

Abstract
Nitric oxide, a signaling molecule, inhibits mitochondrial respiration by binding with cytochrome c oxidase, resulting in elevated production of reactive superoxide species (reactive oxygen and nitrogen) in the mitochondria and increased susceptibility to cell death. Generation of mitochondrial superoxide species can be suppressed by natural compounds such as resveratrol, a dietary polyphenol found in the skin of red fruits. In various cancer cells, resveratrol shows anti-oxidant and cancer preventive properties. Since, the effect of resveratrol on reactive superoxide species–independent apoptosis in prostate cancer cells is not well illustrated; therefore, we investigated this phenomenon in TRAMP murine prostate cancer cells. To accomplish this, TRAMP cells were incubated with resveratrol, resveratrol + DETA-NONOate, DETA-NONOate (nitric oxide donor), resveratrol + L-NMMA, or L-NMMA (nitric oxide inhibitor) for 48 h, and reactive superoxide species in the mitochondria and culture supernatant were measured. In addition, the mitochondrial membrane potential, cell viability, expression of apoptotic markers (Bax and Bcl2), γ-H2A.x, p53, and caspase-3 was determined. We found that resveratrol suppressed reactive superoxide species such as reactive oxygen species in the mitochondria and nitric oxide in culture supernatant when compared to the DETA-NONOate treatment and disrupted the mitochondrial membrane potential. Resveratrol also reduced cell viability, altered the expression of apoptotic markers (Bax and Bcl2), and increased expression of γ-H2A.x (indicative marker of DNA fragmentation) and p53 (a critical DNA damage response protein). However, there was no appreciable modulation of the caspase-3. Therefore, our data suggest that resveratrol induces superoxide species–independent apoptosis and may act as a therapeutic agent against prostate cancer.
Keywords: Resveratrol, DETA-NONOate, L-NMMA, apoptosis, cancer therapeutics
Introduction
Prostate cancer is the most common cancer in American men and is one of the leading causes of cancer-related deaths among men of all races (second leading cause of cancer death among White, Black, and Hispanic men).1 Prostate cancer, like many other cancers, poses a unique problem to affected populations and there is an increasing need for alternative treatment therapies due to the ineffectiveness and toxic side effects of traditional methods. In recent years, there has been a noticeable shift in the thinking of physicians and researchers alike from the use of synthetic/chemical treatments to more natural remedies. Recent findings suggest that the use of dietary compounds could be an alternate strategy to control prostate cancer progression.2,3 In this study, we investigated the prolonged effects of resveratrol (RES) on reactive superoxide species. RES is a naturally occurring polyphenol found in the skin of red fruits, including grapes. The most abundant source of RES is thought to be Polygonum cuspidatum (Japanese knotweed), a plant used to treat inflammation and other ailments.4 RES also demonstrated healing properties, and, in cell cultures and in animals, it has anti-oxidant and anti-cancer effects.4,5 In organ cultures of mouse mammary glands, RES inhibits the formation of preneo-plastic lesions and, in human hepatoma cells, induces the expression of messenger RNA (mRNA) of CYP1A1 (a gene encoding a member of the cytochrome P450 super-family of enzymes) and CYP1A1 activity by preventing interactions between CYP1A1 and the aryl hydrocarbon receptor.5,6 In monocyte endothelial cells, RES reduces proliferation by modulating the activation of protein kinases C and D.7–10 Furthermore, in MCF-7 (Michigan Cancer Foundation-7) cells, RES induces apoptosis by suppressing activation of nuclear factor kappa B (NF-κB) and activator protein-1 (AP1).11 In addition, RES blocks the development of adenomas in the small intestine of mice and contributes to the formation of cryptic foci in rat colon.12,13 These preclinical results indicate that RES can have favorable effects on public health.14–24 Clinical trials have been undertaken to assess the use of RES as a cancer preventive and therapeutic agent.14,25–27
Apoptosis is a natural process of cell/tissue remodeling, replacement and organ size control, process that remove damaged and infected cells.20,21,28,29 During apoptosis membrane blebbing, chromatin condensation, DNA fragmentation, formation of apoptotic bodies, and activation of caspases can be observed.30–35 Mitochondrial dysfunction is involved in caspase activation,36–38 and proteins of the Bcl2 family (Bax and Bcl2) that are localized in the mitochondria suggest their role in the regulation of mitochondria-mediated apoptosis.30–35
Furthermore, nitric oxide (NO), a potent signaling molecule contributes to mitochondrial dysfunction by inhibiting cytochrome c oxidase, resulting in overproduction of reactive superoxide species.39,40 Overproduction of these species in the mitochondria increases susceptibility to cell death.39,40 However, RES suppresses production of reactive superoxide species41,42 and prevents the expression of NO synthase, a process that blocks tumor growth and migration. It also acts as an anti-oxidant and can prevent tumor formation.4,43,44 Previous report from our lab suggested that TRAMP cells incubated with 100 μM of RES for 16 h showed mitochondria-mediated caspase-independent apoptosis.45 However, the long-term effect of RES in relation to the modulation of superoxide species when cells remain in stress due to high reactive superoxide species is not well illustrated. Thus, in this investigation, we hypothesized that how RES induces superoxide species–independent apoptosis in TRAMP cells. To test this, we aimed to determine whether prolonged exposure of RES induces reactive superoxide species–independent apoptosis in murine prostate cancer cells. Our findings demonstrated that RES suppressed reactive superoxide species such as reactive oxygen species (ROS) in the mitochondria and NO in culture supernatant when compared to DETA-NONOate treatment and disrupted mitochondrial membrane potential (Δψm). RES also reduced cell viability, altered the expression of apoptotic markers (Bax and Bcl2), and increased expression of γ-H2A.x (indicative marker of DNA fragmentation) and p53 (a critical DNA damage response protein). However, there was no appreciable modulation of the caspase-3.
