10 portions at 10 min interval. The reaction mixture was stirred at room temperature for 12 h. The reaction progress was monitored by TLC. The mixture was co-evaporated with MeOH in vacuo at 25 °C. The resulting dark residue was dry-loaded onto silica gel and purified by column chromatography (1:9 MeOH/EtOAc) to afford 2 as a white solid. Isolated yield: 2.81g, 66.3%. 1H NMR (400 MHz, CD3OD) δ 5.81 (d, J = 3.9 Hz, 1H), 5.39 (s, 1H), 5.12 (s, 1H), 4.44–4.37 (m, 1H), 4.31 (s, 1H), 4.19–4.09 (m, 2H), 4.05–3.98 (m, 1H), 3.93 (s, 1H), 3.88 (d, J = 9.6 Hz, 1H), 3.83 (d, J = 9.2 Hz, 1H), 3.67 (td, J = 12.4, 12.0, 4.6 Hz, 5H), 3.53 (d, J = 12.5 Hz, 2H), 3.47–3.39 (m, 5H), 3.37 (d, J = 4.4 Hz, 1H), 3.34 (s, 2H), 3.30 (s, 3H), 3.10 (dd, J = 10.5, 3.5 Hz, 1H), 2.22 (d, J = 12.8 Hz, 1H), 1.38 (q, J = 12.2 Hz, 1H). 13C NMR (151 MHz, d2o) δ 109.83, 96.61, 95.70, 84.78, 81.11, 80.68, 75.07, 73.29, 70.27, 67.88, 67.35, 66.96, 61.68, 61.04, 60.02, 55.59, 50.93, 50.22, 49.13, 41.83, 40.36, 31.00, 27.46, 23.15, 21.95.
1,2′,2‴,3,6‴-Pentaazido-6′,5″-di-O-tert-butyldimethylsilyl-1,2′,2‴,3,6‴-pentadeamino-paromomycin (3).
To a solution of intermediate 1 (4.28 g, 5.74 mmol) in pyridine (28 mL) at 0 °C were added DMAP (0.281 g, 2.29 mmol) and tert-butyl silyl methyl chloride (1.903 g, 12.62 mmol). After the resulting solution was allowed to warm to room temperature for 2 h and monitored by TLC (3:1 EtOAc/hexanes). It was diluted with EtOAc and washed with a saturated aqueous NaHCO3 solution, followed by a saturated aqueous NaCl solution. The resulting organic layer was dried over Na2SO4 and purified by silica gel column chromatography to afford the corresponding product 3 as white crystals. Isolated yield: 78.9%. 1H NMR (500 MHz, CDCl3) δ 5.65 (d, J = 3.7 Hz, 1H), 5.30 (d, J = 3.2 Hz, 1H), 5.17–5.10 (m, 1H), 4.61–4.56 (m, 1H), 4.46 (t, J = 4.7 Hz, 1H), 4.23 (dt, J = 7.2, 3.9 Hz, 2H), 4.14–3.97 (m, 3H), 3.95–3.81 (m, 5H), 3.78 (dd, J = 11.5, 3.4 Hz, 1H), 3.75–3.49 (m, 8H), 3.49–3.31 (m, 5H), 3.13 (dd, J = 10.2, 3.8 Hz, 2H), 2.20 (dq, J = 13.6, 4.5 Hz, 1H), 1.36 (d, J = 12.9 Hz, 1H), 0.92 (d, J = 2.2 Hz, 18H), 0.11 (d, J = 7.3 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 105.40, 98.43, 96.51, 83.87, 82.93, 75.92, 74.81, 74.78, 74.66, 73.93, 73.18, 70.88, 70.73, 69.22, 68.95, 64.38, 62.82, 62.23, 60.67, 60.38, 59.79, 59.16, 51.04, 31.97, 30.82, 25.79, 25.73, 18.30, 18.16, −5.51, −5.65, −5.70.
1,2′,2‴,3,6′,6‴-Hexaazido-5″-O-tert-butyldiphenylsilyl-1,2′,2‴,3, 6′,6‴-hexadeamino-neomycin (4).
To a solution of intermediate 2 (2.8 g, 3.63 mmol) in pyridine (28 mL) at 0 °C were added DMAP (0.177 g, 1.45 mmol) and tert-butyl diphenyl silyl chloride (1.41 mL, 5.45 mmol). After the resulting solution was allowed to warm to room temperature and stirred for 12 h, it was monitored by TLC (3:1 EtOAc/Hexanes). It was diluted with EtOAc and washed with a 1 M HCl solution, followed by a saturated aqueous NaCl solution. The resulting organic layer was dried over Na2SO4 and purified by silica gel column chromatography to afford the corresponding product 4 as a white solid. Isolated yield: 2.47g, 67.3%. Isolated yield: 67%. 1H NMR (400 MHz, CD3OD) δ 7.74–7.66 (m, 4H), 7.43 (qd, J = 2.8, 1.2 Hz, 6H), 5.97 (d, J = 3.9 Hz, 1H), 5.37 (d, J = 2.6 Hz, 1H), 5.08 (d, J = 2.0 Hz, 1H), 4.41–4.34 (m, 2H), 4.25–4.11 (m, 2H), 3.95–3.90 (m, 2H), 3.89–3.82 (m, 2H), 3.82–3.78 (m, 1H), 3.70–3.62 (m, 3H), 3.50 (dd, J = 13.2, 2.5 Hz, 2H), 3.47–3.38 (m, 5H), 3.29–3.26 (m, 1H), 2.97 (dd, J = 10.4, 3.9 Hz, 1H), 2.21 (dt, J = 12.9, 4.2 Hz, 1H), 1.44–1.22 (m, 2H), 1.08 (s, 9H). 13C NMR (101 MHz, CD3OD) δ 136.95, 136.91, 134.63, 134.45, 130.92, 128.91, 128.87, 110.62, 100.20, 98.01, 86.26, 84.36, 78.10, 77.48, 77.45, 75.42, 75.26, 73.01, 72.64, 72.47, 70.93, 69.64, 65.93, 65.19, 61.91, 61.58, 61.22, 52.82, 52.26, 33.08, 27.59, 20.15.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-6′,5″-di-O-tert-butyldimethylsilyl-1,2′,2‴,3,6‴-pentadeamino-paromomycin (5).
Sodium hydride (60% dispersion, 0.786 g, 19.64 mmol) was added in portions to a stirring solution of intermediate 3 (1.06 g, 1.19 mmol) in anhydrous DMF (10 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.322 mg, 0.872 mmol) and 2-(bromomethyl)naphthalene (3.377 g, 15.27 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ethyl acetate (EtOAc), washed with a saturated aqueous NaHCO3 solution, followed by a saturated aqueous NaCl solution, and then dried over sodium sulfate (Na2SO4). The crude residue was purified by silica gel column chromatography to afford the corresponding product 5. Isolated yield: 76.4%. 1H NMR (500 MHz, CDCl3) δ 7.87–7.74 (m, 16H), 7.73–7.65 (m, 7H), 7.63 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.50 (dddd, J = 10.2, 8.4, 5.4, 1.5 Hz, 9H), 7.46–7.38 (m, 10H), 7.32 (ddd, J = 10.2, 8.4, 1.6 Hz, 2H), 7.08 (dd, J = 8.4, 1.7 Hz, 1H), 6.02 (d, J = 3.8 Hz, 1H), 5.65 (d, J = 4.3 Hz, 1H), 5.16 (d, J = 11.6 Hz, 1H), 5.11–5.07 (m, 2H), 5.04 (d, J = 11.4 Hz, 2H), 4.91 (d, J = 11.6 Hz, 1H), 4.88–4.85 (m, 2H), 4.83 (s, 1H), 4.73–4.69 (m, 2H), 4.68 (s, 1H), 4.41 (t, J = 4.3 Hz, 1H), 4.37 (s, 1H), 4.31 (q, J = 3.6 Hz, 1H), 4.23–4.16 (m, 2H), 4.13–4.06 (m, 3H), 3.90 (dd, J = 10.5, 2.9 Hz, 3H), 3.81 (dd, J = 11.4, 3.9 Hz, 1H), 3.76 (ddd, J = 7.4, 4.7, 2.2 Hz, 1H), 3.74–3.63 (m, 5H), 3.59 (t, J = 8.7 Hz, 1H), 3.38 (dd, J = 10.3, 3.8 Hz, 1H), 3.29–3.18 (m, 4H), 3.15–3.09 (m, 2H), 2.15 (dt, J = 13.0, 4.6 Hz, 1H), 1.36 (q, J = 12.5 Hz, 1H), 0.94 (d, J = 9.2 Hz, 19H), 0.10 (d, J = 2.7 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 136.25, 135.80, 135.75, 135.35, 134.63, 134.52, 133.46, 133.45, 133.43, 133.23, 133.20, 133.17, 133.08, 133.07, 133.01, 128.57, 128.48, 128.39, 128.26, 128.19, 128.16, 128.15, 128.09, 128.06, 128.03, 128.00, 127.90, 127.87, 127.82, 127.76, 127.32, 127.14, 126.81, 126.71, 126.56, 126.42, 126.41, 126.39, 126.34, 126.31, 126.21, 126.18, 126.17, 126.14, 126.04, 125.94, 125.91, 125.88, 125.77, 125.72, 125.51, 125.38, 107.18, 98.64, 96.33, 83.66, 83.25, 82.04, 81.79, 80.50, 78.36, 75.68, 75.59, 75.11, 75.05, 74.83, 73.94, 73.43, 73.21, 72.54, 72.52, 72.34, 72.14, 64.02, 63.27, 62.42, 60.46, 59.95, 58.06, 51.30, 32.46, 31.72, 26.17, 26.16, 25.42, 22.79, 21.18, 18.48, 18.44, 14.34, 14.26, −4.90, −4.98, −5.16, −5.29.
1,2′,2‴,3,6′,6‴-Hexaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″-O-tert-butyldiphenylsilyl-1,2′,2‴,3,6′,6‴-hexadeamino-neomycin (6).
Sodium hydride (60% dispersion, 0.394 g, 9.86 mmol) was added in portions to a stirring solution of intermediate 4 (0.83 g, 1.21 mmol) in anhydrous DMF (3 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.304 mg, 0.822 mmol) and 2-(bromomethyl) naphthalene (1.82 g, 8.23 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ethyl acetate (EtOAc), washed with a saturated aqueous NaHCO3 solution, followed by a saturated aqueous NaCl solution, and then dried over sodium sulfate (Na2SO4). The crude residue was purified by silica gel column chromatography to afford the corresponding product 6. Isolated yield: 76%. 1H NMR (400 MHz, CDCl3) δ 7.85–7.79 (m, 6H), 7.78–7.70 (m, 12H), 7.69–7.63 (m, 9H), 7.51–7.46 (m, 9H), 7.41 (ddt, J = 10.8, 5.3, 2.1 Hz, 8H), 7.33 (tdd, J = 4.7, 3.4, 1.7 Hz, 8H), 6.05 (d, J = 3.8 Hz, 1H), 5.62 (d, J = 4.0 Hz, 1H), 5.09 (dd, J = 11.5, 9.7 Hz, 2H), 5.06–4.98 (m, 2H), 4.80 (dd, J = 11.6, 2.4 Hz, 2H), 4.70–4.64 (m, 4H), 4.38 (d, J = 12.0 Hz, 1H), 4.33 (d, J = 3.5 Hz, 2H), 4.30–4.23 (m, 2H), 4.19 (d, J = 12.2 Hz, 1H), 4.13–4.04 (m, 2H), 3.96–3.85 (m, 2H), 3.68 (t, J = 3.4 Hz, 1H), 3.64–3.57 (m, 2H), 3.53–3.46 (m, 2H), 3.45–3.38 (m, 2H), 3.36–3.30 (m, 1H), 3.25–3.20 (m, 1H), 3.18 (q, J = 2.8 Hz, 2H), 3.16–3.12 (m, 2H), 3.09 (dd, J = 9.3, 4.8 Hz, 1H), 2.12 (dt, J = 13.0, 4.6 Hz, 1H), 1.36 (q, J = 12.6 Hz, 1H), 1.27 (s, 1H), 1.08 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 135.79, 135.78, 135.54, 135.52, 135.32, 135.30, 134.61, 134.41, 133.40, 133.31, 133.28, 133.23, 133.11, 133.08, 133.06, 133.00, 132.96, 132.90, 129.84, 129.75, 128.45, 128.28, 128.22, 128.11, 128.08, 128.00, 127.99, 127.97, 127.95, 127.91, 127.85, 127.78, 127.77, 127.73, 127.70, 127.69, 127.67, 127.07, 126.74, 126.70, 126.67, 126.50, 126.44, 126.29, 126.22, 126.17, 126.12, 126.06, 126.05, 126.01, 125.90, 125.88, 125.80, 125.74, 125.67, 125.40, 125.29, 107.35, 98.75, 96.11, 83.32, 83.15, 81.67, 81.28, 80.05, 78.58, 77.21, 76.13, 75.32, 75.11, 74.91, 73.26, 72.48, 72.13, 71.97, 70.85, 63.62, 63.53, 60.31, 59.66, 57.87, 51.52, 50.53, 32.23, 27.01, 19.27.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-paromomycin (7).
A solution of tetrabutylammonium fluoride (TBAF) (1.0 M in THF) (4 equiv) was added dropwise to a solution of intermediate 5 (1.13 g, 0.623 mmol) in anhydrous THF (3 mL) at 0 °C. The reaction progress was monitored by TLC (2:3 EtOAc/hexanes). The reaction mixture was slowly quenched with H2O, diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography to afford product 7 as white crystals. Isolated yield: 81%. 1H NMR (500 MHz, CDCl3) δ 7.88–7.79 (m, 16H), 7.77 (dq, J = 12.0, 3.0 Hz, 5H), 7.73 (d, J = 4.7 Hz, 1H), 7.71 (s, 1H), 7.67 (d, J = 4.2 Hz, 2H), 7.65 (s, 1H), 7.57 (d, J = 1.7 Hz, 1H), 7.56 (d, J = 1.6 Hz, 2H), 7.55–7.52 (m, 7H), 7.52 (d, J = 3.0 Hz, 1H), 7.51–7.48 (m, 6H), 7.48–7.45 (m, 3H), 7.44 (d, J = 1.5 Hz, 1H), 7.35 (ddd, J = 8.5, 3.3, 1.7 Hz, 2H), 7.16 (d, J = 1.7 Hz, 1H), 7.14 (d, J = 1.7 Hz, 1H), 5.95 (d, J = 3.7 Hz, 1H), 5.71 (d, J = 3.8 Hz, 1H), 5.17–5.07 (m, 5H), 4.96–4.92 (m, 3H), 4.76–4.69 (m, 3H), 4.55 (t, J = 5.1 Hz, 1H), 4.42–4.35 (m, 2H), 4.33 (d, J = 12.2 Hz, 1H), 4.29–4.22 (m, 2H), 4.17 (dd, J = 10.1, 3.1 Hz, 1H), 4.06 (dd, J = 5.2, 3.8 Hz, 1H), 3.96 (dd, J = 12.4, 2.5 Hz, 2H), 3.92–3.84 (m, 4H), 3.82–3.76 (m, 3H), 3.75–3.70 (m, 2H), 3.39–3.33 (m, 2H), 3.33–3.24 (m, 4H), 3.22 (t, J = 2.5 Hz, 1H), 3.12 (dd, J = 12.8, 4.2 Hz, 1H), 2.18 (dt, J = 13.2, 4.8 Hz, 1H), 1.41 (q, J = 12.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 171.25, 135.58, 135.47, 135.34, 135.27, 134.50, 134.35, 133.43, 133.41, 133.37, 133.22, 133.20, 133.19, 133.17, 133.15, 133.10, 133.04, 128.63, 128.53, 128.34, 128.32, 128.27, 128.13, 128.10, 128.02, 128.01, 127.97, 127.90, 127.88, 127.85, 127.82, 127.80, 127.77, 127.32, 126.97, 126.82, 126.61, 126.52, 126.47, 126.43, 126.35, 126.34, 126.27, 126.23, 126.15, 126.12, 126.10, 126.02, 125.81, 125.73, 125.51, 125.44, 106.71, 98.90, 96.74, 83.58, 83.17, 81.74, 81.13, 79.30, 77.99, 76.11, 75.43, 75.12, 75.10, 74.96, 74.26, 73.28, 73.16, 72.52, 72.37, 72.15, 71.98, 63.15, 61.92, 61.64, 60.51, 60.13, 59.67, 57.80, 51.36, 32.19, 31.02, 21.16, 14.32.
1,2′,2‴,3,6′,6‴-Hexaazido-2″,3′,3‴,4′,4‴,6-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6′,6‴-hexadeamino-neomycin (8).
A solution of tetrabutylammonium fluoride (TBAF) (1.0 M in THF) (2 equiv) was added dropwise to a solution of intermediate 6 (0.78 g, 0.424 mmol) in anhydrous THF (3 mL) at 0 °C. The reaction progress was monitored by TLC (2:3 EtOAc/hexanes). The reaction mixture was slowly quenched with H2O, diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution and then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography to afford product 8 as white crystals. Isolated yield: 83%. 1H NMR (500 MHz, CDCl3) δ 7.71 (ddt, J = 19.3, 10.8, 5.9 Hz, 15H), 7.57 (dd, J = 12.7, 4.1 Hz, 3H), 7.51 (d, J = 8.2 Hz, 2H), 7.42–7.35 (m, 15H), 7.34–7.29 (m, 4H), 7.24–7.20 (m, 2H), 7.00 (dd, J = 8.4, 1.3 Hz, 1H), 5.83 (d, J = 3.7 Hz, 1H), 5.62 (d, J = 4.4 Hz, 1H), 5.03 (dd, J = 21.9, 11.3 Hz, 3H), 4.98–4.92 (m, 2H), 4.83–4.77 (m, 3H), 4.73 (d, J = 11.5 Hz, 1H), 4.63 (d, J = 3.8 Hz, 2H), 4.36 (t, J = 5.0 Hz, 1H), 4.26 (d, J = 12.3 Hz, 1H), 4.23 (dd, J = 5.1, 2.7 Hz, 1H), 4.20–4.15 (m, 2H), 4.12–4.05 (m, 2H), 3.88 (t, J = 4.9 Hz, 1H), 3.81–3.76 (m, 1H), 3.75–3.65 (m, 3H), 3.62 (d, J = 7.4 Hz, 2H), 3.58–3.52 (m, 2H), 3.49 (dd, J = 13.2, 2.1 Hz, 1H), 3.33 (dd, J = 13.2, 4.9 Hz, 1H), 3.20 (td, J = 9.0, 3.0 Hz, 3H), 3.14 (s, 1H), 3.07 (s, 1H), 2.96 (dd, J = 12.7, 4.3 Hz, 1H), 2.82 (dd, J = 8.2, 5.3 Hz, 1H), 2.12 (dt, J = 12.8, 4.3 Hz, 1H), 1.32 (q, J = 12.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 135.21, 133.07, 128.25, 127.93, 127.72, 127.22, 126.90, 126.69, 126.50, 126.20, 126.05, 125.65, 125.44, 106.32, 98.85, 97.08, 83.83, 83.17, 81.88, 80.86, 79.14, 78.72, 76.59, 76.01, 75.15, 74.91, 74.14, 73.35, 73.04, 72.35, 72.02, 71.85, 71.09, 62.95, 61.91, 60.28, 59.67, 57.64, 51.22, 32.32.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴,5″,6′-octa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-paromomycin (9).