Materials and methods
Reagents
Dulbecco’s Modified Eagle’s Medium (DMEM) was obtained from Gibco, Life Technologies (NY, USA), and fetal bovine serum (FBS) was purchased from HyClone Laboratories (CA, USA). RES was purchased from ACROS ORGANICS (NJ, USA). DETA-NONOate (NO donor) and NG-monomethyl-L-arginine acetate (L-NMMA) were obtained from Cayman Chemical (MI, USA). 5,5′,6,6′-Tetrachoro-1,1′,3,3′-tetraethybenzimidazolylcarb ocyanine iodide (JC-1) was obtained from AnaSpec (CA, USA). MitoSox (a mitochondrial ROS stain), Bax, Bcl2, p53, caspase-3 antibodies, trypan blue, trypsin, bovine insulin, penicillin/streptomycin, Dulbecco’s phosphate-buffered saline (DPBS), and dehydroisoandrosterone were purchased from Thermo Fisher Scientific (MA, USA). Protein ladder and nitrocellulose membranes were obtained from Bio-Rad (CA, USA). The γ-H2A.x antibody was purchased from Cell Signaling Technology (MA, USA). Griess reagent was purchased from Promega (WI, USA).
Cell culture
The Biosafety Committee of Alabama State University (ASU) approved all experiments with TRAMP cells, which were obtained from the American Type Culture Collection (ATCC, VA, USA). TRAMP cells were maintained in DMEM, and supplemented with 10% (v/v) FBS, 0.005 mg/mL bovine insulin, 10 nM dehydroisoandrosterone, and 1% antibiotics/antimycotics at 37°C under 5% CO2. Cells were passaged every 3–4 days when they reached 70%–80% confluency, and after trypsinization, the cells were used for experiments. Before the cells were used in experiments, the cell viability was determined by trypan blue exclusion method.
Estimation of reactive superoxide species
ROS in the mitochondria of TRAMP-C1 and TRAMP-C3 cells was accomplished by using MitoSox stain (1:500 dilution) after exposure of cells (1 × 105) to RES (50 μM), with or without DETA-NONOate (750 μM) and L-NMMA, for 48 h under 5% CO2 at 37°C. After incubation, the cells were harvested by trypsinization (0.25%), washed, and stained for mitochondrial ROS at 37°C for 10 min as recommended.46 After staining, the cells were washed and observed under a flow cytometer (NOVOCyte; ACEA Biosciences, CA, USA).
In a separate experiment, NO level in culture supernatants was measured by Griess reagent (Promega). Briefly, 50 μL of culture supernatant was added to each well of a 96-well plate with appropriate standard bovine serum albumin (BSA). A volume of 50 μL of sulfonamide solution was added to each well, and the plates were incubated for 10 min at room temperature. Thereafter, 50 μL of N-1-napthalenediamine dihydrochloride solution was added, and absorbance was read at 570 nm with a microplate reader (ChroMate, FL, USA).
Assessment of Δψm
To measure the Δψm, TRAMP cells (1 × 105) were exposed to RES (50 μM), with or without DETA-NONOate (750 μM) and L-NMMA, for 48 h under 5% CO2 at 37°C. Cells were then stained with JC-1 cationic dye for 10 min at 37°C, washed with DPBS, and analyzed by flow cytometry. The JC-1 dye enters into the cells and, in its multimeric form, fluoresces red within healthy mitochondria. In contrast, for cells with disrupted Δψm, JC-1, in its monomeric form, remains in the cytoplasm and produces a green fluorescence.
Assessment of Bax and Bcl2 expression
Total protein was extracted using radioimmunoprecipitation assay (RIPA) buffer from TRAMP-C1 and TRAMP-C3 cells; protein concentration was estimated by Bradford assay; and estimated proteins were run on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 30–40 μg/lane) using a 1:1 ratio of the sample-loading buffer at 70V for 2–3 h. The proteins were transferred onto nitrocellulose membrane and blocked with 5% skimmed milk in Tris-buffered saline (TBS) containing 0.1% Tween 20 (T20) for 30 min at room temperature. The membranes were then incubated with monoclonal antibodies against murine Bax, p53, and Bcl2 (Bax: 1:100, p53: 1:50, and Bcl2: 1 μg/mL) for overnight at 4°C. Furthermore, membranes were incubated with goat anti-mouse antibody (Goat anti-mouse; 1:1000), and protein bands were detected by the chemiluminescence liquid substrate system (Bio-Rad). Microphotographs were captured by gel documentation system (Bio-Rad), and densitometric analyses were performed with Quantity One 1-D analysis software (Bio-Rad). For densitometry, protein bands were marked with a square and density values in INT/mm2 were used for further analysis. Densitometry data show an average of three independent western blots.