Sodium hydride (60% dispersion, 0.19 g, 8.05 mmol) was added in portions to a stirring solution of intermediate 1 (0.3 g, 0.402 mmol) in anhydrous DMF (4 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.237 g, 0.643 mmol) and 2-(bromomethyl)naphthalene (1.78 g, 8.05 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ice-cold EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The crude residue was purified by silica gel column chromatography to afford the corresponding product 9. Isolated yield: 59%. 1H NMR (500 MHz, CDCl3) δ 7.79–7.55 (m, 24H), 7.55–7.46 (m, 5H), 7.45 (s, 1H), 7.42–7.32 (m, 14H), 7.32–7.27 (m, 6H), 7.27–7.19 (m, 4H), 7.19–7.16 (m, 1H), 7.15–7.10 (m, 2H), 7.10–7.01 (m, 1H), 6.93 (dd, J = 8.4, 1.7 Hz, 1H), 6.06 (d, J = 3.8 Hz, 1H), 5.59 (d, J = 4.9 Hz, 1H), 5.02–4.74 (m, 4H), 4.73 (s, 3H), 4.71–4.45 (m, 8H), 4.38–4.23 (m, 2H), 4.18 (d, J = 12.2 Hz, 1H), 4.16–4.09 (m, 1H), 4.09–3.99 (m, 2H), 3.92 (d, J = 12.3 Hz, 1H), 3.86–3.75 (m, 1H), 3.75–3.48 (m, 6H), 3.44 (dd, J = 12.8, 8.3 Hz, 1H), 3.38–3.28 (m, 1H), 3.24–2.99 (m, 4H), 2.79–2.70 (m, 1H), 2.00 (dt, J = 13.1, 4.6 Hz, 1H), 1.24–1.00 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 138.48, 136.01, 135.88, 135.82, 135.77, 135.74, 135.24, 134.60, 134.46, 133.52, 133.44, 133.40, 133.38, 133.32, 133.22, 133.20, 133.18, 133.16, 133.11, 133.09, 133.06, 133.02, 133.01, 132.99, 128.57, 128.49, 128.48, 128.33, 128.32, 128.29, 128.21, 128.19, 128.15, 128.14, 128.07, 128.05, 128.02, 128.00, 127.99, 127.91, 127.87, 127.85, 127.82, 127.79, 127.77, 127.75, 127.34, 126.95, 126.80, 126.59, 126.55, 126.47, 126.44, 126.41, 126.33, 126.29, 126.26, 126.24, 126.21, 126.15, 126.12, 126.10, 126.05, 126.03, 126.00, 125.99, 125.92, 125.88, 125.84, 125.77, 125.64, 125.56, 125.48, 125.30, 106.85, 98.71, 96.28, 83.96, 82.23, 82.11, 81.94, 80.41, 78.44, 75.67, 75.47, 75.25, 75.09, 74.79, 74.32, 73.61, 73.44, 73.40, 73.06, 72.38, 72.07, 72.01, 71.18, 70.42, 68.97, 65.62, 63.51, 60.32, 60.06, 57.65, 51.10, 32.48.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴,5″,6′-octa-O-benzyl-1,2′,2‴,3,6‴-pentadeamino-paromomycin (10).
Sodium hydride (60% dispersion, 0.11 g, 4.83 mmol) was added in portions to a stirring solution of intermediate 1 (0.2 g, 0.268 mmol) in anhydrous DMF (3 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.08 g, 0.214 mmol) and (bromomethyl)benzene (0.44 mL, 3.76 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 5 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ice-cold EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The crude residue was purified by silica gel column chromatography to afford the corresponding product 10. Isolated yield: 68%. 1H NMR (400 MHz, CDCl3) δ 7.37–7.31 (m, 10H), 7.28 (ddd, J = 12.2, 7.5, 4.9 Hz, 17H), 7.24–7.16 (m, 8H), 7.16 (s, 4H), 4.86–4.81 (m, 3H), 4.81–4.65 (m, 2H), 4.65–4.55 (m, 3H), 4.55–4.48 (m, 2H), 4.48–4.36 (m, 3H), 4.34–4.23 (m, 4H), 4.23–4.00 (m, 2H), 3.99–3.85 (m, 2H), 3.81–3.63 (m, 5H), 3.63–3.47 (m, 3H), 3.47–3.30 (m, 3H), 3.17–3.05 (m, 2H), 1.27–1.22 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 134.42, 134.40, 134.33, 134.16, 134.02, 133.72, 133.14, 133.07, 124.74, 124.63, 124.57, 124.47, 124.45, 124.44, 124.42, 124.38, 124.33, 124.23, 124.18, 123.92, 123.91, 123.82, 123.79, 123.75, 123.70, 123.68, 123.63, 123.61, 123.58, 123.55, 123.53, 123.04, 102.24, 94.71, 92.18, 80.06, 78.29, 78.02, 77.87, 76.33, 74.25, 73.43, 73.12, 72.80, 71.63, 71.41, 71.09, 71.05, 70.70, 70.27, 69.33, 69.28, 69.09, 68.49, 67.87, 67.66, 67.16, 66.21, 64.94, 61.41, 59.37, 56.46, 56.03, 53.43, 47.08, 28.47.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴,5″,6′-octa-O-(1,1′-biphenyl-4-methyl)-1,2′,2‴,3,6‴-pentadeamino-paromomycin (11).
Sodium hydride (60% dispersion, 0.58 g, 24.14 mmol) was added in portions to a stirring solution of intermediate 1 (1.0 g, 1.34 mmol) in anhydrous DMF (13 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.39 g, 1.07 mmol) and 4-(bromomethyl)-1,1′-biphenyl (4.64g, 18.78 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ice-cold EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The crude residue was purified by silica gel column chromatography to afford the corresponding product 11. Isolated yield: 55%. 1H NMR (600 MHz, CDCl3) δ 7.64–7.55 (m, 19H), 7.55–7.52 (m, 10H), 7.47 (ddt, J = 20.9, 8.3, 2.1 Hz, 22H), 7.44–7.32 (m, 27H), 7.25–7.19 (m, 3H), 7.07 (d, J = 7.9 Hz, 2H), 6.22 (d, J = 3.6 Hz, 1H), 5.75 (d, J = 5.7 Hz, 1H), 5.08–5.00 (m, 2H), 4.98–4.82 (m, 3H), 4.80–4.53 (m, 12H), 4.48 (d, J = 11.5 Hz, 1H), 4.42 (ddd, J = 13.6, 5.2, 2.7 Hz, 2H), 4.39–4.27 (m, 2H), 4.24 (dt, J = 9.9, 3.3 Hz, 1H), 4.19–4.05 (m, 3H), 3.95–3.84 (m, 2H), 3.83–3.73 (m, 4H), 3.57–3.45 (m, 2H), 3.37–3.25 (m, 2H), 3.13 (t, J = 2.5 Hz, 1H), 3.07 (dd, J = 10.3, 3.7 Hz, 1H), 2.97 (dd, J = 12.9, 4.0 Hz, 1H), 2.25 (dq, J = 12.6, 4.2, 3.7 Hz, 1H), 1.40 (q, J = 12.8 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 141.18, 141.11, 141.08, 141.05, 141.04, 140.93, 140.92, 140.81, 140.79, 140.78, 140.75, 140.71, 140.67, 140.66, 140.64, 140.51, 140.49, 140.46, 140.12, 140.05, 137.49, 137.43, 137.34, 137.31, 137.23, 136.70, 136.10, 135.99, 128.97, 128.94, 128.92, 128.91, 128.90, 128.88, 128.88, 128.87, 128.85, 128.84, 128.83, 128.81, 128.77, 128.69, 128.55, 128.50, 128.41, 128.38, 128.37, 128.30, 128.23, 128.08, 127.94, 127.90, 127.61, 127.55, 127.45, 127.43, 127.42, 127.39, 127.35, 127.33, 127.31, 127.28, 127.24, 127.22, 127.20, 127.18, 127.16, 127.15, 127.14, 127.13, 127.11, 127.05, 106.32, 98.85, 96.18, 84.38, 82.62, 82.17, 82.11, 80.38, 78.42, 77.37, 77.16, 76.95, 75.66, 75.20, 74.97, 74.69, 74.51, 74.41, 73.28, 73.19, 73.10, 73.02, 72.16, 72.01, 71.67, 71.42, 71.15, 70.46, 68.86, 65.17, 63.39, 60.50, 60.44, 60.18, 57.53, 51.23, 32.59, 31.70, 22.77, 21.15, 14.31, 14.25.
1,2′,2‴,3,6′,6‴-Hexaazido-2″,3′,3‴,4′,4‴,6-hexa-O-(1,1′-biphenyl-4-methyl)-5″-O-tert-butyldiphenylsilyl-1,2′,2‴,3,6′,6‴-hexadeamino-neomycin (12).
Sodium hydride (60% dispersion, 0.6 g, 14.68 mmol) was added in portions to a stirring solution of intermediate 4 (1.26 g, 1.25 mmol) in anhydrous DMF (13 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.455 g, 1.25 mmol) and 4-(bromomethyl)-1,1′-biphenyl (3.04g, 12.29 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ice-cold EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The crude residue was purified by silica gel column chromatography to afford the corresponding product 12. Isolated yield: 66%. 1H NMR (500 MHz, CDCl3) δ 7.68 (ddt, J = 5.9, 4.3, 2.3 Hz, 3H), 7.58 (tdd, J = 11.0, 6.6, 3.2 Hz, 19H), 7.50–7.43 (m, 20H), 7.40–7.34 (m, 21H), 7.11 (d, J = 2.1 Hz, 1H), 6.13 (d, J = 3.9 Hz, 1H), 5.70 (d, J = 5.0 Hz, 1H), 5.07–5.00 (m, 1H), 4.95 (d, J = 11.9 Hz, 2H), 4.89 (d, J = 11.0 Hz, 1H), 4.80 (d, J = 2.1 Hz, 1H), 4.79–4.72 (m, 2H), 4.68 (d, J = 11.5 Hz, 1H), 4.66–4.62 (m, 2H), 4.56 (d, J = 11.8 Hz, 1H), 4.36–4.30 (m, 4H), 4.28–4.24 (m, 1H), 4.21 (d, J = 12.1 Hz, 1H), 4.08–4.03 (m, 2H), 3.96–3.89 (m, 2H), 3.85 (t, J = 8.9 Hz, 1H), 3.73 (dd, J = 6.6, 3.3 Hz, 2H), 3.67 (td, J = 6.8, 2.3 Hz, 1H), 3.53 (dd, J = 13.1, 2.4 Hz, 1H), 3.45 (td, J = 9.5, 8.8, 4.3 Hz, 3H), 3.41 (t, J = 4.2 Hz, 1H), 3.37 (dd, J = 11.7, 4.7 Hz, 1H), 3.32 (d, J = 9.4 Hz, 1H), 3.29 (d, J = 6.5 Hz, 1H), 3.28–3.26 (m, 1H), 3.20 (t, J = 2.7 Hz, 1H), 3.06 (dd, J = 10.3, 3.8 Hz, 1H), 2.27 (dt, J = 13.1, 4.6 Hz, 1H), 1.48 (q, J = 12.7 Hz, 1H), 1.12 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 141.24, 141.07, 140.97, 140.93, 140.85, 140.78, 140.69, 140.55, 140.21, 137.24, 137.21, 137.05, 136.86, 136.31, 136.10, 135.97, 135.94, 133.50, 133.21, 130.08, 129.98, 128.97, 128.92, 128.89, 128.87, 128.59, 128.43, 128.34, 128.28, 128.03, 127.94, 127.81, 127.63, 127.53, 127.48, 127.39, 127.36, 127.33, 127.31, 127.27, 127.24, 127.21, 127.17, 127.14, 106.79, 99.14, 96.21, 83.99, 83.39, 82.24, 81.86, 80.21, 78.77, 77.36, 76.02, 75.62, 75.12, 74.91, 74.76, 73.38, 73.34, 72.76, 72.27, 71.70, 71.57, 71.02, 63.83, 63.48, 60.63, 59.99, 57.75, 51.67, 50.63, 32.47, 27.23, 19.43.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(1,1′-biphenyl-4-methyl)-6′,5″-di-O-tert-butyldimethylsilyl-1,2′,2‴,3,6‴-pentadeamino-paromomycin (13).
Sodium hydride (60% dispersion, 0.10 g, 4.05 mmol) was added in portions to a stirring solution of intermediate 3 (0.219 g, 0.225 mmol) in anhydrous DMF (2.25 mL) at 0 °C under argon. After the reaction mixture had been stirred at 0 °C for 30 min, tetrabutylammonium iodide (TBAI) (0.06 g, 0.180 mmol) and 4-(bromomethyl)-1,1′-biphenyl (0.78g, 3.15 mmol) were sequentially added to the reaction mixture. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:4 EtOAc/hexanes) and stopped upon completion. The reaction mixture was diluted with ice-cold EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The crude residue was purified by silica gel column chromatography to afford the corresponding product 13. Isolated yield: 81%. 1H NMR (600 MHz, CDCl3) δ 7.60–7.49 (m, 21H), 7.49–7.28 (m, 43H), 7.25 (d, J = 9.1 Hz, 14H), 7.07–7.02 (m, 2H), 6.04 (d, J = 3.7 Hz, 1H), 5.67 (d, J = 5.3 Hz, 1H), 5.04–4.98 (m, 2H), 4.96–4.87 (m, 4H), 4.76 (dd, J = 11.4, 9.2 Hz, 2H), 4.70–4.56 (m, 4H), 4.32–4.24 (m, 4H), 4.17–4.09 (m, 3H), 3.94–3.83 (m, 5H), 3.83–3.71 (m, 4H), 3.71–3.58 (m, 4H), 3.50–3.41 (m, 2H), 3.36–3.24 (m, 4H), 3.21–3.12 (m, 2H), 2.25 (dt, J = 13.0, 4.6 Hz, 1H), 1.43 (q, J = 12.7 Hz, 1H), 0.92 (dd, J = 9.3, 2.5 Hz, 21H), 0.11–0.08 (m, 13H). 13C NMR (151 MHz, CDCl3) δ 141.07, 140.71, 140.51, 137.49, 137.42, 136.78, 136.09, 129.00, 128.96, 128.94, 128.92, 128.90, 128.88, 128.85, 128.40, 128.24, 128.03, 127.66, 127.62, 127.56, 127.50, 127.36, 127.31, 127.28, 127.25, 127.22, 127.19, 127.15, 127.08, 106.45, 98.89, 96.21, 84.17, 83.10, 82.39, 80.54, 78.38, 77.37, 77.16, 76.95, 75.29, 74.54, 73.94, 73.04, 72.56, 72.24, 71.46, 65.66, 63.50, 62.33, 60.60, 60.15, 57.78, 51.29, 37.98, 32.57, 31.73, 26.20, 22.80, 18.49, 18.43, 14.64, 12.14.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4′,4‴,6-hexa-O-(1,1′-biphenyl-4-methyl)-1,2′,2‴,3,6′,6‴-pentadeamino-paromomycin (14).
A solution of tetrabutylammonium fluoride (TBAF) (1.0 M in THF) (4 equiv) was added dropwise to a solution of intermediate 13 (0.36 g, 0.182 mmol) in anhydrous THF (1 mL) at 0 °C. The reaction progress was monitored by TLC (2:3 EtOAc/hexanes). The reaction mixture was slowly quenched with H2O, diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution and then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography to afford product 14. Isolated yield: 82%. 1H NMR (400 MHz, CDCl3) δ 7.63–7.50 (m, 17H), 7.50–7.29 (m, 34H), 7.26 (d, J = 4.1 Hz, 5H), 7.13–7.07 (m, 2H), 5.92 (d, J = 3.7 Hz, 1H), 5.69 (d, J = 4.6 Hz, 1H), 5.05–4.89 (m, 5H), 4.83 (d, J = 11.0 Hz, 1H), 4.76 (d, J = 11.3 Hz, 1H), 4.66–4.58 (m, 3H), 4.47 (t, J = 4.8 Hz, 1H), 4.34 (dd, J = 18.1, 12.1 Hz, 2H), 4.27–4.05 (m, 4H), 3.96 (t, J = 8.4 Hz, 1H), 3.92–3.84 (m, 4H), 3.82 (d, J = 3.7 Hz, 1H), 3.81–3.72 (m, 4H), 3.67 (dd, J = 10.0, 8.8 Hz, 1H), 3.51 (tdd, J = 11.3, 9.3, 4.8 Hz, 2H), 3.40 (t, J = 9.1 Hz, 1H), 3.34 (d, J = 2.7 Hz, 1H), 3.23 (dd, J = 10.4, 3.8 Hz, 1H), 3.17 (d, J = 2.7 Hz, 1H), 3.11 (dd, J = 12.8, 4.2 Hz, 1H), 2.30 (dt, J = 13.2, 4.9 Hz, 1H), 1.53–1.40 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 138.60, 138.24, 138.13, 137.95, 137.86, 137.75, 134.44, 134.27, 134.05, 133.36, 133.22, 126.24, 126.21, 126.18, 126.14, 125.94, 125.64, 125.56, 125.53, 125.35, 124.93, 124.86, 124.81, 124.71, 124.66, 124.63, 124.55, 124.51, 124.47, 124.42, 124.39, 103.44, 96.29, 94.19, 81.32, 80.47, 79.60, 78.60, 76.45, 74.74, 74.43, 74.11, 73.27, 72.40, 72.19, 72.10, 71.67, 70.52, 70.32, 69.66, 69.55, 69.02, 68.95, 60.31, 59.33, 57.56, 57.07, 54.95, 48.64, 29.59.
1,2′,2‴,3,6′,6‴-Hexazido-2″,3′,3‴,4′,4‴,6-hexa-O-(1,1′-biphenyl-4-methyl)-1,2′,2‴,3,6′,6‴-Hexadeamino-neomycin (15).
A solution of tetrabutylammonium fluoride (TBAF) (1.0 M in THF) (4 equiv) was added dropwise to a solution of intermediate 12 (0.38 g, 0.187 mmol) in anhydrous THF (1 mL) at 0 °C. The reaction progress was monitored by TLC (2:3 EtOAc/hexanes). The reaction mixture was slowly quenched with H2O, diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution and then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography to afford product 15. Isolated yield 82%. 1H NMR (500 MHz, CDCl3) δ 7.65–7.52 (m, 18H), 7.52–7.32 (m, 37H), 7.11 (d, J = 8.1 Hz, 2H), 5.98 (d, J = 3.7 Hz, 1H), 5.77 (d, J = 5.4 Hz, 1H), 5.07–4.91 (m, 5H), 4.83 (d, J = 10.9 Hz, 1H), 4.68 (dd, J = 21.0, 12.2 Hz, 4H), 4.49–4.44 (m, 1H), 4.40–4.29 (m, 3H), 4.22 (d, J = 12.2 Hz, 2H), 4.19–4.14 (m, 1H), 4.01 (t, J = 9.0 Hz, 1H), 3.95–3.90 (m, 1H), 3.90–3.86 (m, 2H), 3.82–3.69 (m, 4H), 3.62–3.56 (m, 2H), 3.56–3.37 (m, 5H), 3.34 (s, 1H), 3.23 (dd, J = 10.4, 3.7 Hz, 1H), 3.18 (s, 1H), 3.11 (dd, J = 12.8, 4.3 Hz, 1H), 3.04 (dd, J = 7.9, 5.6 Hz, 1H), 1.51 (q, J = 12.7 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 141.16, 141.06, 140.91, 140.72, 140.56, 140.35, 136.78, 136.74, 136.66, 135.98, 135.84, 128.82, 128.79, 128.75, 128.55, 128.20, 128.14, 127.99, 127.51, 127.45, 127.37, 127.29, 127.21, 127.13, 127.07, 127.02, 105.59, 98.93, 97.22, 84.32, 83.18, 82.37, 81.00, 78.92, 78.79, 75.79, 75.00, 74.81, 74.74, 74.28, 73.03, 72.08, 71.54, 71.48, 71.07, 67.99, 62.73, 62.03, 60.38, 59.76, 57.44, 51.19, 43.35, 30.93.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(N-1,1′-biphenyl-4-yl)-paromomycindicarboxamide (16).
To a solution of intermediate 7 (0.580 g, 0.357 mmol) in DCM/H2O (1:1) (3 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (22.37 mg, 0.143 mmol) and iodobenzene diacetate (BAIB) (576.5 mg, 1.79 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with saturated aqueous NaHCO3 solution and saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude carboxylate in anhydrous DCM (4 mL), oxalyl chloride (0.042 mL, 0.495 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (3 mL). [1,1′-Biphenyl]-4-amine (0.159g, 0.94 mmol) and triethylamine (0.013 mL, 0.940 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product 16. Isolated yield: 72%. 1H NMR (600 MHz, CDCl3) δ 8.67 (s, 1H), 8.13 (s, 1H), 7.83 (ddt, J = 13.3, 7.9, 3.9 Hz, 4H), 7.81–7.66 (m, 22H), 7.66–7.62 (m, 3H), 7.62–7.56 (m, 8H), 7.56–7.36 (m, 34H), 7.36–7.26 (m, 5H), 7.25–7.16 (m, 2H), 5.91 (d, J = 3.8 Hz, 1H), 5.78 (d, J = 4.1 Hz, 1H), 5.09–4.88 (m, 9H), 4.75 (t, J = 10.5 Hz, 2H), 4.71–4.62 (m, 1H), 4.54 (d, J = 11.8 Hz, 1H), 4.51–4.39 (m, 4H), 4.25 (t, J = 4.4 Hz, 1H), 4.17–4.05 (m, 1H), 3.91 (ddd, J = 8.1, 5.4, 2.5 Hz, 1H), 3.86–3.79 (m, 2H), 3.75–3.68 (m, 2H), 3.66 (t, J = 9.0 Hz, 1H), 3.44 (dd, J = 10.1, 3.8 Hz, 1H), 3.42–3.34 (m, 2H), 3.34–3.30 (m, 2H), 3.26 (ddt, J = 18.1, 12.5, 5.0 Hz, 3H), 2.27 (dq, J = 15.7, 6.6, 5.7 Hz, 1H), 1.52 (q, J = 12.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 164.98, 137.89, 137.80, 134.77, 134.15, 132.66, 132.53, 131.94, 131.77, 130.69, 130.64, 130.55, 130.50, 130.41, 126.30, 126.25, 126.15, 125.98, 125.86, 125.80, 125.66, 125.62, 125.45, 125.35, 125.21, 125.19, 125.09, 124.98, 124.59, 124.57, 124.49, 124.34, 124.29, 124.26, 124.24, 123.91, 123.84, 123.77, 123.67, 123.48, 123.41, 123.36, 123.02, 122.89, 117.63, 117.55, 117.48, 104.58, 97.05, 95.07, 80.76, 79.56, 79.46, 77.35, 76.83, 76.57, 75.66, 74.71, 74.50, 74.29, 72.78, 72.63, 71.29, 70.85, 70.54, 70.39, 69.88, 69.62, 60.20, 58.04, 56.58, 56.14, 48.30, 29.44, 27.16.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(N-1-naphthyl)-paromomycindicarboxamide (17).
To a solution of intermediate 7 (0.580 g, 0.357 mmol) in DCM/H2O (1:1) (3 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (22.37 mg, 0.143 mmol) and iodobenzene diacetate (BAIB) (576.5 mg, 1.79 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude carboxylate in anhydrous DCM (4 mL), oxalyl chloride (0.042 mL, 0.495 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (3 mL). Naphthalene-1-amine (0.052g, 0.309 mmol) and triethylamine (0.043 mL, 0.309 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product S17. Isolated yield: 74.5%. 1H NMR (600 MHz, CDCl3) δ 10.02 (s, 1H), 9.24 (s, 1H), 8.36 (s, 1H), 8.25 (d, J = 7.4 Hz, 1H), 8.20 (t, J = 7.2 Hz, 1H), 8.05 (t, J = 7.6 Hz, 2H), 8.02 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.85 (dd, J = 12.7, 7.9 Hz, 2H), 7.80 (dd, J = 11.0, 7.4 Hz, 6H), 7.75 (dq, J = 9.0, 5.4, 4.3 Hz, 4H), 7.71 (td, J = 9.8, 9.4, 4.6 Hz, 13H), 7.66 (d, J = 5.5 Hz, 2H), 7.66–7.62 (m, 3H), 7.62–7.60 (m, 1H), 7.58 (ddd, J = 10.5, 7.2, 2.8 Hz, 5H), 7.56 (d, J = 3.4 Hz, 1H), 7.53 (s, 2H), 7.52–7.49 (m, 2H), 7.49–7.36 (m, 20H), 7.32 (ddd, J = 14.5, 8.1, 4.7 Hz, 3H), 7.29–7.27 (m, 1H), 7.27–7.23 (m, 2H), 7.21 (dd, J = 8.5, 1.7 Hz, 1H), 5.92–5.85 (m, 1H), 5.80 (d, J = 3.9 Hz, 1H), 5.19 (d, J = 2.9 Hz, 1H), 5.02 (dd, J = 10.9, 2.3 Hz, 2H), 4.98 (d, J = 11.1 Hz, 1H), 4.96–4.85 (m, 3H), 4.81 (d, J = 11.0 Hz, 1H), 4.71 (d, J = 12.0 Hz, 1H), 4.67–4.61 (m, 2H), 4.53–4.37 (m, 5H), 4.34–4.29 (m, 2H), 4.16–4.07 (m, 2H), 3.88 (dd, J = 10.3, 7.2 Hz, 2H), 3.81 (t, J = 4.1 Hz, 1H), 3.75–3.66 (m, 2H), 3.50 (dd, J = 10.2, 3.9 Hz, 1H), 3.43 (t, J = 9.4 Hz, 1H), 3.31–3.27 (m, 1H), 3.27–3.24 (m, 2H), 3.21 (t, J = 9.5 Hz, 1H), 3.10 (dp, J = 10.0, 6.9, 4.6 Hz, 2H), 2.23–2.15 (m, 1H), 1.42 (q, J = 12.7 Hz, 1H).13C NMR (151 MHz, CDCl3) δ 168.20, 135.31, 135.10, 134.69, 134.56, 134.19, 133.39, 133.31, 133.27, 133.22, 133.19, 133.13, 131.96, 131.81, 130.98, 129.05, 128.96, 128.91, 128.61, 128.50, 128.32, 128.26, 128.16, 128.08, 128.03, 127.87, 127.80, 127.72, 127.22, 127.11, 126.98, 126.90, 126.66, 126.55, 126.52, 126.47, 126.40, 126.35, 126.30, 126.27, 126.13, 126.02, 125.95, 125.84, 125.62, 125.52, 120.54, 120.40, 120.29, 120.21, 119.35, 119.02, 108.05, 99.78, 98.22, 84.16, 82.16, 81.85, 80.18, 80.12, 79.77, 79.65, 79.08, 75.57, 75.45, 73.90, 73.67, 73.26, 73.16, 72.66, 72.54, 63.28, 60.63, 59.41, 59.16, 51.03, 32.35.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(N-phenyl)-paromomycindicarboxamide (18).
To a solution of intermediate 7 (0.580 g, 0.357 mmol) in DCM/H2O (1:1) (3 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (22.37 mg, 0.143 mmol) and iodobenzene diacetate (BAIB) (576.5 mg, 1.79 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude carboxylate in anhydrous DCM (1.38 mL), oxalyl chloride (0.189 mL, 2.21 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (0.6 mL). Aniline (0.075 mL, 0.828 mmol) and triethylamine (0.115 mL, 0.828 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product 18. Isolated yield: 43%. 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 8.3 Hz, 1H), 7.96 (d, J = 14.9 Hz, 1H), 7.79–7.12 (m, 41H), 7.15–7.00 (m, 3H), 7.04–6.91 (m, 1H), 6.68 (t, J = 7.4 Hz, 0H), 6.60 (d, J = 7.8 Hz, 1H), 5.76 (dd, J = 10.5, 4.0 Hz, 1H), 5.65 (d, J = 4.2 Hz, 1H), 4.95–4.81 (m, 5H), 4.84–4.71 (m, 3H), 4.68–4.55 (m, 2H), 4.44–4.22 (m, 4H), 4.14–3.93 (m, 2H), 3.85–3.64 (m, 3H), 3.61–3.53 (m, 2H), 3.56–3.45 (m, 1H), 3.38–3.24 (m, 1H), 3.25–3.15 (m, 4H), 3.15 (s, 0H), 3.12 (dt, J = 13.9, 7.0 Hz, 2H), 2.15 (dq, J = 9.0, 5.9, 4.6 Hz, 1H), 1.46–1.32 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 167.58, 166.52, 146.34, 138.86, 137.45, 137.28, 135.26, 135.12, 134.91, 134.90, 134.53, 134.37, 133.93, 133.29, 133.27, 133.22, 133.13, 133.12, 133.09, 133.08, 133.05, 133.00, 132.97, 130.76, 129.31, 129.18, 129.10, 129.06, 128.54, 128.41, 128.35, 128.21, 128.20, 128.08, 128.04, 128.02, 128.00, 127.92, 127.91, 127.78, 127.77, 127.74, 127.69, 127.66, 127.58, 127.13, 127.11, 127.01, 126.86, 126.82, 126.79, 126.47, 126.38, 126.33, 126.31, 126.22, 126.20, 126.04, 126.02, 125.98, 125.93, 125.63, 125.47, 124.67, 124.57, 124.47, 123.36, 120.02, 119.86, 119.74, 118.60, 115.15, 107.47, 99.61, 97.67, 83.42, 82.26, 82.16, 80.71, 79.94, 79.51, 79.20, 78.30, 75.39, 75.32, 73.81, 73.48, 73.10, 72.95, 72.43, 72.35, 72.17, 62.91, 60.63, 60.41, 59.20, 58.67, 50.83, 32.08, 29.72.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(1,1′-biphenyl-4-methyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(N-1,1′-biphenyl-4-yl)-paromomycindicarboxamide (19).
To a solution of intermediate 14 (0.15 g, 0.086 mmol) in DCM/H2O (1:1) (0.5 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (5.8 mg, 0.034 mmol) and iodobenzene diacetate (BAIB) (138 mg, 0.043 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH:DCM). The reaction mixture was diluted with DCM, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude carboxylate in anhydrous DCM (0.4 mL), oxalyl chloride (0.064 mL, 0.069 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (0.6 mL). [1,1′-Biphenyl]-4-amine (0.072 mg, 0.430 mmol) and triethylamine (0.059 mL, 0.430 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc:Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product S19. Isolated yield: 68%. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.16 (s, 1H), 7.72–7.62 (m, 5H), 7.60–7.50 (m, 36H), 7.46–7.33 (m, 36H), 7.29–7.22 (m, 3H), 6.84–6.72 (m, 3H), 6.01 (d, J = 3.9 Hz, 1H), 5.88 (d, J = 4.9 Hz, 1H), 5.11 (d, J = 2.0 Hz, 1H), 5.05 (d, J = 2.2 Hz, 1H), 5.01–4.90 (m, 3H), 4.88–4.81 (m, 3H), 4.80–4.66 (m, 3H), 4.59–4.38 (m, 6H), 4.23 (t, J = 4.7 Hz, 1H), 4.19–4.06 (m, 3H), 4.06–3.97 (m, 2H), 3.89 (t, J = 3.4 Hz, 1H), 3.86–3.71 (m, 4H), 3.56–3.44 (m, 4H), 3.39 (dd, J = 10.2, 3.9 Hz, 1H), 3.36–3.24 (m, 2H), 2.39 (dt, J = 13.3, 4.3 Hz, 1H), 2.35 (s, 1H), 1.63 (q, J = 12.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 167.41, 166.51, 145.88, 141.25, 141.18, 141.11, 140.97, 140.85, 140.84, 140.82, 140.71, 140.63, 140.56, 140.44, 140.42, 140.40, 137.48, 137.35, 136.80, 136.79, 136.66, 136.59, 136.53, 136.34, 136.07, 135.89, 131.56, 128.88, 128.87, 128.85, 128.82, 128.81, 128.80, 128.76, 128.69, 128.53, 128.52, 128.28, 128.24, 128.02, 127.68, 127.60, 127.56, 127.52, 127.48, 127.36, 127.28, 127.26, 127.24, 127.22, 127.20, 127.18, 127.12, 127.11, 127.09, 127.06, 127.05, 126.87, 126.86, 126.40, 126.27, 120.22, 120.12, 115.44, 106.55, 99.83, 97.65, 83.41, 82.63, 82.40, 81.60, 79.88, 79.47, 79.00, 77.91, 75.08, 74.98, 74.80, 73.94, 73.47, 72.75, 72.56, 72.18, 71.96, 62.74, 60.83, 60.42, 59.23, 58.49, 50.90.
1,2′,2‴,3,6′,6‴-Hexaazido-2″,3′,3‴,4′,4‴,6-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6′,6‴-hexadeamino-5″-(N-1,1′-biphenyl-4-yl)-neomycincarboxamide (20).
To a solution of intermediate 8 (0.28 g, 0.175 mmol) in DCM/H2O (1:1) (0.5 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (5.5 mg, 0.035 mmol) and iodobenzene diacetate (BAIB) (141 mg, 0.043 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with saturated aqueous NaHCO3 solution and saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude carboxylate in anhydrous DCM (0.4 mL), oxalyl chloride (0.056 mL, 0.065 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (0.6 mL). [1,1′-Biphenyl]-4-amine (0.109g, 0.649 mmol) and triethylamine (0.06 mL, 0.44 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product 20. Isolated yield: 72% over three steps. 1H NMR (500 MHz, CDCl3) δ 8.70 (s, 1H), 7.84–7.64 (m, 24H), 7.57 (d, J = 7.4 Hz, 2H), 7.52–7.38 (m, 22H), 7.34–7.31 (m, 2H), 7.21 (dd, J = 8.4, 1.8 Hz, 1H), 5.80 (d, J = 3.9 Hz, 1H), 5.75 (d, J = 4.4 Hz, 1H), 5.02 (td, J = 10.6, 7.4 Hz, 4H), 4.97–4.92 (m, 2H), 4.88 (d, J = 11.0 Hz, 1H), 4.77 (d, J = 11.5 Hz, 1H), 4.71 (d, J = 12.0 Hz, 1H), 4.61 (dd, J = 4.8, 2.7 Hz, 1H), 4.53 (d, J = 11.8 Hz, 1H), 4.42 (ddd, J = 29.0, 12.0, 3.1 Hz, 4H), 4.29 (ddd, J = 10.1, 5.2, 2.5 Hz, 1H), 4.20 (t, J = 4.6 Hz, 1H), 4.07 (dd, J = 10.3, 8.8 Hz, 1H), 3.87 (ddd, J = 8.3, 5.6, 2.8 Hz, 1H), 3.79 (t, J = 4.0 Hz, 1H), 3.71–3.63 (m, 2H), 3.60–3.50 (m, 3H), 3.40 (dd, J = 13.2, 5.2 Hz, 1H), 3.37–3.32 (m, 2H), 3.28 (dt, J = 16.9, 4.4 Hz, 3H), 3.25–3.17 (m, 2H), 2.22 (dt, J = 13.1, 4.6 Hz, 1H), 1.47 (q, J = 12.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 167.61, 140.48, 137.34, 136.63, 135.27, 135.21, 134.97, 134.94, 134.59, 134.45, 133.29, 133.25, 133.13, 133.12, 133.10, 133.07, 133.04, 132.99, 132.98, 128.72, 128.50, 128.39, 128.33, 128.29, 128.23, 128.17, 128.07, 128.06, 128.02, 127.98, 127.92, 127.77, 127.76, 127.72, 127.67, 127.65, 127.61, 127.11, 127.06, 126.92, 126.86, 126.85, 126.76, 126.53, 126.42, 126.34, 126.29, 126.22, 126.18, 126.15, 126.05, 126.04, 125.94, 125.68, 125.65, 125.49, 120.01, 107.40, 99.63, 97.35, 83.50, 82.26, 82.16, 80.89, 79.85, 79.15, 78.67, 77.49, 75.36, 75.13, 73.79, 73.60, 73.13, 73.01, 72.46, 71.28, 63.42, 60.76, 60.40, 59.36, 58.88, 51.27, 50.83, 32.23, 25.63, 21.06.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(O-benzyl)-paromomycindicarboxylate (21).