Assessment of apoptosis
To access the number of apoptotic cells, annexin V–fluorescein isothiocyanate (FITC)–propidium iodide (PI) staining was carried out. FITC–annexin V is a sensitive probe for identifying apoptotic cells (early apoptotic: FITC+, late apoptotic cells: FITC+/PI+); however, PI+ cells were considered as dead. To accomplished this, cells (1 × 105) were exposed to RES (50 μM), RES + DETA-NONOate (750 μM), DETA-NONOate, RES + L-NMMA (750 μM), or L-NMMA for 48 h under 5% CO2 at 37°C. Cells were harvested and incubated with annexin V–FITC–PI for 15 min at room temperature. Cells were washed and analyzed by a flow cytometer.
Assessment of caspase-3 expression
Caspase-3 expression was examined in TRAMP-C1 and TRAMP-C3 cells. For caspase-3 expression, TRAMP cells (1 × 105 cells/mL) were exposed to RES (50 μM), with or without DETA-NONOate (750 μM) and L-NMMA, for 48 h under 5% CO2 at 37°C. Cells were then harvested and lysed in RIPA buffer. Estimated protein (by Bradford assay) was resolved on 10% SDS-PAGE. Proteins were transferred to nitrocellulose membranes. Membranes were blocked with 5% skimmed milk (freshly prepared in TBS-T20) and incubated with the anti-caspase-3 antibody (2 μg/mL) for 1 h at room temperature. After primary antibody incubation, membranes were hybridized with secondary antibody (horse anti-mouse: 1:2000). Caspase-3 expression was determined by using a chemiluminescent liquid substrate system (Millipore Sigma, MA, USA).
DNA fragmentation analysis
To assess DNA fragmentation, γ-H2A.x expression was measured by Western blot analyses. Cells were exposed to RES in the presence or absence of DETA-NONOate or L-NMMA. After incubation, the cells were lysed in RIPA buffer at 4°C, and estimated concentration of total proteins was run on 10% SDS-PAGE. The protein complexes were transferred onto nitrocellulose membranes and incubated with mouse anti-γ-H2A.x (1:1000) and internal control β-actin (1:2000) antibodies, followed by secondary antibody (1:2000) incubation (goat anti-rabbit and horse anti-mouse). The expression of γ-H2A.x was detected using a chemiluminescent liquid substrate system (Millipore Sigma, MA, USA).
Statistical analyses
The results represent at least three independent experiments, and numerical data are presented as mean ± standard error of the mean (SEM). Data were analyzed with Sigma plot version 12. Statistical significance was evaluated with analysis of variance (ANOVA), followed by a Bonferroni post hoc test. Data were considered significant when p < 0.05.
Results
RES suppresses reactive superoxide species
To assess the effect of RES on the reactive superoxide species such as ROS in the mitochondria, MitoSox staining of TRAMP-C3 and TRAMP-C1 cells was accomplished. In TRAMP (TRAMP-C1 and TRAMP-C3) cells, RES, in the presence and/or absence of DETA-NONOate or L-NMMA, suppressed ROS in the mitochondria relative to the DETA-NONOate treatment (Figure 1(a)). Furthermore, mitochondrial ROS was suppressed by RES in TRAMP-C1 and TRAMP-C3 cells; however, more effectively in TRAMP-C1 than in TRAMP-C3 (Figure 1(a)). Incubated with L-NMMA, TRAMP-C1 cells, but not TRAMP-C3 cells, showed a decrease in mitochondrial ROS (Figure 1(a)). TRAMP-C1 cells exposed to RES, RES + L-NMMA, and L-NMMA had lower levels of ROS than TRAMP-C3 cells (Figure 1(a)). Interestingly, RES treatment showed low mitochondrial ROS in TRAMP (TRAMP-C1 and TRAMP-C3) cells when compared to the RES + DETA-NONOate and DETA-NONOate treatment (Figure 1(a)).
Figure 1.
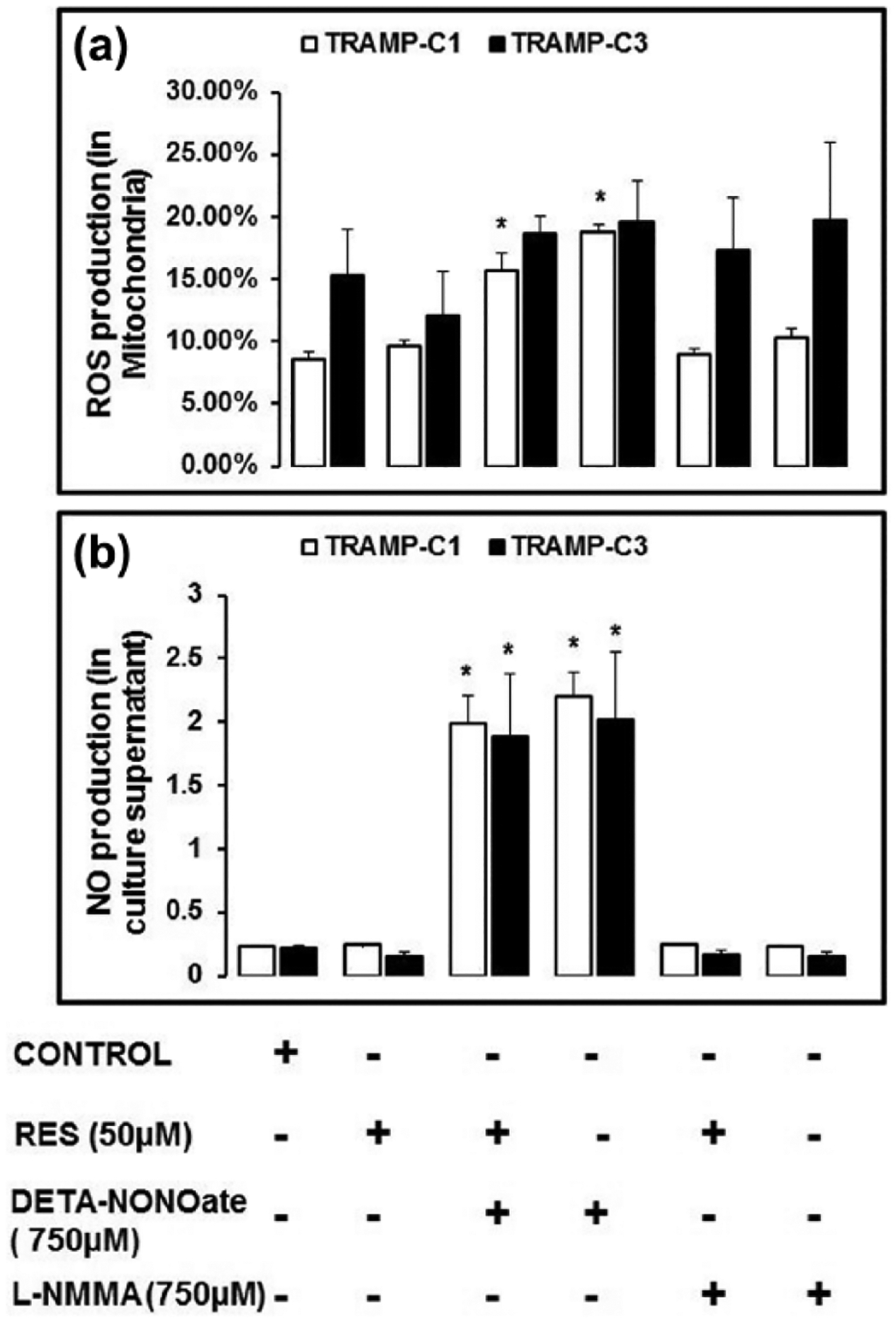
RES reduces reactive superoxide species. (a) TRAMP cells were incubated with RES in the absence or in the presence of DETA-NONOate and L-NMMA for 48 h in 5% CO2 at 37°C. After incubation, the cells were harvested, washed, and stained with MitoSox stain (1:500 dilution) for flow cytometric analysis. RES treatment suppressed reactive superoxide species such as ROS in the mitochondria as compared DETA-NONOate treatment. (b) Nitric oxide (NO) production in culture supernatant was examined using Griess Reagent colorimetric assay. We found that DETA-NONOate treated cells showed high level of NO in culture supernatant when compared to the RES treatment. However, cells incubated with RES, RES + DETA-NONOate, and L-NMMA showed decreased level of NO compared to DETA-NONOate treatment. Furthermore, TRAMP-C1 cells showed a higher level of NO in culture supernatant compared to TRAMP-C3. These findings suggest that RES can suppress reactive superoxide species such as ROS in the mitochondria and NO in culture supernatant. Data showed three independent experiment (*p < 0.05).
In the culture supernatants, there was higher level of NO for TRAMP-C1 and TRAMP-C3 cells when exposed to RES + DETA-NONOate or DETA-NONOate (Figure 1(b)). RES treatment with DETA-NONOate exhibited lower level of NO in the culture supernatant of TRAMP (TRAMP-C1 and TRAMP-C3) cells relative to DETA-NONOate treated cells (Figure 1(b)). In TRAMP-C1 and TRAMP-C3 cells, RES treatment, with or without L-NMMA, resulted in basal level production of NO (Figure 1(b)). In addition, TRAMP-C3 cells incubated with RES + DETA-NONOate or DETA-NONOate demonstrated low level of NO in the culture supernatant than TRAMP-C1. These findings suggest that RES can suppress reactive superoxide species such as ROS in the mitochondria and NO in culture supernatant.
RES treatment modulates the Δψm
RES treatment, with or without DETA-NONOate or L-NMMA, disrupted Δψm compared to the control cells (Figure 2(a) and (b)). TRAMP-C3 cells showed more disrupted Δψm as compared to TRAMP-C1 cells when treated with RES, RES + DETA-NONOate, DETA-NONOate, and RES + L-NMMA. Furthermore, results revealed that cells incubated with RES + DETA-NONOate and DETA-NONOate showed higher number of TRAMP-C3 cells with disrupted Δψm than other experimental groups (Figure 2(b)). In addition, in TRAMP-C3 cells, RES + DETA-NONOate treatment results in less number of cells with disrupted Δψm than DETA-NONOate treatment. L-NMMA did not show a significant difference between treated and control cells. Data suggest that TRAMP-C3 cells are more sensitive to disrupted Δψm compared to the TRAMP-C1 cells.
Figure 2.