To a solution of intermediate 7 (0.15 g, 0.086 mmol) in DCM/H2O (1:1) (0.5 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (5.8 mg, 0.034 mmol) and iodobenzene diacetate (BAIB) (138 mg, 0.043 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude carboxylate in anhydrous DCM (0.4 mL), oxalyl chloride (0.064 mL, 0.069 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in DMF (0.6 mL), and potassium carbonate (182 mg, 1.32 mmol) and benzyl bromide (0.183 mL, 1.54 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product S21. Isolated yield: 54%. 1H NMR (500 MHz, CDCl3) δ 7.75–7.56 (m, 18H), 7.55–7.49 (m, 2H), 7.46 (dd, J = 5.9, 1.6 Hz, 2H), 7.43–7.31 (m, 14H), 7.30–7.24 (m, 5H), 7.23–7.16 (m, 7H), 7.15–7.12 (m, 3H), 7.10 (dd, J = 8.4, 1.7 Hz, 1H), 6.99 (dd, J = 8.4, 1.7 Hz, 1H), 6.20 (d, J = 3.8 Hz, 1H), 5.69 (d, J = 5.1 Hz, 1H), 5.10 (d, J = 8.9 Hz, 3H), 5.00 (dd, J = 11.8, 6.9 Hz, 2H), 4.96–4.84 (m, 2H), 4.82–4.71 (m, 5H), 4.64–4.45 (m, 4H), 4.33 (dd, J = 4.4, 2.4 Hz, 1H), 4.24 (d, J = 12.2 Hz, 1H), 4.10 (d, J = 12.1 Hz, 1H), 4.03 (t, J = 4.8 Hz, 1H), 4.01–3.94 (m, 2H), 3.83 (t, J = 9.3 Hz, 1H), 3.68–3.57 (m, 3H), 3.57–3.52 (m, 1H), 3.44–3.33 (m, 2H), 3.25–3.10 (m, 3H), 3.04 (dt, J = 5.4, 2.7 Hz, 2H), 2.89 (dd, J = 12.8, 5.2 Hz, 1H), 2.10 (dt, J = 13.1, 4.5 Hz, 1H), 1.30 (q, J = 12.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 170.43, 169.60, 135.70, 135.53, 135.45, 135.29, 135.25, 134.73, 134.50, 134.38, 133.34, 133.33, 133.28, 133.14, 133.14, 133.12, 133.10, 133.08, 132.99, 132.97, 132.90, 128.66, 128.61, 128.58, 128.52, 128.45, 128.40, 128.39, 128.36, 128.30, 128.20, 128.09, 128.07, 128.04, 128.02, 127.95, 127.92, 127.90, 127.84, 127.79, 127.74, 127.70, 127.23, 127.02, 126.95, 126.81, 126.77, 126.54, 126.40, 126.32, 126.27, 126.24, 126.23, 126.17, 126.15, 126.12, 126.04, 126.02, 125.92, 125.88, 125.82, 125.78, 125.68, 125.46, 125.32, 107.03, 98.98, 96.88, 83.93, 82.29, 82.24, 81.08, 79.79, 76.06, 75.64, 74.95, 74.74, 74.03, 73.75, 73.21, 72.50, 72.16, 71.88, 71.24, 67.34, 67.25, 63.20, 60.32, 59.36, 57.73, 50.65, 32.09.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(pyrolidinyl)-paromomycindicarboxamide (22).
To a solution of intermediate 7 (0.250 g, 0.158 mmol) in DCM/H2O (1:1) (5 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (9.83 mg, 0.0630 mmol) and iodobenzene diacetate (BAIB) (254 mg, 0.788 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude residue in DCM (4 mL), oxalyl chloride (56.5 μL, 0.619 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (3 mL). Pyrrolidine (101 μL, 1.21 mmol) and triethylamine (211 μL, 1.51 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (3:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product 22. Isolated yield: 60.4%. 1H NMR (500 MHz, CDCl3) δ 7.83–7.61 (m, 21H), 7.56–7.54 (m, 1H), 7.52–7.42 (m, 13H), 7.40–7.32 (m, 5H), 7.27 (s, 1H), 7.07 (dd, J = 8.4, 1.7 Hz, 1H), 6.42 (d, J = 3.8 Hz, 1H), 5.73 (d, J = 5.2 Hz, 1H), 5.19 (d, J = 11.5 Hz, 1H), 5.06 (s, 2H), 4.99 (d, J = 11.5 Hz, 1H), 4.85 (dd, J = 7.8, 2.1 Hz, 2H), 4.81–4.69 (m, 5H), 4.65 (d, J = 12.2 Hz, 1H), 4.42 (dd, J = 4.5, 2.4 Hz, 1H), 4.35 (t, J = 4.8 Hz, 1H), 4.28 (d, J = 12.2 Hz, 1H), 4.19–4.13 (m, 2H), 4.08 (t, J = 9.4 Hz, 1H), 3.96 (d, J = 12.3 Hz, 1H), 3.72–3.62 (m, 4H), 3.60–3.36 (m, 11H), 3.31–3.17 (m, 3H), 3.07 (t, J = 2.3 Hz, 1H), 3.05–2.99 (m, 2H), 2.13 (dt, J = 13.0, 4.5 Hz, 1H), 1.86 (dd, J = 13.6, 6.5 Hz, 3H), 1.82–1.73 (m, 4H), 1.69–1.57 (m, 2H), 1.26 (q, J = 12.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 167.73, 167.44, 136.32, 136.13, 135.94, 135.41, 134.84, 134.74, 133.70, 133.64, 133.47, 133.41, 133.29, 133.27, 133.19, 128.86, 128.71, 128.60, 128.51, 128.49, 128.40, 128.37, 128.26, 128.19, 128.15, 128.12, 128.05, 127.98, 127.62, 127.42, 127.34, 127.11, 126.86, 126.71, 126.65, 126.59, 126.55, 126.52, 126.48, 126.45, 126.41, 126.34, 126.24, 126.09, 126.02, 125.89, 125.83, 125.66, 107.00, 99.41, 96.99, 84.33, 82.88, 82.19, 81.75, 80.79, 80.03, 75.89, 75.52, 75.40, 75.34, 74.53, 74.06, 73.44, 72.65, 72.45, 71.89, 69.58, 63.71, 60.64, 60.53, 57.86, 51.10, 46.90, 46.85, 46.63, 46.58, 33.13, 26.37, 26.33, 24.40.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(piperidinyl)-paromomycindicarboxamide (23).
To a solution of intermediate 7 (0.200 g, 0.126 mmol) in DCM/H2O (1:1) (4 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (7.89 mg, 0.0504 mmol) and iodobenzene diacetate (BAIB) (203 mg, 0.630 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH/DCM). The reaction mixture was diluted with DCM, washed with saturated aqueous NaHCO3 solution and saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude residue in DCM (4 mL), oxalyl chloride (56.5 μL, 0.495 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was dissolved in THF (3 mL). Piperidine (95.7 μL, 0.968 mmol) and triethylamine (169 μL, 1.21 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (1:1 EtOAc/Hex), it was diluted with EtOAc, washed with saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product 23. Isolated yield: 70.1%. 1H NMR (500 MHz, CDCl3) δ 7.82–7.77 (m, 7H), 7.76–7.73 (m, 6H), 7.70 (dd, J = 6.5, 2.6 Hz, 5H), 7.66 (d, J = 6.3 Hz, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.49 (dd, J = 3.7, 1.7 Hz, 4H), 7.48 (s, 1H), 7.47–7.46 (m, 3H), 7.46–7.44 (m, 4H), 7.43–7.39 (m, 4H), 7.37 (dd, J = 8.4, 1.7 Hz, 1H), 7.33 (dd, J = 8.4, 1.7 Hz, 1H), 7.29 (dd, J = 8.4, 1.7 Hz, 1H), 7.13 (dd, J = 8.4, 1.7 Hz, 1H), 6.18 (d, J = 3.8 Hz, 1H), 5.55 (d, J = 3.8 Hz, 1H), 5.11 (d, J = 11.7 Hz, 1H), 5.07 (s, 2H), 4.99 (d, J = 11.6 Hz, 1H), 4.93–4.90 (m, 2H), 4.88 (d, J = 3.0 Hz, 1H), 4.84–4.78 (m, 4H), 4.63 (t, J = 11.9 Hz, 2H), 4.49 (t, J = 4.2 Hz, 1H), 4.36 (d, J = 12.1 Hz, 1H), 4.27–4.25 (m, 1H), 4.23 (s, 1H), 4.18–4.09 (m, 4H), 3.78–3.72 (m, 1H), 3.66 (dtd, J = 12.5, 6.5, 2.6 Hz, 3H), 3.61 (t, J = 3.2 Hz, 1H), 3.55–3.48 (m, 4H), 3.44 (dt, J = 8.4, 4.8 Hz, 4H), 3.38 (dd, J = 12.3, 6.8 Hz, 2H), 3.25 (dd, J = 12.3, 6.8 Hz, 1H), 3.20–3.12 (m, 4H), 3.05–2.99 (m, 1H), 2.98–2.92 (m, 2H), 2.07–2.02 (m, 1H), 1.61–1.58 (m, 7H), 1.53–1.49 (m, 6H), 1.17 (q, J = 12.7 Hz, 1H).13C NMR (126 MHz, CDCl3) δ 166.67, 166.63, 136.04, 135.49, 135.37, 135.25, 134.49, 134.34, 133.21, 133.19, 133.17, 132.98, 132.95, 132.84, 132.77, 132.70, 128.35, 128.18, 128.15, 128.04, 127.98, 127.91, 127.81, 127.79, 127.68, 127.61, 127.59, 127.55, 127.21, 127.01, 126.93, 126.60, 126.42, 126.32, 126.16, 126.10, 126.04, 126.01, 125.98, 125.91, 125.89, 125.85, 125.77, 125.68, 125.65, 125.58, 125.37, 125.27, 124.98, 106.89, 98.77, 96.75, 83.13, 81.63, 80.70, 80.38, 79.42, 78.78, 75.46, 75.39, 74.90, 74.84, 73.96, 73.19, 72.95, 72.33, 72.09, 71.42, 67.35, 63.50, 60.07, 59.61, 57.94, 50.21, 46.79, 46.40, 43.54, 42.99, 32.48, 26.40, 26.27, 25.38, 25.23, 24.36.
1,2′,2‴,3,6‴-Pentaazido-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-1,2′,2‴,3,6‴-pentadeamino-5″,6′-(4-(pyrimidin-2-yl)piperazinyl)-paromomycindicarboxamide (24).
To a solution of intermediate 7 (0.250 g, 0.158 mmol) in DCM/H2O (1:1) (5 mL) at room temperature, 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) (9.83 mg, 0.0630 mmol) and iodobenzene diacetate (BAIB) (254 mg, 0.788 mmol) were added. The resulting mixture was stirred at room temperature for 2 h. The reaction progress was monitored by TLC (1:9 MeOH:DCM). The reaction mixture was diluted with DCM, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo to give the crude residue. To a solution of the crude residue in DCM (4 mL), oxalyl chloride (56.5 μL, 0.619 mmol) was added, and the resulting solution was stirred at room temperature for 12 h under argon. The reaction progress was monitored by TLC (1:5 EtOAc/hexanes) and concentrated in vacuo upon completion. The crude acyl chloride intermediate was then dissolved in THF (3 mL). 2-(1-Piperazinyl)pyrimidine (198 mg, 0.969 mmol) and triethylamine (169 μL, 1.21 mmol) were added to the solution. After the reaction was stirred for 3 h and monitored by TLC (1:1 EtOAc/Hex), it was diluted with EtOAc, washed with a saturated aqueous NaHCO3 solution, then a saturated aqueous NaCl solution, and dried over Na2SO4. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel column chromatography to afford product 24. Isolated yield: 64.8%.1H NMR (500 MHz, CDCl3) δ 8.32 (d, J = 4.7 Hz, 2H), 8.28 (d, J = 4.7 Hz, 2H), 7.82 (q, J = 3.7 Hz, 3H), 7.78–7.75 (m, 4H), 7.74–7.72 (m, 5H), 7.71–7.69 (m, 5H), 7.66 (d, J = 8.4 Hz, 3H), 7.63–7.59 (m, 3H), 7.50–7.46 (m, 9H), 7.41 (dd, J = 3.4, 2.0 Hz, 5H), 7.35 (t, J = 1.9 Hz, 2H), 7.30–7.28 (m, 2H), 7.11 (dd, J = 8.4, 1.7 Hz, 1H), 6.52–6.50 (m, 2H), 6.31 (d, J = 3.8 Hz, 1H), 5.70 (d, J = 4.5 Hz, 1H), 5.15 (d, J = 11.6 Hz, 1H), 5.07–5.01 (m, 4H), 4.98 (d, J = 3.2 Hz, 2H), 4.95–4.91 (m, 2H), 4.90–4.85 (m, 3H), 4.83 (d, J = 1.8 Hz, 2H), 4.76 (d, J = 11.8 Hz, 1H), 4.68–4.63 (m, 3H), 4.44 (dd, J = 4.5, 3.1 Hz, 1H), 4.34 (d, J = 12.1 Hz, 1H), 4.26 (d, J = 4.5 Hz, 1H), 4.21 (s, 1H), 4.18–4.11 (m, 4H), 4.04 (s, 1H), 3.81 (t, J = 5.5 Hz, 5H), 3.77 (t, J = 6.0 Hz, 4H), 3.72 (d, J = 4.2 Hz, 4H), 3.68–3.64 (m, 5H), 3.59 (dd, J = 6.0, 3.0 Hz, 2H), 3.55 (ddd, J = 10.2, 6.4, 3.2 Hz, 3H), 3.47–3.40 (m, 3H), 3.24 (t, J = 9.4 Hz, 2H), 3.20–3.14 (m, 3H), 3.14–3.08 (m, 3H), 2.97 (t, J = 2.6 Hz, 1H), 2.13 (dt, J = 13.2, 4.6 Hz, 1H), 1.27–1.26 (m, 2H).13C NMR (126 MHz, CDCl3) δ 167.56, 167.52, 161.63, 157.88, 157.81, 136.04, 135.75, 135.48, 135.15, 134.65, 134.52, 133.46, 133.42, 133.34, 133.22, 133.07, 133.00, 132.96, 128.61, 128.45, 128.33, 128.24, 128.18, 128.15, 128.04, 128.01, 127.94, 127.87, 127.84, 127.77, 127.40, 127.29, 127.19, 126.87, 126.60, 126.46, 126.41, 126.38, 126.34, 126.29, 126.21, 126.17, 126.09, 126.00, 125.92, 125.72, 125.60, 125.26, 110.56, 110.52, 106.89, 98.98, 97.18, 83.77, 82.21, 81.44, 80.54, 79.65, 78.99, 75.74, 75.69, 75.24, 75.17, 74.31, 73.55, 73.32, 72.54, 72.32, 71.63, 67.67, 63.60, 60.39, 60.26, 57.84, 50.63, 45.60, 45.34, 43.94, 43.86, 43.45, 43.36, 42.45, 42.05, 32.85, 31.06, 29.85, 14.35.
1,2′,2‴,3,6‴-Penta-N-sulfo-paramomycin (L1).
The intermediate 9 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L10. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L1. The desired product was obtained as a white powder. Isolated yield: 94% 1H NMR (600 MHz, D2O) δ 5.27 (d, J = 4.3 Hz, 1H), 5.25 (d, J = 3.4 Hz, 1H), 4.95 (d, J = 2.2 Hz, 1H), 4.30–4.20 (m, 2H), 4.11–4.05 (m, 1H), 3.98 (ddd, J = 7.6, 5.4, 2.0 Hz, 1H), 3.82–3.75 (m, 3H), 3.71 (d, J = 4.2 Hz, 1H), 3.67–3.59 (m, 1H), 3.52 (t, J = 9.4 Hz, 1H), 3.42 (t, J = 9.5 Hz, 1H), 3.39–3.33 (m, 1H), 3.18–3.11 (m, 2H), 3.07–2.99 (m, 1H), 2.73 (dt, J = 13.4, 4.2 Hz, 1H), 1.24 (q, J = 12.5 Hz, 1H). 13C NMR (151 MHz, D2O) δ 107.99, 99.70, 98.78, 83.20, 82.75, 82.11, 76.86, 74.38, 73.37, 73.11, 72.72, 70.94, 69.51, 68.65, 68.29, 61.43, 59.96, 58.24, 54.95, 53.97, 53.67, 43.65, 32.66. HRMS: m/z calc. for C23H40N5Na5O29S5 [M + Na]+: 1147.9793; found: 1147.9794.
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-di-O-sulfo-paramomycin (L2).
The intermediate 7 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L11. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L2. The desired product was obtained as a white powder. Isolated yield: 71% 1H NMR (600 MHz, D2O) δ 5.34 (d, J = 4.2 Hz, 1H), 5.19 (d, J = 3.4 Hz, 1H), 4.95 (d, J = 2.1 Hz, 1H), 4.35–4.16 (m, 5H), 4.06 (dd, J = 10.0, 2.3 Hz, 1H), 3.97 (td, J = 6.6, 1.7 Hz, 1H), 3.66 (t, J = 9.3 Hz, 1H), 3.62–3.55 (m, 1H), 3.50 (td, J = 9.4, 4.8 Hz, 2H), 3.39–3.30 (m, 2H), 3.14 (ddd, J = 13.8, 8.4, 2.5 Hz, 2H), 3.10–2.99 (m, 2H), 2.77–2.68 (m, 1H), 1.24 (q, J = 12.5 Hz, 1H). 13C NMR (151 MHz, D2O) δ 107.67, 100.02, 98.62, 83.24, 82.53, 79.27, 76.44, 74.48, 73.52, 72.83, 71.11, 70.39, 68.98, 68.75, 68.20, 68.01, 66.39, 58.13, 54.80, 53.63, 53.56, 43.13, 33.01. HRMS: m/z calc. for C23H38N5Na7O35S7 [M + Na]+: 1351.8568; found: 1351.8566
1,2′,2‴,3,6‴-Penta-N-sulfo-paramomycindicarboxylate (L3).
The intermediate 21 was subjected to azide reduction followed by a subsequent sulfation and ion exchange. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L3. The desired product was obtained as a white powder. Isolated yield: 58%. 1H NMR (600 MHz, D2O) δ 5.49 (d, J = 3.7 Hz, 1H), 5.45 (dd, J = 14.5, 3.9 Hz, 2H), 5.03 (s, 2H), 4.48–4.27 (m, 3H), 4.22 (s, 2H), 3.81–3.67 (m, 2H), 3.65–3.58 (m, 2H), 3.41 (t, J = 9.4 Hz, 2H), 3.36 (d, J = 27.9 Hz, 2H), 3.21–2.98 (m, 4H), 2.66 (s, 2H), 1.30 (q, J = 12.6 Hz, 1H). 13C NMR (151 MHz, D2O) δ 183.18, 172.68, 106.73, 98.88, 97.99, 84.90, 84.01, 79.70, 76.61, 73.61, 73.54, 72.31, 71.54, 71.43, 68.65, 68.43, 60.40, 57.90, 55.43, 55.05, 53.44, 53.41, 53.02, 43.55, 33.33. HRMS: m/z calc. for C23H34N5Na7O31S5 [M + Na]+: 1219.9017; found: 1219.9009
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-(N-1,1′-biphenyl-4-yl)-paromomycindicarboxamide (L4).