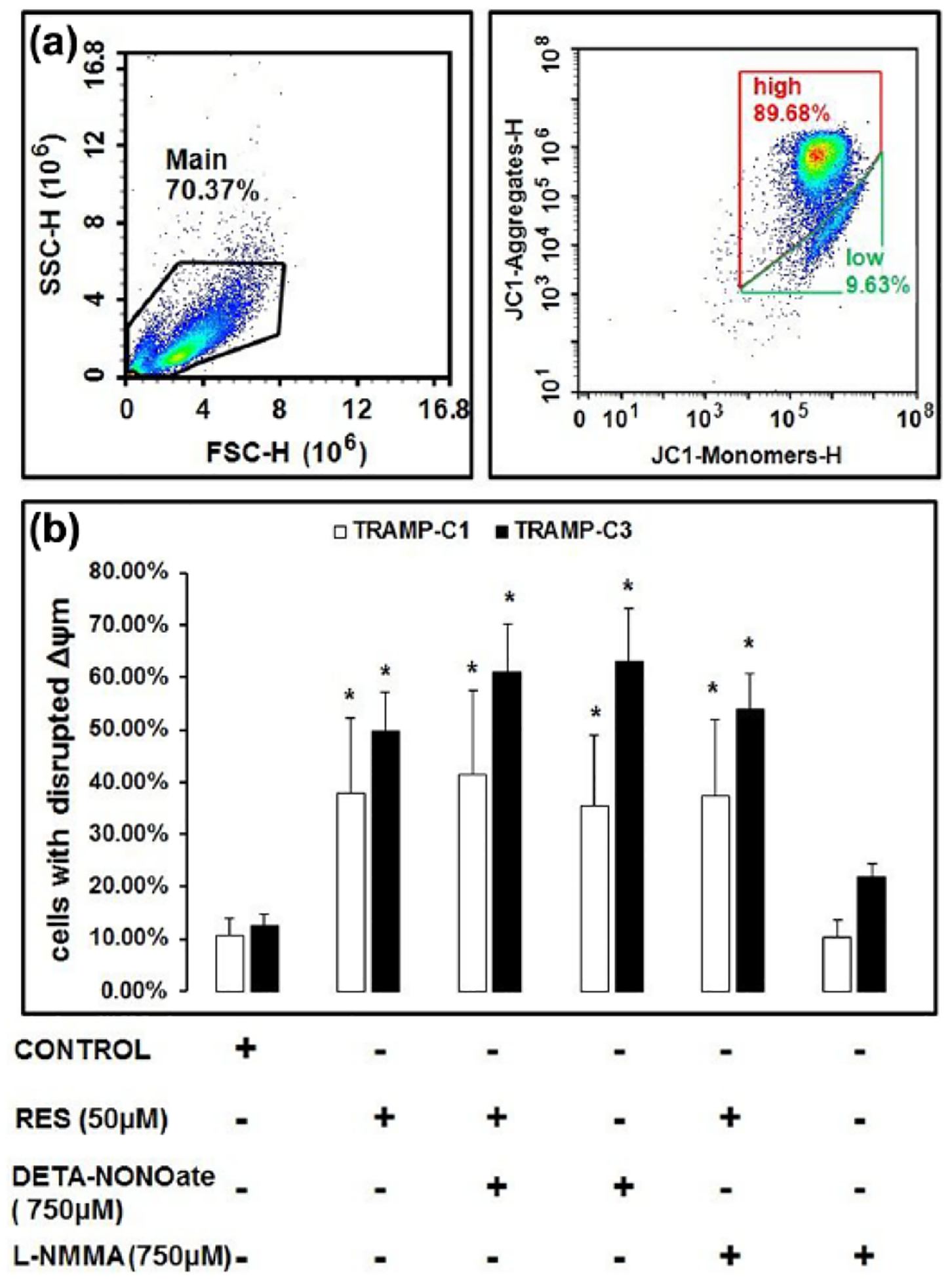
RES modulates mitochondrial membrane potential (Δψm). TRAMP-C1 and TRAMP-C3 cells were exposed to RES, with or without DETA-NONOate or L-NMMA, for 48 h at 37°C under 5% CO2; thereafter, the cells were stained with JC-1 (cationic dye) for 10 min in DPBS and analyzed by a flow cytometry. Results indicated that treatment of RES, with or without DETA-NONOate and L-NMMA, showed increased number of cells with disrupted Δψm as compared to the control. The number of cells with disrupted Δψm was higher in TRAMP-C3 than TRAMP-C1 which suggested that TRAMP-C3 cells were more sensitive to death than TRAMP-C1. (a) Representative figure of experiment. (b) Histogram showing the percentages of cells with disrupted Δψm, derived as averages of three independent experiments (*p < 0.05).
RES modulates expression of Bax and Bcl2
As determined by Western blotting, RES, with or without DETA-NONOate or L-NMMA, modulated the expression of Bax and Bcl2 in TRAMP (TRAMP-C1 and TRAMP-C3) cells (Figure 3(a)). However, treatment with L-NMMA did not cause a significant change in the expression of Bax or Bcl2 (Figure 3(a)). Densitometric analysis of the results revealed that, in TRAMP-C3 cells, RES, in the absence and/or presence of DETA-NONOate, showed higher expression of Bax but not Bcl2 (Figure 3(b)). Furthermore, there was a higher expression of Bax in four of the five groups of treated TRAMP-C3 cells (Figure 3(b)). In TRAMP-C1 cells, the levels of Bcl2 were higher than Bax (Figure 3(c)). Higher expression of Bax in TRAMP-C3 cells suggested that TRAMP-C3 cells are more sensitive to apoptosis than TRAMP-C1 cells.