The intermediate 16 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L15. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L4. Isolated yield: 50.1% 1H NMR (500 MHz, CD3OD) δ 7.69–7.60 (m, 7H), 7.59–7.50 (m, 5H), 7.43 (t, J = 7.3 Hz, 4H), 5.72 (s, 1H), 5.70 (s, 1H), 5.28 (s, 1H), 4.34 (s, 2H), 4.10–4.06 (m, 1H), 3.99–3.93 (m, 4H), 3.89 (s, 1H), 3.82–3.69 (m, 17H), 3.69–3.62 (m, 11H), 3.52 (s, 1H), 3.39 (d, J = 10.1 Hz, 3H), 3.29–3.17 (m, 3H), 2.78 (s, 1H), 2.43 (s, 1H), 2.26 (d, J = 1.4 Hz, 1H), 2.20 (t, J = 7.3 Hz, 2H).13C NMR (126 MHz, CD3OD) δ 168.90, 154.57, 134.21, 127.55, 126.41, 126.16, 125.80, 125.42, 124.94, 123.80, 123.29, 122.85, 126.51, 122.85, 125.51, 125.18, 104.92, 98.95, 98.61, 81.35, 81.35, 75.04, 68.73, 74.37, 84.67, 73.05, 53.45, 69.73, 56.11, 47.81, 29.21, 22.57. HRMS: m/z calc. for C47H54N7Na5O29S5 [M–4Na + 2H]2−: 683.1379; found: 683.1313
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-(N-phenyl)-paromomycindicarboxamide (L5).
The intermediate 18 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L16. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L5. Isolated yield: 71.4%. 1H NMR (600 MHz, CD3OD) δ 7.77–7.65 (m, 4H), 7.26 (dt, J = 11.5, 7.7 Hz, 4H), 7.05 (t, J = 7.4 Hz, 2H), 5.65 (d, J = 3.3 Hz, 1H), 5.38 (d, J = 3.8 Hz, 1H), 5.27 (d, J = 2.3 Hz, 1H), 5.02 (d, J = 10.0 Hz, 1H), 4.69 (t, J = 4.4 Hz, 1H), 4.53 (t, J = 3.9 Hz, 1H), 4.27 (s, 1H), 4.13 (s, 1H), 3.89–3.82 (m, 2H), 3.78 (t, J = 9.2 Hz, 1H), 3.69–3.61 (m, 7H), 2.83–2.80 (m, 0H), 1.46 (q, J = 12.2 Hz, 1H). HRMS: m/z calc. for C35H45N7Na5O29S5 [M–5Na + 1H]2–: 594.5216; found: 594.4762
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-(N-pyrrolidinyl)-paromomycindicarboxamide (L6).
The intermediate 22 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L18. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L6. Isolated yield: 73.8%. 1H NMR (500 MHz, D2O) δ 5.59 (d, J = 3.6 Hz, 1H), 5.49 (d, J = 2.3 Hz, 1H), 5.12 (d, J = 2.1 Hz, 1H), 4.46 (dd, J = 5.3, 2.3 Hz, 1H), 4.38 (t, J = 3.6 Hz, 1H), 4.03 (t, J = 6.9 Hz, 1H), 3.91 (t, J = 8.7 Hz, 1H), 3.84 (d, J = 9.6 Hz, 2H), 3.81 (d, J = 4.7 Hz, 2H), 3.78 (s, 2H), 3.76–3.73 (m, 2H), 3.71 (d, J = 9.5 Hz, 2H), 3.61 (t, J = 9.3 Hz, 2H), 3.54 (t, J = 7.0 Hz, 3H), 3.49 (d, J = 9.2 Hz, 4H), 3.31 (d, J = 3.3 Hz, 1H), 3.29 (d, J = 3.6 Hz, 1H), 3.26 (d, J = 4.1 Hz, 1H), 3.22 (dd, J = 12.4, 6.3 Hz, 2H), 3.11 (dd, J = 12.7, 6.6 Hz, 1H), 2.83–2.78 (m, 1H), 2.02 (d, J = 6.5 Hz, 5H), 1.97–1.92 (m, 4H), 1.47 (q, J = 12.4 Hz, 2H). 13C NMR (126 MHz, D2O) δ 168.58, 106.49, 98.29, 97.64, 83.26, 78.56, 78.33, 77.70, 73.52, 73.35, 73.18, 72.37, 70.79, 70.00, 68.64, 67.89, 58.15, 54.90, 53.54, 53.48, 47.94, 47.21, 46.79, 42.98, 32.86, 25.39, 25.35, 23.68, 23.63. HRMS: m/z calc. for C31H50N7Na5O29S5 [M–2Na + 2H]2−: 607.5400; found: 607.5499
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-(N-piperidinyl)-paromomycindicarboxamide (L7).
The intermediate 23 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L19. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L7. Isolated yield: 60.5%. 1H NMR (500 MHz, D2O) δ 5.63 (d, J = 3.6 Hz, 1H), 5.51 (d, J = 2.3 Hz, 1H), 5.14 (d, J = 2.2 Hz, 1H), 5.08 (d, J = 9.8 Hz, 1H), 5.03 (d, J = 6.1 Hz, 1H), 4.46 (dd, J = 5.2, 2.3 Hz, 1H), 4.37 (t, J = 3.7 Hz, 1H), 4.05 (td, J = 6.6, 1.9 Hz, 1H), 3.93 (t, J = 8.7 Hz, 2H), 3.88 (dd, J = 10.3, 9.3 Hz, 2H), 3.82–3.75 (m, 5H), 3.74 (s, 1H), 3.72–3.67 (m, 4H), 3.62 (t, J = 9.1 Hz, 3H), 3.56–3.51 (m, 2H), 3.49 (ddd, J = 3.6, 2.3, 1.0 Hz, 1H), 3.45–3.39 (m, 2H), 3.33 (d, J = 3.6 Hz, 1H), 3.31 (d, J = 3.7 Hz, 1H), 3.28 (t, J = 3.6 Hz, 1H), 3.26–3.24 (m, 1H), 3.24–3.20 (m, 2H), 3.17 (dd, J = 12.9, 6.9 Hz, 2H), 2.78 (dt, J = 13.2, 4.3 Hz, 1H), 1.71 (s, 8H), 1.65 (s, 4H), 1.51–1.44 (m, 1H). 13C NMR (126 MHz, D2O) δ 168.39, 168.23, 106.16, 98.38, 97.32, 83.08, 78.95, 77.41, 77.19, 73.73, 73.44, 73.35, 72.26, 70.94, 68.69, 68.09, 67.77, 58.14, 55.02, 53.58, 53.45, 47.85, 47.26, 44.22, 44.10, 43.16, 32.96, 26.25, 26.11, 25.27, 23.74, 23.68. HRMS: m/z calc. for C33H54N7Na5O29S5 [M–5Na + 3H]3–: 391.7271; found: 391.7253
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-(N-4-(tetrahydropyrimidin-2-yl)piperazin-1-yl)-paromomycindicarboxamide (L8).
The intermediate 24 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L20. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L8. Isolated yield: 61.7%. 1H NMR (500 MHz, D2O) δ 5.66 (d, J = 3.8 Hz, 1H), 5.43 (d, J = 1.3 Hz, 1H), 5.13 (d, J = 2.2 Hz, 1H), 5.02 (d, J = 9.8 Hz, 2H), 4.97 (d, J = 6.7 Hz, 2H), 4.94–4.91 (m, 2H), 4.51 (dd, J = 4.5, 1.3 Hz, 1H), 4.33 (t, J = 3.9 Hz, 1H), 4.04 (t, J = 7.1 Hz, 1H), 3.97–3.91 (m, 4H), 3.89 (d, J = 10.0 Hz, 2H), 3.85–3.78 (m, 2H), 3.78–3.71 (m, 4H), 3.70 (s, 1H), 3.70–3.65 (m, 2H), 3.65–3.61 (m, 2H), 3.61 (s, 1H), 3.60–3.53 (m, 4H), 3.51 (dd, J = 7.4, 4.8 Hz, 3H), 3.47 (d, J = 3.3 Hz, 1H), 3.42 (td, J = 5.9, 3.5 Hz, 9H), 3.31 (ddd, J = 12.8, 8.6, 4.5 Hz, 1H), 3.29–3.18 (m, 3H), 3.13 (dd, J = 12.7, 7.3 Hz, 1H), 2.72 (dt, J = 13.1, 4.4 Hz, 1H), 1.97 (h, J = 5.9 Hz, 4H), 1.51 (q, J = 12.4 Hz, 1H). 13C NMR (126 MHz, D2O) δ 169.41, 153.58, 106.78, 98.51, 96.73, 82.85, 79.12, 77.41, 77.10, 73.77, 73.47, 72.24, 70.72, 68.68, 68.19, 67.94, 62.50, 58.08, 55.12, 53.65, 53.33, 44.80, 44.57, 43.75, 43.50, 43.28, 41.68, 38.82, 38.80, 32.74, 19.75. HRMS: m/z calc. for C39H64N13Na5O29S5 [M–5Na + 3H]3−: 447.0927; found: 447.0910
1,2′,2‴,3,6′,6‴-Hexa-N-sulfo-5″-O-sulfo-neomycin (L9).
The intermediate 8 was subjected to azide reduction followed by a subsequent sulfation and ion exchange to afford L12. Global deprotection of the naphthalene (Nap) was achieved according to the outlined hydrogenolysis procedure above to afford the final L9. Isolated yield: 44.0%. 1H NMR (500 MHz, D2O) δ 5.50 (d, J = 4.0 Hz, 1H), 5.43 (d, J = 3.6 Hz, 1H), 5.13 (d, J = 2.1 Hz, 1H), 4.51–4.44 (m, 2H), 4.41 (q, J = 3.3, 2.6 Hz, 2H), 4.39–4.29 (m, 2H), 4.16–4.12 (m, 1H), 4.09 (ddd, J = 9.6, 5.0, 2.9 Hz, 1H), 3.83 (t, J = 9.3 Hz, 1H), 3.80–3.68 (m, 3H), 3.60 (d, J = 1.6 Hz, 1H), 3.59–3.54 (m, 1H), 3.54–3.45 (m, 2H), 3.44 (d, J = 2.6 Hz, 1H), 3.35–3.23 (m, 5H), 3.19 (ddd, J = 12.0, 10.1, 3.9 Hz, 1H), 2.88 (dt, J = 13.6, 3.9 Hz, 1H), 1.43 (q, J = 12.4 Hz, 1H). 13C NMR (126 MHz, D2O) δ 105.67, 96.99, 96.47, 81.28, 79.20, 77.13, 74.50, 72.32, 71.39, 70.92, 68.90, 68.87, 68.50, 66.65, 66.08, 65.92, 56.06, 52.69, 51.59, 51.45, 41.20, 41.06, 30.73.
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴,5″,6′-octa-O-(2-naphthylmethyl)-paromomycin (L10).
L10 was synthesized from the free amine product of intermediate 9 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 62.7%. 1H NMR (600 MHz, CD3OD) δ 7.86 (s, 1H), 7.80–7.69 (m, 7H), 7.72–7.65 (m, 6H), 7.68–7.60 (m, 4H), 7.62–7.57 (m, 1H), 7.59–7.53 (m, 4H), 7.48–7.27 (m, 14H), 7.28–7.18 (m, 3H), 7.16 (q, J = 3.4 Hz, 1H), 6.99 (dd, J = 8.4, 1.7 Hz, 1H), 5.57 (d, J = 3.5 Hz, 1H), 5.41 (d, J = 2.2 Hz, 1H), 4.98–4.91 (m, 1H), 4.91–4.85 (m, 2H), 4.79–4.66 (m, 5H), 4.66–4.57 (m, 1H), 4.59–4.52 (m, 1H), 4.50 (dd, J = 11.6, 2.3 Hz, 2H), 4.49–4.42 (m, 2H), 4.39 (q, J = 5.4 Hz, 1H), 4.28 (dd, J = 5.3, 2.4 Hz, 1H), 4.09–3.95 (m, 2H), 3.88–3.81 (m, 3H), 3.71–3.66 (m, 1H), 3.65–3.57 (m, 2H), 3.52 (dt, J = 3.1, 1.3 Hz, 1H), 3.38 (s, 1H), 3.33 (s, 33H), 2.78–2.73 (m, 1H), 1.31–1.25 (m, 1H).13C NMR (126 MHz, CD3OD) δ 136.68, 136.05, 135.96, 135.84, 135.74, 135.56, 135.14, 134.93, 133.35, 133.33, 133.29, 133.27, 133.20, 133.15, 133.05, 133.02, 132.97, 132.92, 132.85, 128.09, 127.95, 127.86, 127.80, 127.78, 127.75, 127.70, 127.68, 127.60, 127.55, 127.38, 127.26, 127.22, 127.20, 127.18, 127.16, 127.11, 126.91, 126.79, 126.71, 126.50, 126.33, 126.27, 126.22, 126.20, 126.06, 125.88, 125.84, 125.63, 125.60, 125.58, 125.50, 125.46, 125.42, 125.37, 125.35, 125.23, 125.20, 125.08, 106.31, 99.05, 98.57, 81.38, 81.01, 80.73, 79.60, 78.06, 77.72, 77.44, 77.18, 74.58, 74.11, 74.02, 73.80, 73.11, 73.03, 72.97, 72.63, 72.21, 71.62, 71.40, 70.73, 68.15, 60.14, 58.35, 53.29, 52.97, 52.32, 51.90, 48.76, 47.78, 47.60, 47.43, 47.26, 47.09, 44.18, 43.85, 29.37, 19.46, 13.06. HRMS: m/z calc. for C111H104N5Na5O29S5 [M–5Na + 4H]3–: 711.5824; found: 711.5856
1,2′,2‴,3,6‴-Penta-N-sulfo-6′,5″-di-O-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-paromomycin (L11).
L11 was synthesized from the free amine product of intermediate 7 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 52.4%. 1H NMR (600 MHz, CD3OD) δ 7.82–7.78 (m, 3H), 7.76 (td, J = 8.1, 4.3 Hz, 7H), 7.70 (ddd, J = 15.5, 5.7, 3.2 Hz, 6H), 7.66–7.63 (m, 3H), 7.60–7.54 (m, 4H), 7.48–7.45 (m, 3H), 7.42 (dtd, J = 9.0, 3.6, 1.8 Hz, 5H), 7.40–7.36 (m, 4H), 7.35–7.32 (m, 1H), 7.31–7.29 (m, 2H), 7.24 (dd, J = 8.4, 1.7 Hz, 1H), 5.29 (d, J = 3.4 Hz, 1H), 5.25 (d, J = 2.6 Hz, 1H), 5.10 (d, J = 5.2 Hz, 1H), 5.08 (s, 1H), 5.00 (s, 1H), 4.98 (d, J = 2.3 Hz, 1H), 4.96 (s, 1H), 4.89 (d, J = 11.1 Hz, 1H), 4.75–4.67 (m, 4H), 4.56–4.48 (m, 3H), 4.46–4.42 (m, 2H), 4.05 (dd, J = 5.2, 2.6 Hz, 1H), 4.00–3.96 (m, 1H), 3.94–3.89 (m, 3H), 3.84 (dd, J = 12.0, 5.1 Hz, 1H), 3.76 (dd, J = 10.1, 8.8 Hz, 1H), 3.66 (dd, J = 6.3, 2.8 Hz, 1H), 3.24 (d, J = 9.3 Hz, 1H), 3.15 (d, J = 9.5 Hz, 1H), 2.97 (d, J = 12.5 Hz, 1H), 2.86–2.81 (m, 2H), 2.68 (s, 1H), 2.61–2.55 (m, 1H), 2.42–2.35 (m, 1H), 1.84 (dt, J = 13.1, 4.2 Hz, 1H), 1.14 (t, J = 12.5 Hz, 1H). 13C NMR (151 MHz, CD3OD) δ 134.65, 134.24, 133.69, 133.60, 133.11, 131.80, 131.75, 131.59, 131.45, 126.61, 126.51, 126.43, 126.39, 126.22, 126.18, 126.10, 125.99, 125.90, 125.79, 125.75, 125.37, 125.13, 124.84, 124.50, 124.44, 124.40, 124.33, 124.27, 124.22, 124.18, 124.10, 123.96, 123.87, 123.58, 105.80, 98.43, 97.78, 84.62, 83.75, 80.76, 79.56, 79.38, 76.99, 73.34, 73.26, 72.94, 72.45, 71.74, 70.42, 70.19, 59.24, 59.07, 54.43, 49.82, 49.32, 46.50, 46.35, 46.21, 46.07, 45.93, 45.79, 45.65, 39.97, 34.45. HRMS: m/z calc. for C89H86N5Na7O35S7 [M + 2Na]2+: 1107.6108; found: 1107.6194
1,2′,2‴,3,6′,6‴-Henta-N-sulfo-5″-O-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-neomycin (L12).
L12 was synthesized from the free amine product of intermediate 8 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 56.0%. 1H NMR (500 MHz, CD3OD) δ 7.96 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.82 (s, 1H), 7.73–7.59 (m, 15H), 7.58 (s, 1H), 7.55 (s, 1H), 7.53–7.44 (m, 5H), 7.43–7.37 (m, 3H), 7.36–7.23 (m, 12H), 7.23–7.15 (m, 3H), 7.04–6.96 (m, 2H), 5.44 (d, J = 3.6 Hz, 1H), 5.20 (s, 1H), 5.00 (d, J = 11.9 Hz, 1H), 4.89–4.78 (m, 4H), 4.76 (d, J = 3.1 Hz, 1H), 4.66–4.58 (m, 3H), 4.58–4.53 (m, 2H), 4.47 (s, 2H), 4.43 (d, J = 11.4 Hz, 1H), 4.32 (ddd, J = 15.0, 9.1, 3.6 Hz, 4H), 4.25 (d, J = 4.1 Hz, 1H), 4.10 (d, J = 5.0 Hz, 1H), 4.02 (t, J = 4.2 Hz, 1H), 3.99–3.91 (m, 3H), 3.79 (s, 1H), 3.73 (s, 1H), 3.54 (s, 1H), 3.48 (dd, J = 12.5, 2.8 Hz, 1H), 3.41 (d, J = 8.3 Hz, 1H), 3.37–3.34 (m, 2H), 3.33–3.28 (m, 1H), 3.12–3.00 (m, 2H), 2.47 (dt, J = 13.8, 4.6 Hz, 1H), 1.82 (dt, J = 13.3, 6.4 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 136.60, 136.22, 136.02, 135.54, 135.35, 134.84, 133.45, 133.38, 133.29, 133.23, 133.18, 133.15, 133.07, 132.97, 132.88, 128.18, 127.90, 127.84, 127.81, 127.77, 127.74, 127.68, 127.63, 127.62, 127.36, 127.33, 127.25, 127.20, 127.17, 127.16, 127.10, 127.09, 126.97, 126.61, 126.53, 126.42, 126.37, 126.34, 126.01, 125.74, 125.71, 125.62, 125.49, 125.46, 125.41, 125.37, 125.27, 125.12, 125.01, 104.86, 99.06, 98.29, 81.05, 80.47, 79.82, 79.63, 77.81, 77.28, 75.22, 74.32, 74.21, 72.70, 72.46, 72.08, 71.94, 71.76, 70.10, 68.31, 58.62, 53.11, 52.40, 50.77, 45.00, 44.33. 26.71 HRMS: m/z calc. for C89H87N6Na7O34S7 [M–4Na + 3H]3–: 685.4333; found: 685.4349
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-paromomycindicarboxylate (L13).
L13 was synthesized from the free amine product of intermediate 21 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. We observed the transesterification of the OBn to OMe during the MeOH Sephadex as such the OMe was deprotected using LiOH (5 equiv of 0.2M) in 4:1 THF: H2O to afford L13. Isolated yield: 72.5%.1H NMR (600 MHz, D2O) δ 7.99–7.91 (m, 4H), 7.88 (d, J = 6.9 Hz, 3H), 7.81–7.72 (m, 6H), 7.66 (d, J = 8.5 Hz, 5H), 7.62–7.54 (m, 8H), 7.52–7.42 (m, 9H), 7.41–7.34 (m, 5H), 7.27 (d, J = 6.6 Hz, 5H), 7.19–7.08 (m, 5H), 5.66 (s, 1H), 5.04 (s, 2H), 4.48 (d, J = 9.2 Hz, 4H), 4.25 (dd, J = 22.7, 10.4 Hz, 4H), 4.17 (d, J = 11.3 Hz, 3H), 4.08 (s, 2H), 3.90 (s, 4H), 3.80 (t, J = 9.4 Hz, 3H), 3.60 (s, 3H), 3.36 (dd, J = 19.9, 11.4 Hz, 6H), 3.17 (s, 1H), 2.95 (d, J = 13.5 Hz, 2H), 2.29 (s, 1H), 1.56 (d, J = 7.5 Hz, 1H). HRMS: m/z calc. for C89H82N5Na7O31S5 [M–7Na + 4H]3–: 626.7627; found: 626.7604
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(O-methyl)-paromomycindicarboxylate (L14).
L14 was synthesized from the free amine product of intermediate 21 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. OBn transesterification to OMe was observed during the MeOH Sephadex to afford L14. Isolated yield: 52.8%. 1H NMR (500 MHz, CD3OD) δ 7.99–7.88 (m, 2H), 7.85–7.70 (m, 6H), 7.68–7.53 (m, 13H), 7.54–7.37 (m, 15H), 7.38–7.28 (m, 6H), 7.26–7.10 (m, 5H), 7.01 (s, 1H), 6.88–6.76 (m, 1H), 5.95 (s, 1H), 5.36 (d, J = 11.7 Hz, 1H), 5.28 (d, J = 12.1 Hz, 1H), 5.18 (d, J = 12.2 Hz, 2H), 5.09 (d, J = 10.1 Hz, 2H), 5.03 (d, J = 11.5 Hz, 1H), 5.01–4.88 (m, 5H), 4.78 (d, J = 11.1 Hz, 1H), 4.69–4.54 (m, 5H), 4.27 (qd, J = 10.5, 9.1, 4.7 Hz, 3H), 4.03 (td, J = 8.9, 3.6 Hz, 3H), 3.88 (s, 1H), 3.77 (dd, J = 10.1, 9.1 Hz, 1H), 3.64 (s, 1H), 3.54–3.44 (m, 4H), 1.46 (q, J = 12.2 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 174.96, 170.76, 136.44, 135.81, 135.58, 135.56, 134.84, 133.24, 133.00, 132.75, 128.70, 127.92, 127.78, 127.67, 127.61, 127.53, 127.22, 126.97, 126.45, 126.02, 125.61, 125.42, 125.16, 125.10, 107.53, 101.44, 98.43, 85.86, 84.26, 82.05, 80.87, 79.60, 78.74, 74.70, 74.21, 74.02, 73.24, 72.34, 71.75, 70.89, 67.36, 53.50, 53.20, 53.05, 48.10, 47.93, 47.76, 47.59, 47.42, 47.25, 47.08, 44.09, 34.64, 29.26. HRMS: m/z calc. for C91H88N5Na5O31S5 [M–NaSO3–4Na + 4H]–:1784.4432; found: 1784.4412
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(N-1,1′-biphenyl-4-yl)-paromomycindicarboxamide (L15).
L15 was synthesized from the free amine product of intermediate 16 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 73.4%. 1H NMR (500 MHz, CD3OD) δ 8.07 (s, 1H), 7.83 (t, J = 6.6 Hz, 2H), 7.78–7.72 (m, 6H), 7.73–7.67 (m, 8H), 7.66 (d, J = 3.9 Hz, 2H), 7.64 (d, J = 2.4 Hz, 1H), 7.63–7.59 (m, 3H), 7.58 (s, 1H), 7.55 (dt, J = 7.0, 3.9 Hz, 1H), 7.52–7.48 (m, 3H), 7.46 (d, J = 1.7 Hz, 1H), 7.45–7.36 (m, 13H), 7.35 (q, J = 1.5 Hz, 1H), 7.34–7.31 (m, 3H), 7.31–7.28 (m, 3H), 7.27–7.24 (m, 3H), 7.23 (dd, J = 3.4, 1.9 Hz, 1H), 7.21–7.19 (m, 1H), 7.17 (ddd, J = 8.7, 4.4, 1.6 Hz, 4H), 5.72 (d, J = 3.4 Hz, 1H), 5.62 (s, 1H), 5.12 (d, J = 11.7 Hz, 1H), 5.07–5.03 (m, 2H), 4.94–4.89 (m, 2H), 4.87 (dd, J = 5.0, 3.1 Hz, 1H), 4.76–4.71 (m, 2H), 4.69 (d, J = 6.4 Hz, 1H), 4.65 (dd, J = 7.7, 4.7 Hz, 2H), 4.60–4.52 (m, 2H), 4.49 (d, J = 11.0 Hz, 2H), 4.43–4.37 (m, 2H), 4.22 (q, J = 6.9 Hz, 2H), 4.04 (s, 1H), 3.88 (dd, J = 16.2, 7.0 Hz, 2H), 3.81 (t, J = 9.4 Hz, 2H), 3.72 (dd, J = 11.3, 5.8 Hz, 1H), 3.64 (dd, J = 9.8, 3.4 Hz, 1H), 3.51 (d, J = 2.8 Hz, 1H), 3.10 (d, J = 6.4 Hz, 2H), 2.82–2.73 (m, 1H), 1.68 (q, J = 11.9 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 171.98, 170.52, 142.23, 142.02, 141.97, 139.33, 138.73, 138.68, 138.51, 138.24, 138.19, 138.15, 138.10, 138.04, 137.86, 137.53, 137.17, 137.12, 137.07, 137.02, 136.98, 136.54, 136.45, 135.44, 135.40, 135.36, 135.32, 135.28, 135.23, 135.20, 135.15, 135.11, 135.07, 135.02, 134.98, 134.90, 134.86, 134.81, 134.73, 134.68, 134.63, 134.60, 130.19, 130.15, 130.09, 130.04, 129.99, 129.94, 129.89, 129.85, 129.69, 129.65, 129.60, 129.56, 129.51, 129.43, 129.36, 129.28, 129.22, 129.17, 129.13, 129.07, 129.01, 128.96, 128.92, 128.84, 128.79, 128.74, 128.71, 128.67, 128.58, 128.52, 128.47, 128.43, 128.38, 128.30, 128.24, 128.20, 128.14, 128.08, 128.02, 127.99, 127.93, 127.87, 127.82, 127.77, 127.73, 127.69, 127.65, 127.54, 127.45, 127.40, 127.35, 127.29, 127.24, 127.19, 127.13, 127.09, 127.04, 127.00, 126.96, 126.91, 126.87, 126.82, 122.99, 122.94, 122.89, 122.54, 122.45, 122.40, 108.63, 99.61, 99.51, 82.46, 82.18, 81.75, 81.58, 81.49, 80.86, 79.25, 76.91, 76.21, 75.87, 75.63, 74.21, 74.12, 73.89, 73.40, 59.99, 55.00, 54.77, 54.72, 45.67, 33.33. HRMS: m/z calc. for C113H102N7Na5O29S5 [M–5Na + 3H]2–: 727.8524; found: 727.8521
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(N-phenyl)-paromomycindicarboxamide (L16).
L16 was synthesized from the free amine product of intermediate 18 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 55.1%.1H NMR (500 MHz, CD3OD) δ 7.99 (d, J = 11.1 Hz, 1H), 7.76–7.48 (m, 13H), 7.45 (d, J = 4.4 Hz, 1H), 7.38–7.18 (m, 7H), 7.18–7.09 (m, 2H), 7.10–7.02 (m, 2H), 7.01–6.89 (m, 2H), 5.54 (dd, J = 12.7, 9.1 Hz, 1H), 5.01 (t, J = 11.0 Hz, 1H), 4.97–4.90 (m, 1H), 4.64 (d, J = 3.3 Hz, 1H), 4.50–4.38 (m, 2H), 4.38–4.29 (m, 1H), 4.28–4.17 (m, 1H), 4.12 (t, J = 6.1 Hz, 0H), 3.97 (d, J = 23.6 Hz, 1H), 3.80–3.65 (m, 3H), 3.49 (dd, J = 10.0, 3.3 Hz, 1H), 3.43–3.31 (m, 1H), 2.99 (d, J = 6.5 Hz, 1H), 2.68 (s, 1H), 1.64–1.49 (m, 1H). 13C NMR (151 MHz, cd3od) δ 170.95, 170.01, 136.47, 136.02, 135.47, 135.34, 134.80, 134.04, 133.84, 133.31, 133.25, 133.00, 132.92, 132.84, 132.54, 131.76, 128.71, 128.03, 127.97, 127.86, 127.76, 127.69, 127.58, 127.19, 127.09, 127.02, 126.77, 126.33, 126.25, 126.16, 125.78, 125.69, 125.57, 125.52, 125.46, 125.35, 125.30, 125.09, 124.97, 124.84, 122.98, 122.23, 107.01, 100.01, 98.16, 80.06, 79.39, 78.02, 75.11, 74.02, 72.49, 72.18, 71.58, 58.30, 53.27, 43.78. HRMS: m/z calc. for C101H94N7Na5O29S5 [M–5Na + 2H]3−: 663.8062; found: 663.8076
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(N-1-naphthyl)-paromomycindicarboxamide (L17).
L17 was synthesized from the free amine product of intermediate 17 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 68.5%. 1H NMR (500 MHz, CD3OD) δ 8.11 (d, J = 8.6 Hz, 1H), 7.96 (d, J = 7.9 Hz, 2H), 7.85 (dd, J = 8.7, 3.3 Hz, 2H), 7.82–7.79 (m, 3H), 7.76 (d, J = 8.5 Hz, 4H), 7.73 (d, J = 2.7 Hz, 2H), 7.71 (d, J = 2.5 Hz, 1H), 7.69 (d, J = 3.5 Hz, 1H), 7.68 (d, J = 3.3 Hz, 1H), 7.66 (d, J = 3.0 Hz, 1H), 7.64 (s, 1H), 7.62 (s, 2H), 7.61–7.60 (m, 1H), 7.59 (s, 1H), 7.56 (d, J = 7.1 Hz, 2H), 7.53 (d, J = 3.5 Hz, 3H), 7.51 (s, 1H), 7.50–7.48 (m, 2H), 7.47 (d, J = 1.3 Hz, 2H), 7.47–7.44 (m, 3H), 7.43–7.42 (m, 1H), 7.41 (dd, J = 4.0, 2.5 Hz, 2H), 7.40 (dt, J = 3.9, 1.7 Hz, 1H), 7.38 (dt, J = 2.8, 1.2 Hz, 2H), 7.36 (dp, J = 3.1, 1.6 Hz, 2H), 7.35–7.33 (m, 2H), 7.33–7.31 (m, 1H), 7.30–7.27 (m, 1H), 7.27–7.25 (m, 1H), 7.25–7.23 (m, 2H), 7.21 (dt, J = 6.9, 1.3 Hz, 1H), 7.20–7.17 (m, 1H), 6.97–6.91 (m, 2H), 5.78 (d, J = 3.4 Hz, 1H), 5.76 (s, 1H), 5.12 (d, J = 7.8 Hz, 1H), 5.10–5.05 (m, 3H), 5.01–4.94 (m, 2H), 4.93–4.89 (m, 3H), 4.77 (d, J = 12.0 Hz, 1H), 4.73 (d, J = 11.4 Hz, 1H), 4.69 (d, J = 3.1 Hz, 1H), 4.59 (t, J = 11.9 Hz, 2H), 4.55–4.51 (m, 2H), 4.43 (d, J = 10.7 Hz, 2H), 4.36–4.31 (m, 1H), 4.25 (t, J = 6.6 Hz, 1H), 3.99 (s, 1H), 3.97–3.92 (m, 3H), 3.76 (dd, J = 10.7, 5.5 Hz, 1H), 3.68 (s, 2H), 3.54 (s, 1H), 3.10 (dd, J = 13.3, 5.1 Hz, 1H), 3.04 (dd, J = 13.3, 7.6 Hz, 1H), 2.88–2.79 (m, 1H), 1.73 (q, J = 11.9 Hz, 1H). 13C NMR (151 MHz, CD3OD) δ 170.96, 170.01, 136.47, 136.02, 135.47, 134.05, 133.85, 133.37, 133.31, 133.25, 133.21, 133.00, 132.92, 132.84, 128.71, 128.03, 127.97, 127.86, 127.76, 127.69, 127.59, 127.19, 127.10, 127.03, 126.77, 126.33, 126.26, 126.16, 125.69, 125.57, 125.52, 125.46, 125.34, 125.30, 125.09, 123.17, 123.03, 122.98, 122.24, 122.13, 107.00, 100.13, 98.25, 98.16, 81.03, 81.03, 80.24, 80.06, 79.40, 78.94, 78.02, 75.11, 74.00, 72.55, 72.49, 72.18, 71.58, 58.30, 53.28, 53.00, 47.99, 47.85, 47.70, 47.56, 47.42, 47.28, 47.14, 43.77, 34.50, 31.60, 24.74, 24.29, 23.20, 21.14, 18.76, 18.11, 17.94, 15.57, 14.90, 13.36, 12.69, 12.27, 10.52, 9.53, 7.79, 4.66, 3.43, 3.10, 2.66, 2.31, 1.76, 1.37, 1.17. HRMS: m/z calc. for C109H98N7Na5O29S5 [M–5Na + 3H]3−: 710.5086; found: 710.5043
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(N-pyrrolidinyl)-paromomycindicarboxamide (L18).
L18 was synthesized from the free amine product of intermediate 22 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 50.5%. 1H NMR (500 MHz, CD3OD) δ 8.03 (s, 1H), 7.90 (d, J = 3.7 Hz, 2H), 7.80–7.77 (m, 7H), 7.74 (d, J = 4.2 Hz, 2H), 7.71 (d, J = 6.7 Hz, 3H), 7.66 (d, J = 1.3 Hz, 3H), 7.61 (t, J = 4.3 Hz, 2H), 7.55 (d, J = 6.4 Hz, 2H), 7.48 (dt, J = 8.4, 1.9 Hz, 3H), 7.45–7.42 (m, 7H), 7.38–7.33 (m, 7H), 7.18–7.13 (m, 4H), 5.57 (d, J = 3.3 Hz, 1H), 5.44 (s, 1H), 5.06 (d, J = 11.7 Hz, 1H), 4.99 (d, J = 11.6 Hz, 2H), 4.72 (s, 1H), 4.70 (d, J = 4.8 Hz, 2H), 4.65 (dd, J = 11.9, 3.2 Hz, 5H), 4.54 (d, J = 11.2 Hz, 1H), 4.48 (d, J = 10.8 Hz, 1H), 4.39 (d, J = 11.3 Hz, 1H), 4.28 (d, J = 4.6 Hz, 1H), 4.17 (s, 1H), 4.04 (d, J = 6.3 Hz, 1H), 3.90 (dd, J = 11.3, 5.8 Hz, 3H), 3.78 (q, J = 3.1 Hz, 2H), 3.77–3.73 (m, 3H), 3.69 (d, J = 9.1 Hz, 2H), 3.56 (q, J = 3.9 Hz, 2H), 3.52 (dd, J = 10.5, 4.2 Hz, 4H), 3.47–3.41 (m, 2H), 3.20–3.10 (m, 3H), 2.74–2.68 (m, 1H), 1.84–1.72 (m, 3H), 1.62–1.58 (m, 4H), 1.49–1.44 (m, 1H), 1.11 (t, J = 7.3 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 170.08, 170.00, 161.64, 138.02, 137.66, 137.37, 137.07, 136.75, 136.24, 134.93, 134.88, 134.85, 134.73, 134.67, 134.56, 134.53, 134.49, 129.34, 129.26, 129.07, 128.97, 128.84, 128.80, 128.77, 128.71, 128.45, 128.38, 127.95, 127.71, 127.60, 127.26, 127.15, 127.10, 127.02, 126.97, 126.91, 126.79, 100.45, 100.28, 82.46, 82.08, 79.90, 76.99, 76.56, 75.71, 75.33, 74.29, 74.07, 73.80, 73.39, 70.74, 60.28, 54.57, 47.78, 47.65, 47.16, 45.18, 26.89, 26.69, 25.00, 11.20. HRMS: m/z calc. for C97H98N7Na5O29S5 [M–3Na + H]3–: 667.1632; found: 667.1608
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(N-piperidinyl)-paromomycindicarboxamide (L19).