Figure 3.
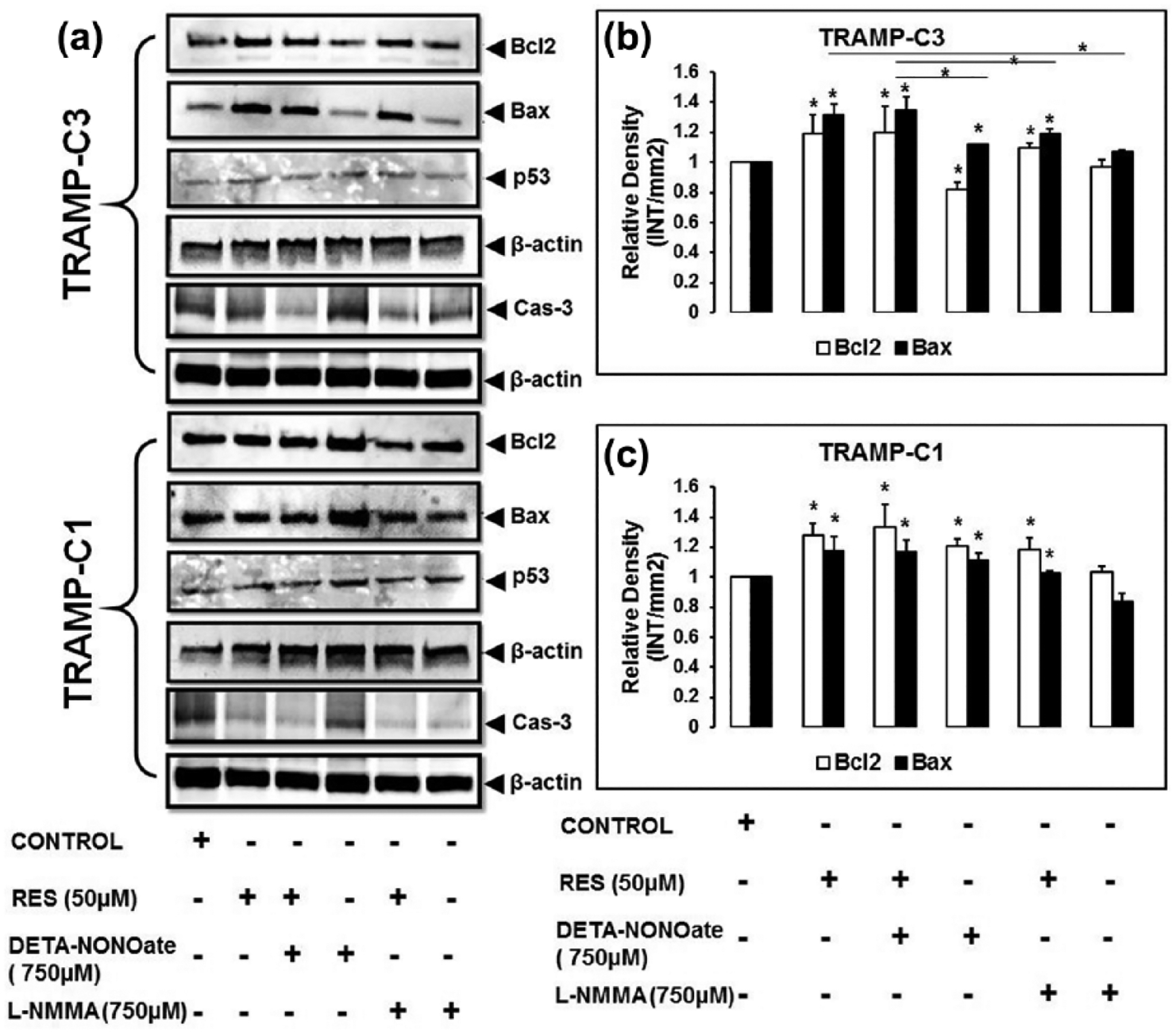
In TRAMP cells, RES modulates the expression of Bax and Bcl2. (a–c) To examine the expression of Bax and Bcl2, cells were incubated with RES in the presence or in the absence of DETA-NONOate and L-NMMA for 48 h. After incubation, the cells were lysed in RIPA buffer and estimated concentration of lysates was resolved on 10% SDS-PAGE for western blot analysis. For TRAMP-C3 cells, Bax expression was higher after exposure to RES, RES + DETA-NONOate, DETA-NONOate, or RES + L-NMMA compared to Bcl2. However, Bcl2 expression was less as compared to Bax. (c) For treated TRAMP-C1 cells, Bcl2 level was higher than that of Bax. (a) The expression of p53 (a DNA damage response protein) was also higher in treated TRAMP cells in comparison to the control. In TRAMP cells, there was no appreciable change in expression of caspase-3 as compared to the control. (a) These findings suggest that increased expression of Bax and p53 is playing important role in the induction of apoptosis (*p < 0.05).
RES modulates expression of caspase-3 and p53
Apoptosis was assessed by examining the expression of caspase-3 and p53. RES, with DETA-NONOate, in TRAMP-C3 cells, caused slight change in caspase-3 expression relative to the control (Figure 3(a)), while other treatment group(s) in TRAMP-C3 cells did not show a significant difference in caspase-3 expression. TRAMP-C1 cells demonstrated that RES treatment in the presence of both DETA-NONOate and L-NMMA resulted in slight change in caspase-3 as compared to the control (Figure 3(a)); however, no significant difference was evident in the DETA-NONOate treated group (Figure 3(a)). Furthermore, the expression of p53 in the TRAMP cells exposed to RES, with or without DETA-NONOate or L-NMMA, was higher relative to the control (Figure 3(a)). Increased expression of p53 suggested sign of DNA damage and apoptosis.
DNA fragmentation analysis
To confirm, reactive superoxide species–independent apoptosis in TRAMP cells, DNA fragmentation was determined by testing γ-H2A.x expression. RES, with or without DETA-NONOate or L-NMMA, caused higher expression of γ-H2A.x relative to the control (Figure 4). RES, alone or in combination with DETA-NONOate, caused high expression of γ-H2A.x in TRAMP (TRAMP-C1 and TRAMP-C3) cells compared to the control (Figure 4). TRAMP-C1 and TRAMP-C3 cells demonstrated the same pattern of γ-H2A.x expression when incubated with RES, RES + DETA-NONOate, DETA-NONOate, and RES + L-NMMA (Figure 4). However, DETA-NONOate treatment showed higher expression of γ-H2A.x in TRAMP-C3 cells relative to TRAMP-C1 cells (Figure 4). In TRAMP-C1, RES and RES + L-NMMA treatment resulted in higher expression of γ-H2A.x than other treatment groups (Figure 4). These findings suggest that increased expression of γ-H2A.x can be used as an indicator of DNA fragmentation.
Figure 4.

RES induces DNA fragmentation. Cells were treated in similar experimental condition as previously mentioned and expression of γ-H2A.x (indicative of DNA fragmentation) was examined. Relative to the control, γ-H2A.x expression was higher after treatment of cells with RES, RES + DETA-NONOate, DETA-NONOate, or RES + L-NMMA. Further analysis revealed that RES, DETA-NONOate, and RES + L-NMMA treatment resulted in high expression of γ-H2A.x than control and RES + DETA-NONOate, suggesting its role in DNA fragmentation.
RES induces apoptosis
To test RES-induced reactive superoxide species–independent apoptosis of TRAMP cells, cell killing was examined using a flow cytometer. In TRAMP cells, RES, with or without DETA-NONOate or L-NMMA, increased killing as compared to control. The cell killing was found higher when TRAMP cells were incubated with RES, RES + DETA-NONOate, DETA-NONOate, and L-NMMA. RES + DETA-NONOate treatment resulted in higher cell killing in TRAMP-C1 and TRAMP-C3 cells compared to the control cells (Figure 5(b)). TRAMP-C3 cells were more susceptible to cell killing than TRAMP-C1 cells (Figure 5(b)). In addition, TRAMP cells showed statistically significant differences when compared to their respective control (Figure 5(b)). Our results suggested that TRAMP-C3 cells were more sensitive to RES + DETA-NONOate and DETA-NONOate than TRAMP-C1 cells.
Figure 5.

RES decreases cell viability in TRAMP cells. Cells were exposed to RES, RES + DETA-NONOate, DETA-NONOate, RES + L-NMMA, or L-NMMA for 48 h in 5% CO2 at 37°C. After incubation, cells were harvested and stained with propidium iodide and annexin V conjugated with FITC for flow cytometric analysis. Our data suggested that RES treatment, with or without DETA-NONOate and L-NMMA, increased cell killing relative to the control. Further analysis revealed that the cell killing was higher in TRAMP-C3 cells compared to TRAMP-C1 cells. This suggests that TRAMP-C3 cells were more sensitive to undergo apoptosis than TRAMP-C1. (a) Representative figure of gating strategies. (b) Average values of cell killing derived from three independent experiments (*p < 0.05).