L19 was synthesized from the free amine product of intermediate 23 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 60.7%. 1H NMR (500 MHz, CD3OD) δ 8.03 (s, 1H), 7.90 (d, J = 8.0 Hz, 2H), 7.78 (dd, J = 5.7, 3.3 Hz, 5H), 7.74 (s, 1H), 7.72 (s, 2H), 7.70 (s, 1H), 7.68 (d, J = 1.3 Hz, 2H), 7.66 (d, J = 2.9 Hz, 1H), 7.64 (s, 3H), 7.61–7.60 (m, 1H), 7.59 (d, J = 3.6 Hz, 2H), 7.57–7.55 (m, 1H), 7.55–7.51 (m, 2H), 7.48 (s, 1H), 7.47–7.45 (m, 2H), 7.43 (ddd, J = 6.4, 3.2, 1.7 Hz, 6H), 7.40 (d, J = 4.8 Hz, 2H), 7.38 (d, J = 1.3 Hz, 1H), 7.36 (t, J = 1.7 Hz, 1H), 7.34 (d, J = 1.4 Hz, 1H), 7.33 (t, J = 1.4 Hz, 1H), 7.30–7.27 (m, 2H), 7.17 (dd, J = 8.3, 1.7 Hz, 1H), 7.11 (td, J = 7.1, 1.7 Hz, 2H), 5.55 (d, J = 3.4 Hz, 1H), 5.52 (s, 1H), 5.06 (d, J = 11.6 Hz, 1H), 4.99 (d, J = 5.6 Hz, 3H), 4.96 (d, J = 2.2 Hz, 2H), 4.90 (d, J = 10.9 Hz, 2H), 4.70 (d, J = 11.3 Hz, 2H), 4.63–4.55 (m, 6H), 4.37 (s, 1H), 4.33 (d, J = 4.4 Hz, 1H), 4.17–4.06 (m, 4H), 3.84–3.77 (m, 5H), 3.75–3.64 (m, 4H), 3.63–3.56 (m, 5H), 3.54–3.44 (m, 5H), 3.42 (s, 2H), 3.23 (d, J = 6.7 Hz, 3H), 2.60 (d, J = 14.0 Hz, 1H), 2.02 (s, 1H), 1.60 (s, 2H), 1.45 (s, 9H), 1.38 (s, 2H). 13C NMR (126 MHz, CD3OD) δ 169.75, 169.66, 161.80, 138.15, 137.80, 137.68, 137.19, 136.88, 136.47, 135.12, 135.08, 135.04, 135.02, 134.96, 134.93, 134.84, 134.79, 134.69, 134.66, 134.64, 129.77, 129.64, 129.56, 129.53, 129.42, 129.38, 129.34, 129.22, 129.19, 129.09, 128.98, 128.94, 128.92, 128.90, 128.84, 128.71, 128.46, 128.25, 128.18, 128.14, 128.05, 128.01, 127.67, 127.37, 127.36, 127.33, 127.31, 127.26, 127.22, 127.17, 127.11, 126.96, 126.89, 126.85, 108.73, 100.60, 100.45, 82.52, 82.30, 81.69, 80.43, 79.83, 79.47, 78.90, 77.02, 76.47, 75.75, 74.37, 74.21, 74.02, 73.42, 68.72, 60.32, 54.84, 54.47, 52.81, 45.48, 45.08, 44.98, 27.75, 27.68, 27.09, 27.03, 25.71, 25.45, 24.55. HRMS: m/z calc. for C99H102N7Na5O29S5 [M–3Na + H]3–: 686.5070; found: 686.5048
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴-hexa-O-(2-naphthylmethyl)-5″,6′-(N-4-(pyrimidin-2-yl)piperazinyl)-paromomycindicarboxamide (L20).
L20 was synthesized from the free amine product of intermediate 24 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 65.7%. 1H NMR (500 MHz, CD3OD) δ 8.20 (d, J = 4.8 Hz, 2H), 8.16 (d, J = 4.8 Hz, 2H), 8.02 (s, 1H), 7.92 (d, J = 8.1 Hz, 2H), 7.78 (dd, J = 8.4, 5.7 Hz, 4H), 7.75–7.71 (m, 3H), 7.67 (d, J = 4.7 Hz, 11H), 7.60–7.56 (m, 4H), 7.52 (s, 1H), 7.48 (s, 2H), 7.44–7.42 (m, 3H), 7.40–7.38 (m, 3H), 7.36–7.33 (m, 5H), 7.31–7.30 (m, 2H), 7.15–7.11 (m, 3H), 6.50–6.45 (m, 2H), 5.59 (d, J = 3.5 Hz, 1H), 5.57 (s, 1H), 5.04 (d, J = 15.0 Hz, 4H), 4.96–4.90 (m, 5H), 4.62 (d, J = 8.4 Hz, 4H), 4.52 (dd, J = 21.4, 11.1 Hz, 3H), 4.35 (s, 1H), 4.25 (d, J = 11.1 Hz, 1H), 4.11 (d, J = 6.7 Hz, 1H), 4.02 (s, 1H), 3.91 (t, J = 5.2 Hz, 1H), 3.81 (d, J = 8.7 Hz, 4H), 3.76–3.70 (m, 8H), 3.64 (q, J = 8.1 Hz, 7H), 3.57–3.51 (m, 5H), 3.45–3.40 (m, 4H), 3.25–3.21 (m, 2H), 3.14 (dd, J = 13.0, 5.0 Hz, 2H), 2.73–2.68 (m, 1H), 1.82 (dd, J = 15.4, 7.4 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 170.12, 170.00, 162.38, 162.23, 158.63, 137.56, 137.25, 136.83, 136.67, 136.37, 136.15, 135.85, 134.60, 134.57, 134.50, 134.41, 134.33, 134.28, 134.13, 134.10, 134.08, 132.16, 129.66, 129.25, 129.14, 129.03, 128.93, 128.90, 128.78, 128.66, 128.47, 128.43, 128.37, 128.34, 128.11, 128.00, 127.69, 127.60, 127.38, 127.26, 126.89, 126.86, 126.74, 126.70, 126.60, 126.50, 126.37, 111.16, 111.00, 100.11, 99.71, 81.86, 81.57, 79.26, 78.72, 76.54, 76.13, 75.44, 75.09, 73.91, 73.38, 73.02, 68.52, 66.46, 59.87, 54.36, 54.10, 53.27, 46.69, 46.43, 45.14, 44.55, 44.26, 43.94, 43.35, 43.04, 31.50, 20.04, 13.85. HRMS: m/z calc. for C105H104N13Na5O29S5 [M–3Na + H]3–: 739.1850; found: 739.1824
1,2′,2‴,3,6′,6‴-Hexa-N-sulfo-2″,3′,3‴,4′,4‴,6-hexa-O-(2-naphthylmethyl)-5″-(N-1,1′-biphenyl-4-yl)-neomycincarboxamide (L21).
L21 was synthesized from the free amine product of intermediate 20 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 48.0%.1H NMR (500 MHz, CD3OD) δ 8.07 (s, 1H), 7.87–7.82 (m, 2H), 7.78 (ddd, J = 7.3, 4.7, 2.5 Hz, 6H), 7.75–7.71 (m, 4H), 7.70–7.62 (m, 8H), 7.60 (d, J = 11.8 Hz, 3H), 7.56–7.50 (m, 4H), 7.42 (dddd, J = 18.0, 16.4, 13.9, 7.3, 5.7 Hz, 15H), 7.35–7.19 (m, 10H), 5.54 (d, J = 3.8 Hz, 1H), 5.51 (s, 1H), 5.10 (d, J = 12.0 Hz, 1H), 5.04 (td, J = 8.5, 7.8, 4.8 Hz, 3H), 4.96 (d, J = 11.5 Hz, 2H), 4.90 (s, 1H), 4.86–4.82 (m, 3H), 4.67 (t, J = 2.9 Hz, 1H), 4.65–4.58 (m, 3H), 4.56–4.51 (m, 2H), 4.39 (d, J = 11.0 Hz, 1H), 4.24–4.18 (m, 1H), 4.06 (ddd, J = 9.6, 6.8, 2.8 Hz, 1H), 4.01 (t, J = 7.0 Hz, 1H), 3.92–3.79 (m, 3H), 3.73–3.67 (m, 1H), 3.63 (d, J = 3.4 Hz, 1H), 3.60 (dd, J = 13.1, 2.9 Hz, 1H), 3.54 (dd, J = 10.4, 3.4 Hz, 1H), 3.51–3.45 (m, 1H), 3.41 (s, 1H), 3.26 (dd, J = 12.5, 6.6 Hz, 1H), 3.14 (s, 1H), 3.01–2.94 (m, 1H), 2.92–2.83 (m, 2H), 1.72–1.64 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 170.40, 160.08, 140.40, 137.02, 136.57, 136.38, 136.12, 135.41, 135.16, 134.74, 133.42, 133.26, 133.22, 133.14, 133.06, 132.98, 132.87, 128.29, 128.11, 127.90, 127.82, 127.76, 127.70, 127.65, 127.40, 127.25, 127.20, 127.06, 126.83, 126.69, 126.50, 126.38, 126.29, 126.24, 126.04, 125.89, 125.79, 125.61, 125.49, 125.43, 125.28, 125.17, 125.04, 121.45, 98.84, 97.87, 80.58, 79.74, 79.35, 79.19, 77.13, 75.37, 74.40, 73.93, 73.80, 73.56, 72.98, 72.41, 72.07, 71.74, 70.94, 58.88, 53.44, 53.27, 52.65, 44.39, 43.55. HRMS: m/z calc. for C101H95N7Na6O31S6 [M–3Na + H]3–: 721.1300; found: 721.1377
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴,5″,6′-octa-O-(1,1′-biphenyl-4-methyl)-paromomycin (L22).
L22 was synthesized from the free amine product of intermediate 11 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 42.6%.1H NMR (500 MHz,CD3OD) δ 8.45 (s, 0H), 7.80 (s, 1H), 7.70 (d, J = 9.9 Hz, 1H), 7.69–7.54 (m, 7H), 7.56–7.48 (m, 6H), 7.45 (q, J = 3.7, 3.0 Hz, 2H), 7.45–7.36 (m, 3H), 7.34–7.19 (m, 18H), 7.18–7.11 (m, 3H), 7.11–7.02 (m, 3H), 5.47 (d, J = 3.5 Hz, 1H), 5.37 (d, J = 2.4 Hz, 1H), 4.94 (d, J = 12.1 Hz, 1H), 4.85–4.78 (m, 4H), 4.67 (d, J = 10.4 Hz, 2H), 4.56 (dd, J = 12.1, 2.3 Hz, 2H), 4.54–4.44 (m, 4H), 4.44–4.26 (m, 4H), 4.25–4.15 (m, 1H), 3.86 (t, J = 6.8 Hz, 1H), 3.78 (t, J = 3.4 Hz, 1H), 3.75–3.68 (m, 2H), 3.65 (d, J = 9.1 Hz, 1H), 3.43 (d, J = 9.6 Hz, 1H), 3.35 (dt, J = 3.4, 1.7 Hz, 0H), 3.07 (p, J = 1.6 Hz, 0H), 1.63 (dq, J = 15.3, 8.5, 6.4 Hz, 1H).13C NMR (101 MHz, CD3OD) δ 136.92, 135.69, 135.48, 134.19, 133.59, 132.98, 132.93, 132.81, 132.76, 132.68, 132.45, 132.36, 132.32, 132.27, 132.22, 132.06, 128.53, 128.34, 128.25, 128.06, 127.89, 127.74, 127.61, 127.49, 127.34, 127.00, 126.74, 126.51, 126.14, 125.83, 125.72, 125.08, 124.84, 109.38, 98.30, 97.86, 80.51, 79.13, 78.28, 76.49, 74.88, 74.06, 71.56, 69.98, 67.74, 66.32, 58.77, 53.68, 53.46, 43.67, 31.29. HRMS: m/z calc. for C127H120N5Na5O29S5 [M–4Na + 3H]3–: 788.2054; found: 788.2090
1,2′,2‴,3,6‴-Penta-N-sulfo-5″,6′-di-O-sulfo-2″,3′,3‴,4,4′,4‴-hexaa-O-(1,1′-biphenyl-4-methyl)-paromomycin (L23).
L23 was synthesized from the free amine product of intermediate 14 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 55.25%.1H NMR (400 MHz, CD3OD) δ 7.60 (dd, J = 7.9, 5.5 Hz, 3H), 7.58–7.46 (m, 11H), 7.47–7.39 (m, 4H), 7.39–7.21 (m, 18H), 7.20–7.12 (m, 3H), 5.60 (s, 1H), 5.34 (s, 1H), 4.95 (q, J = 13.3, 11.3 Hz, 2H), 4.73 (d, J = 11.7 Hz, 2H), 4.67–4.60 (m, 2H), 4.57–4.46 (m, 4H), 4.40 (dd, J = 17.0, 10.3 Hz, 2H), 4.30 (dd, J = 11.0, 6.6 Hz, 2H), 4.18 (t, J = 6.6 Hz, 2H), 3.95 (s, 1H), 3.80 (d, J = 23.3 Hz, 2H), 3.66–3.55 (m, 2H), 3.54–3.38 (m, 4H), 3.19 (d, J = 11.8 Hz, 1H), 2.58 (d, J = 12.8 Hz, 1H), 1.28 (s, 1H). 13C NMR (101 MHz, CD3OD) δ 139.07, 138.98, 138.81, 138.49, 138.21, 138.01, 136.13, 135.75, 135.34, 135.01, 134.50, 127.30, 127.08, 126.93, 126.56, 126.53, 126.49, 126.46, 126.39, 126.20, 124.99, 124.90, 124.74, 124.69, 124.67, 124.62, 124.46, 124.36, 102.84, 97.41, 78.64, 77.74, 73.12, 72.30, 72.15, 70.53, 69.93, 69.74, 69.52, 67.37, 64.42, 56.36, 51.34, 50.47, 46.39, 46.18, 45.97, 45.75, 45.54, 45.33, 45.12, 42.57. HRMS: m/z calc. for C101H98N5Na7O35S7 [M–6Na + 4H]2–: 1095.7154; found: 1095.7194
1,2′,2‴,3,6′,6‴-Hexa-N-sulfo-5″-O-sulfo-2″,3′,3‴,4,4′,4‴-hexaa-O-(1,1′-biphenyl-4-methyl)-neomycin (L24).
L24 was synthesized from the free amine product of intermediate 15 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 62.0%. 1H NMR (500 MHz, CD3OD) δ 7.55–7.03 (m, 54H), 5.47 (s, 1H), 5.24 (s, 1H), 4.94–4.80 (m, 2H), 4.71–4.55 (m, 6H), 4.54–4.23 (m, 11H), 4.21–4.04 (m, 3H), 4.00–3.87 (m, 2H), 3.79 (s, 1H), 3.67 (s, 1H), 3.60 (s, 1H), 3.50–3.29 (m, 5H), 3.20–2.95 (m, 3H), 2.43 (s, 1H), 2.02–1.87 (m, 1H), 1.29–1.09 (m, 1H). 13C NMR (126 MHz, CD3OD) δ 140.93, 140.77, 140.69, 140.63, 140.53, 140.34, 140.05, 139.83, 138.01, 137.66, 137.54, 137.16, 136.85, 136.31, 129.15, 129.07, 128.95, 128.84, 128.73, 128.42, 128.39, 128.36, 128.33, 128.23, 127.95, 126.86, 126.78, 126.60, 126.56, 126.54, 126.51, 126.46, 126.33, 126.24, 126.19, 104.99, 99.08, 98.43, 80.93, 80.25, 79.91, 79.51, 77.73, 77.23, 75.13, 74.14, 72.28, 71.76, 71.57, 71.40, 70.21, 68.38, 58.56, 53.16, 52.32, 50.55, 45.02, 44.47, 31.67, 29.29, 27.19. HRMS: m/z calc. for C101H99N6Na7O34S7 [M–4Na + 3H]2−: 752.1209; found: 752.1292
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4′,4‴,6-hexa-O-(2-naphthylmethyl)-5″,6′-(N-1,1′-biphenyl-4-yl)-paromomycindicarboxamide (L25).
L25 was synthesized from the free amine product of intermediate 19 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 45.3%. 1H NMR (600 MHz, CD3OD) δ 7.82–7.78 (m, 4H), 7.75 (d, J = 3.7 Hz, 1H), 7.74–7.70 (m, 6H), 7.70–7.67 (m, 3H), 7.65 (dd, J = 7.3, 5.1 Hz, 3H), 7.63–7.58 (m, 4H), 7.53 (d, J = 8.8 Hz, 2H), 7.47–7.43 (m, 4H), 7.42–7.38 (m, 3H), 7.38–7.35 (m, 1H), 7.35–7.32 (m, 2H), 7.32–7.30 (m, 1H), 7.30–7.28 (m, 1H), 7.28–7.24 (m, 2H), 7.22 (d, J = 1.7 Hz, 0H), 7.21 (d, J = 1.8 Hz, 0H), 7.19–7.18 (m, 0H), 7.17 (ddd, J = 7.3, 3.8, 1.9 Hz, 1H), 7.14–7.09 (m, 3H), 7.08 (dd, J = 8.5, 1.7 Hz, 1H), 5.58 (d, J = 2.1 Hz, 1H), 5.43 (d, J = 4.6 Hz, 1H), 5.09 (dd, J = 11.9, 9.3 Hz, 2H), 5.01 (d, J = 12.3 Hz, 1H), 4.71 (s, 0H), 4.68 (d, J = 3.3 Hz, 1H), 4.66 (d, J = 4.0 Hz, 1H), 4.64–4.59 (m, 2H), 4.58–4.55 (m, 2H), 4.52–4.41 (m, 3H), 4.35 (dd, J = 5.5, 3.8 Hz, 1H), 4.17–4.13 (m, 1H), 4.09 (s, 1H), 3.92 (t, J = 4.5 Hz, 1H), 3.86 (t, J = 2.8 Hz, 1H), 3.78–3.71 (m, 1H), 3.70–3.65 (m, 1H), 3.58 (dd, J = 10.3, 8.7 Hz, 1H), 3.47 (t, J = 9.3 Hz, 1H), 3.14–3.05 (m, 1H), 2.96–2.87 (m, 1H), 2.69–2.67 (m, 1H), 2.65–2.61 (m, 1H), 2.58 (t, J = 5.9 Hz, 1H), 1.94 (dt, J = 12.7, 4.2 Hz, 1H), 1.19–1.11 (m, 1H).13C NMR (151 MHz, CD3OD) δ 169.33, 167.95, 134.34, 133.96, 133.70, 133.61, 133.30, 133.13, 131.78, 131.69, 131.61, 131.51, 131.43, 126.77, 126.52, 126.44, 126.38, 126.31, 126.10, 126.07, 126.01, 125.96, 125.90, 125.84, 125.50, 125.22, 124.93, 124.55, 124.47, 124.43, 124.32, 124.29, 124.07, 123.92, 123.74, 123.55, 104.90, 98.74, 84.22, 82.11, 79.53, 79.30, 76.33, 75.62, 73.32, 73.20, 72.45, 72.29, 71.72, 71.08, 70.89, 70.69, 70.53, 70.26, 65.37, 60.43, 52.12, 50.26, 49.74, 49.00, 46.94, 46.52, 46.38, 46.24, 46.09, 45.95, 45.81, 45.67, 39.87, 33.37, 29.36, 22.47, 15.30. HRMS: m/z calc. for C125H114N7Na5O29S5 [M–5Na + 2H]2−: 788.8837; found: 788.8828
1,2′,2‴,3,6‴-Penta-N-sulfo-2″,3′,3‴,4,4′,4‴,5″,6′-octa-O-benzyl-paromomycin (L26).
L26 was synthesized from the free amine product of compound 10 according to the general azide reduction and sulfation procedure and subsequent ion exchange to replace the triethylamine complex with sodium ions. Isolated yield: 39.4%. 1H NMR (400 MHz, CD3OD) δ 7.45–7.27 (m, 24H), 7.27–7.07 (m, 19H), 5.31 (d, J = 2.6 Hz, 0H), 5.17 (d, J = 3.4 Hz, 1H), 4.89 (dd, J = 16.6, 11.5 Hz, 1H), 4.71 (d, J = 10.8 Hz, 2H), 4.64–4.43 (m, 11H), 4.43–4.23 (m, 3H), 4.00 (dd, J = 5.1, 2.6 Hz, 1H), 3.95–3.86 (m, 1H), 3.73–3.56 (m, 6H), 3.56–3.43 (m, 2H), 3.35 (d, J = 5.0 Hz, 2H), 3.14–3.05 (m, 1H), 2.84–2.67 (m, 5H), 2.55 (dd, J = 13.4, 4.7 Hz, 1H), 1.91 (dt, J = 12.6, 4.2 Hz, 1H), 1.17 (d, J = 12.5 Hz, 1H). 13C NMR (101 MHz, CD3OD) δ 147.13, 139.45, 138.82, 138.11, 129.06, 128.97, 128.88, 128.86, 128.82, 128.76, 128.61, 128.53, 128.45, 128.39, 128.35, 128.21, 128.18, 127.98, 127.62, 108.79, 101.03, 99.78, 90.50, 87.85, 86.34, 82.31, 81.70, 81.21, 79.25, 76.22, 75.70, 74.85, 73.85, 70.87, 69.49, 56.77, 52.13, 51.60, 48.15, 47.93, 47.72, 42.14, 36.83. HRMS: m/z calc. for C79H88N5Na5O29S5 [M-5Na+3H]2–: 867.1828; found: 867.2155
Supplementary Material
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.5c00937.
Molecular docking; molecular dynamics; synthetic schemes; biological assays; NMR spectra; and HPLC trace (PDF)
Molecular formula strings (CSV)
PDB models of docked structures (ZIP)
ACKNOWLEDGMENTS
This work was supported by NIGMS (R01 GM098285), awarded to H.M.N. and I.V. I.V. was also supported by the US-Israel Binational Science Foundation (BSF-2021059) and the Israel Cancer Research Fund (ICRF). We extend our gratitude to Dr. Patty Thepsuwan for her invaluable assistance with the cell culture assays and to the Pohl group at Indiana University for teaching us how to conduct the flow chemistry experiments. Additionally, the Wayne State University Lumigen Center received partial support from the NIH, specifically S10OD028488 for NMR and R01GM098285 for Mass Spectrometry. We also thank the Wayne State University Grid for computing resources.
ABBREVIATIONS USED
- ATIII
antithrombin III
- BLI
biolayer interferometry
- BM
basement membrane
- CPM
counts per minutes
- (COCl)2
oxalyl chloride
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- ECM
extracellular matrix
- EC50
half-maximal effective concentration
- Et3N
trimethylamine
- FBS
fetal bovine serum
- FIIa
factor II a (thrombin)
- FXa
factor X
- HIT
heparin-induced thrombocytopenia
- HPSE
heparanase
- HS
heparan sulfate
- HSPGs
heparan sulfate proteoglycans
- MeOH
methanol
- MD
molecular dynamics
- MM
multiple myeloma
- mRNA
messenger ribonucleic acid
- M W
molecular weight
- NaH
sodium hydride
- Nap
naphthylmethyl
- PF4
platelet factor 4
- PhI(Ac)2
diacetoxy iodobenzene
- rRNA
ribosomal ribonucleic acid
- SO3Me3N
sulfur trioxidetrimethylamine
- TBAI
tetrabutylammonium iodide
- TEMPO
2,2,6,6-tetramethyl-1-piperidinyloxy
- TFA
trifluoroacetic acid
- TGI
tumor growth inhibition
- TR-FRET
time-resolved fluorescence resonance energy transfer
- NapBr
naphthyl bromide
Footnotes
Accession Codes
PDB code for Heparanase is 5E9C
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.5c00937
The authors declare no competing financial interest.
Contributor Information
Hawau Abdulsalam, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States;.
Livia Philip, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States.
Kartikey Singh, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States;.
Malik Farhoud, Technion Integrated Cancer Center, Rappaport Faculty of Medicine, Technion—Israel Institute of Technology, Haifa 3525422, Israel.
Neta Ilan, Technion Integrated Cancer Center, Rappaport Faculty of Medicine, Technion—Israel Institute of Technology, Haifa 3525422, Israel.
Israel Vlodavsky, Technion Integrated Cancer Center, Rappaport Faculty of Medicine, Technion—Israel Institute of Technology, Haifa 3525422, Israel.
Hien M. Nguyen, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States;
Data Availability Statement
The U-87 glioblastoma, B-16 melanoma, and MPC-11 myeloma data sets that support the findings are publicly available and were obtained from NCBI GEO with the accession codes GSE58644 and GSE9014. The remaining data are available in the Article and Supporting Information.
REFERENCES
- (1).Vlodavsky I; Singh P; Boyango I; Gutter-Kapon L; Elkin M; Sanderson RD; Ilan N Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resistance Updates 2016, 29, 54–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wu L; Viola CM; Brzozowski AM; Davies GJ Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol 2015, 22 (12), 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wilson JC; Laloo AE; Singh S; Ferro V 1H NMR spectroscopic studies establish that heparanase is a retaining glycosidase. Biochem. Biophys. Res. Commun 2014, 443 (1), 185–188. [DOI] [PubMed] [Google Scholar]
- (4).Sarrazin S; Lamanna WC; Esko JD Heparan sulfate proteoglycans. Cold Spring Harbor Perspect. Biol 2011, 3 (7), No. a004952, DOI: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vlodavsky I; Fuks Z; Bar-Ner M; Ariav Y; Schirrmacher V Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: relationship to tumor cell metastasis. Cancer Res. 1983, 43 (6), 2704–2711. [PubMed] [Google Scholar]
- (6).Jayatilleke KM; Hulett MD Heparanase and the hallmarks of cancer. J. Transl. Med 2020, 18 (1), No. 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Vlodavsky I; Ilan N; Sanderson RD Forty Years of Basic and Translational Heparanase Research. Adv. Exp. Med. Biol 2020, 1221, 3–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rivara S; Milazzo FM; Giannini G Heparanase: a rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem 2016, 8 (6), 647–680. [DOI] [PubMed] [Google Scholar]
- (9).Nakajima M; Irimura T; Di Ferrante D; Di Ferrante N; Nicolson GL Heparan sulfate degradation: relation to tumor invasive and metastatic properties of mouse B16 melanoma sublines. Science 1983, 220 (4597), 611–613. [DOI] [PubMed] [Google Scholar]
- (10).Zhang Y; Cui L Discovery and development of small-molecule heparanase inhibitors. Bioorg. Med. Chem 2023, 90, No. 117335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Weissmann M; Arvatz G; Horowitz N; Feld S; Naroditsky I; Zhang Y; Ng M; Hammond E; Nevo E; Vlodavsky I; Ilan N Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc. Natl. Acad. Sci. U.S.A 2016, 113 (3), 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhou H; Roy S; Cochran E; Zouaoui R; Chu CL; Duffner J; Zhao G; Smith S; Galcheva-Gargova Z; Karlgren J; Dussault N; Kwan RY; Moy E; Barnes M; Long A; Honan C; Qi YW; Shriver Z; Ganguly T; Schultes B; Venkataraman G; Kishimoto TK M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS One 2011, 6 (6), No. e21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ritchie JP; Ramani VC; Ren Y; Naggi A; Torri G; Casu B; Penco S; Pisano C; Carminati P; Tortoreto M; Zunino F; Vlodavsky I; Sanderson RD; Yang Y SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin. Cancer Res 2011, 17 (6), 1382–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lewis KD; Robinson WA; Millward MJ; Powell A; Price TJ; Thomson DB; Walpole ET; Haydon AM; Creese BR; Roberts KL; Zalcberg JR; Gonzalez R A phase II study of the heparanase inhibitor PI-88 in patients with advanced melanoma. Invest. New Drugs 2008, 26 (1), 89–94. [DOI] [PubMed] [Google Scholar]
- (15).Chhabra M; Wilson JC; Wu L; Davies GJ; Gandhi NS; Ferro V Structural Insights into Pixatimod (PG545) Inhibition of Heparanase, a Key Enzyme in Cancer and Viral Infections. Chem. - Eur. J 2022, 28 (11), No. e202104222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lemech C; Dredge K; Bampton D; Hammond E; Clouston A; Waterhouse NJ; Stanley AC; Leveque-El Mouttie L; Chojnowski GM; Haydon A; Pavlakis N; Burge M; Brown MP; Goldstein D Phase Ib open-label, multicenter study of pixatimod, an activator of TLR9, in combination with nivolumab in subjects with microsatellite-stable metastatic colorectal cancer, metastatic pancreatic ductal adenocarcinoma and other solid tumors. J. Immunother. Cancer 2023, 11 (1), No. e006136, DOI: 10.1136/jitc-2022-006136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Krause KM; Serio AW; Kane TR; Connolly LE Aminoglycosides: An Overview. Cold Spring Harbor Perspect. Med 2016, 6 (6), No. a027029, DOI: 10.1101/cshperspect.a027029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Patil R; Das S; Stanley A; Yadav L; Sudhakar A; Varma AK Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing. PLoS One 2010, 5 (8), No. e12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sun Q The Hydrophobic Effects: Our Current Understanding. Molecules 2022, 27 (20), No. 7009, DOI: 10.3390/molecules27207009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Loka RS; Sletten ET; Barash U; Vlodavsky I; Nguyen HM Specific Inhibition of Heparanase by a Glycopolymer with Well-Defined Sulfation Pattern Prevents Breast Cancer Metastasis in Mice. ACS Appl. Mater. Interfaces 2019, 11 (1), 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wakpal J; Pathiranage V; Walker AR; Nguyen HM Rational Design and Expedient Synthesis of Heparan Sulfate Mimetics from Natural Aminoglycosides for Structure and Activity Relationship Studies. Angew. Chem., Int. Ed 2023, 62 (32), No. e202304325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vlodavsky I Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells Curr. Protoc. Cell Biol 2001. DOI: 10.1002/0471143030.cb1004s01. [DOI] [PubMed] [Google Scholar]
- (24).Loka RS; Yu F; Sletten ET; Nguyen HM Design, synthesis, and evaluation of heparan sulfate mimicking glycopolymers for inhibiting heparanase activity. Chem. Commun 2017, 53 (65), 9163–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hong X; Jiang F; Kalkanis SN; Zhang ZG; Zhang X; Zheng X; Jiang H; Mikkelsen T; Chopp M Increased chemotactic migration and growth in heparanase-overexpressing human U251n glioma cells. J. Exp. Clin. Cancer Res 2008, 27 (1), No. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Varricchio A; Khan S; Price ZK; Davis RA; Ramesh SA; Yool AJ Pharmacological Inhibition of Membrane Signaling Mechanisms Reduces the Invasiveness of U87-MG and U251-MG Glioblastoma Cells In Vitro. Cancers 2023, 15 (4), No. 1027, DOI: 10.3390/cancers15041027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hong X; Nelson KK; deCarvalho AC; Kalkanis SN Heparanase expression of glioma in human and animal models. J. Neurosurg 2010, 113 (2), 261–269. [DOI] [PubMed] [Google Scholar]
- (28).Mitsiades CS; Rouleau C; Echart C; Menon K; Teicher B; Distaso M; Palumbo A; Boccadoro M; Anderson KC; Iacobelli M; Richardson PG Preclinical studies in support of defibrotide for the treatment of multiple myeloma and other neoplasias. Clin. Cancer Res 2009, 15 (4), 1210–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shteper PJ; Zcharia E; Ashhab Y; Peretz T; Vlodavsky I; Ben-Yehuda D Role of promoter methylation in regulation of the mammalian heparanase gene. Oncogene 2003, 22 (49), 7737–7749. [DOI] [PubMed] [Google Scholar]
- (30).Goldshmidt O; Zcharia E; Abramovitch R; Metzger S; Aingorn H; Friedmann Y; Schirrmacher V; Mitrani E; Vlodavsky I Cell surface expression and secretion of heparanase markedly promote tumor angiogenesis and metastasis. Proc. Natl. Acad. Sci. U.S.A 2002, 99 (15), 10031–10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zetser A; Bashenko Y; Miao HQ; Vlodavsky I; Ilan N Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003, 63 (22), 7733–7741. [PubMed] [Google Scholar]
- (32).de Boer C; Armstrong Z; Lit VAJ; Barash U; Ruijgrok G; Boyango I; Weitzenberg MM; Schröder SP; Sarris AJC; Meeuwenoord NJ; Bule P; Kayal Y; Ilan N; Codée JDC; Vlodavsky I; Overkleeft HS; Davies GJ; Wu L Mechanism-based heparanase inhibitors reduce cancer metastasis in vivo. Proc. Natl. Acad. Sci. U.S.A 2022, 119 (31), No. e2203167119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Greinacher A; Farner B; Kroll H; Kohlmann T; Warkentin TE; Eichler P Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis. Thromb. Haemostasis 2005, 94 (1), 132–135. [DOI] [PubMed] [Google Scholar]
- (34).Arepally GM; Ortel TL Heparin-Induced Thrombocytopenia. N. Engl. J. Med 2006, 355 (8), 809–817. [DOI] [PubMed] [Google Scholar]
- (35).Ahmed I; Majeed A; Powell R Heparin induced thrombocytopenia: diagnosis and management update. Postgrad. Med. J 2007, 83 (983), 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen Q; Li F; Wang H; Bu C; Shi F; Jin L; Zhang Q; Chi L Evaluating the immunogenicity of heparin and heparin derivatives by measuring their binding to platelet factor 4 using biolayer interferometry. Front. Mol. Biosci 2022, 9, No. 966754, DOI: 10.3389/fmolb.2022.966754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhang X; Chen L; Bancroft DP; Lai CK; Maione TE Crystal structure of recombinant human platelet factor 4. Biochemistry 1994, 33 (27), 8361–8366. [DOI] [PubMed] [Google Scholar]
- (38).Huynh A; Arnold DM; Kelton JG; Smith JW; Horsewood P; Clare R; Guarné A; Nazy I Characterization of platelet factor 4 amino acids that bind pathogenic antibodies in heparin-induced thrombocytopenia. J. Thromb. Haemost 2019, 17 (2), 389–399. [DOI] [PubMed] [Google Scholar]
- (39).Olson ST; Chuang Y-J Heparin Activates Antithrombin Anticoagulant Function by Generating New Interaction Sites (Exosites) for Blood Clotting Proteinases. Trends Cardiovasc. Med 2002, 12 (8), 331–338. [DOI] [PubMed] [Google Scholar]
- (40).Chuang YJ; Swanson R; Raja SM; Olson ST Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence - Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin. J. Biol. Chem 2001, 276 (18), 14961–14971. [DOI] [PubMed] [Google Scholar]
- (41).Oh YI; Sheng GJ; Chang SK; Hsieh-Wilson LC Tailored glycopolymers as anticoagulant heparin mimetics. Angew. Chem., Int. Ed 2013, 52 (45), 11796–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wei MY; Ward SM The Anti-Factor Xa Range For Low Molecular Weight Heparin Thromboprophylaxis. Hematol. Rep 2015, 7 (4), No. 5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Vlodavsky I; Ilan N; Sanderson RD Forty Years of Basic and Translational Heparanase Research; Springer International Publishing, 2020; pp 3–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Elkin M; Vlodavsky I Tail vein assay of cancer metastasis Curr. Protoc. Cell Biol 2001; Vol. 12 DOI: 10.1002/0471143030.cb1902s12. [DOI] [PubMed] [Google Scholar]
- (45).Barash U; Rangappa S; Mohan CD; Vishwanath D; Boyango I; Basappa B; Vlodavsky I; Rangappa KS New Heparanase-Inhibiting Triazolo-Thiadiazoles Attenuate Primary Tumor Growth and Metastasis. Cancers 2021, 13 (12), No. 2959, DOI: 10.3390/cancers13122959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Higgs JT; Lee JH; Wang H; Ramani VC; Chanda D; Hardy CY; Sanderson RD; Ponnazhagan S Mesenchymal stem cells expressing osteoprotegerin variants inhibit osteolysis in a murine model of multiple myeloma. Blood Adv. 2017, 1 (25), 2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Marino S; Roodman GD Multiple Myeloma and Bone: The Fatal Interaction. Cold Spring Harbor Perspect. Med 2018, 8 (8), No. a031286, DOI: 10.1101/cshperspect.a031286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yang Y; Macleod V; Miao HQ; Theus A; Zhan F; Shaughnessy JD Jr.; Sawyer J; Li JP; Zcharia E; Vlodavsky I; Sanderson RD Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J. Biol. Chem 2007, 282 (18), 13326–13333. [DOI] [PubMed] [Google Scholar]
- (49).Lovell R; Dunn JA; Begum G; Barth NJ; Plant T; Moss PA; Drayson MT; Pratt G Soluble syndecan-1 level at diagnosis is an independent prognostic factor in multiple myeloma and the extent of fall from diagnosis to plateau predicts for overall survival. Br. J. Hamaetol 2005, 130 (4), 542–548. [DOI] [PubMed] [Google Scholar]
- (50).Seidel C; Sundan A; Hjorth M; Turesson I; Dahl IM; Abildgaard N; Waage A; Borset M Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood 2000, 95 (2), 388–392. [PubMed] [Google Scholar]
- (51).Ramani VC; Vlodavsky I; Ng M; Zhang Y; Barbieri P; Noseda A; Sanderson RD Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016, 55, 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Ramani VC; Zhan F; He J; Barbieri P; Noseda A; Tricot G; Sanderson RD Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget 2016, 7 (2), 1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Belch A; Reece DE; Bahlis NJ; White D; Teixeira B; Camacho F; Plante RK Bortezomib [VELCADE], Pegylated Liposomal Doxorubicin [DOXIL/CAELYX] and Dexamethasone in the Treatment of Previously Untreated Multiple Myeloma Patients: Impact on Quality-of-Life. Blood 2007, 110 (11), No. 3618.17699740 [Google Scholar]
- (54).Tian W; Chen C; Lei X; Zhao J; Liang J CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46 (W1), W363–w367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cochran S; Li CP; Ferro V A surface plasmon resonance-based solution affinity assay for heparan sulfate-binding proteins. Glycoconjugate J. 2009, 26, 577–587. [DOI] [PubMed] [Google Scholar]
- (56).Valente V; Teixeira SA; Neder L; Okamoto OK; Oba-Shinjo SM; Marie SK; Scrideli CA; Paçó-Larson ML; Carlotti CG Jr. Jr., Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC Mol. Biol 2009, 10, No. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The U-87 glioblastoma, B-16 melanoma, and MPC-11 myeloma data sets that support the findings are publicly available and were obtained from NCBI GEO with the accession codes GSE58644 and GSE9014. The remaining data are available in the Article and Supporting Information.