Discussion
In this article, we demonstrated that RES induced reactive superoxide species–independent apoptosis in murine prostate cancer cells by the suppression of reactive superoxide species such as ROS in the mitochondria and NO in culture supernatant when compared to DETA-NONOate treatment and disrupted the Δψm. Further results suggest that RES treatment reduced cell viability, altered the expression of apoptotic markers (Bax and Bcl2), and increased expression of γ-H2A.x (indicative of DNA fragmentation) and p53 (a critical DNA damage response protein). However, there was no appreciable modulation of the caspase-3. Furthermore, RES protects cells, in part, by suppressing superoxide species in the mitochondria and preserves cellular homeostasis.20,21,24,29 RES can modulate a variety of cellular processes such as mitochondria-associated apoptosis.47
Our findings demonstrate that DETA-NONOate-induced level of ROS in the mitochondria and NO in culture supernatant can be suppressed by prolonged exposure of RES, which may result in the inhibition of superoxide species–dependent apoptosis. In contrast, studies suggested that RES treatment induced apoptosis in HT-29 human colorectal carcinoma cells through ROS-dependent mitochondrial pathway.48,49 However, RES treatment showed suppression of ROS in the mitochondria when combined with DETA-NONOate compared to the DETA-NONOate. Also, NO level was observed to be higher in DETA-NONOate treated group of cells compared to RES + DETA-NONOate. Furthermore, the level of ROS in the mitochondria and NO in the culture supernatant in RES treated cells was not higher when compared to RES + DETA-NONOate and DETA-NONOate treated cells. RES, when combined with DETA-NONOate, suppressed ROS in the mitochondria and NO in culture supernatant in TRAMP cells (TRAMP-C1 and TRAMP-C3). These findings suggest that RES may be a possible therapeutic regimen that can be used as an anti-oxidant agent. Studies demonstrated that superoxide species in mitochondria can cause mitochondrial and endoplasmic dysfunction which further leads to the induction of apoptosis.37,50 However, our findings indicate a possible role of RES in superoxide species–independent apoptosis by the suppressing reactive superoxide species such as ROS and NO when compared to DETA-NONOate treatment. In addition, results suggested that cells incubated with DETA-NONOate and RES + DETA-NONOate demonstrated significantly high number of TRAMP-C3 cells with disrupted mitochondria compared to TRAMP-C1. This suggests that TRAMP-C3 cells are more sensitive to undergo mitochondrial dysfunction than TRAMP-C1.
Furthermore, proteins of the Bcl2 family are involved in the control of mitochondria-dependent apoptosis. These proteins, acting upstream to the mitochondria, are proapoptotic (Bax) and anti-apoptotic (Bcl2). The relative ratio of these proteins (Bax/Bcl2) is a determining factor in the induction of apoptosis.51 Thus, in this study, the expression of Bax and Bcl2 was determined which suggested that their expression was altered relative to their respective controls. It was found that Bax expression was relatively higher in TRAMP-C3 cells than Bcl2 and respective control cells. Enhanced expression of Bax plays a significant role in the induction of apoptosis.52 Furthermore, p53, a DNA damage response protein and guardian of the cell, plays a crucial role in the induction of apoptosis. Thus, the level of p53 was examined in TRAMP cells (TRAMP-C1 and TRAMP-C3) which was found to be higher in treatment groups relative to the control. This suggests that apoptosis in TRAMP-C1 cells may be associated with the level of p53. High expression of p53 also indicated fragmentation of genomic DNA into low molecular weight DNA fragments. In view of this, our data suggested that DETA-NONOate and RES + L-NMMA treatment increased the expression of γ-H2A.x; however, RES + DETA-NONOate treatment resulted in less expression of this protein compared to the treatment of RES, RES + L-NMMA, and DETA-NONOate. This suggests that γ-H2A.x could be a reliable indicator of DNA fragmentation. Previous studies demonstrated that histone γ-H2A.x, an evolutionarily conserved variant of histone H2A, has been identified as one of the key histones to undergo various post-translational modifications in response to double-stranded DNA breaks,53,54 and increased expression of γ-H2A.x can be used to determine DNA fragmentation.55,56
The sensitivity of cancer cells to apoptosis induced by anti-cancer agents suggested decreased cell viability and altered cell morphology.18,21 Our data suggested that RES treatment, with or without DETA-NONOate and L-NMMA, showed increased cell killing relative to their respective controls. Further analysis revealed that TRAMP-C3 cells are more sensitive to DETA-NONOate and RES + DETA-NONOate than TRAMP-C1.
In summary, RES induces reactive superoxide species–independent apoptosis in murine prostate cancer cells by the suppression of reactive superoxide species such as ROS in the mitochondria and NO in culture supernatant when compared to DETA-NONOate treatment and disrupted the Δψm. Further results suggest that RES treatment reduced cell viability, altered the expression of apoptotic markers (Bax and Bcl2), and increased expression of γ-H2A.x (indicative of DNA fragmentation) and p53 (a critical DNA damage response protein). However, there was no appreciable modulation of the caspase-3 expression. These findings suggest that RES possesses the therapeutic potential and can be used against prostate cancer cells.
Acknowledgements
The authors thank Dr Donald Hill and Shalie Malik for their critical review of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors have been supported, in part, by National Institutes of Health (Grants: P20CA192976 to M.K.M. and P20CA192973 to U.M.), U.S. Department of Defense (Grants: W911NF-12-1-0073 and W911NF-14-1-0064 to M.K.M.), and National Science Foundation (Grant: 1154214 to M.K.M.).
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Jackson DD, Owens OL, Friedman DB, et al. An intergenerational approach to prostate cancer education: findings from a pilot project in the southeastern USA. J Cancer Educ 2014; 29(4): 649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rai G, Suman S, Mishra S, et al. Evaluation of growth inhibitory response of Resveratrol and Salinomycin combinations against triple negative breast cancer cells. Biomed Pharmacother 2017; 89: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Chen B, Jiang R, et al. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell Immunol 2017; 311: 86–93. [DOI] [PubMed] [Google Scholar]
- 4.Carter LG, D’Orazio JA and Pearson KJ. Resveratrol and cancer: focus on in vivo evidence. Endocr Relat Cancer 2014; 21(3): R209–R225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Androutsopoulos VP, Ruparelia KC, Papakyriakou A, et al. Anticancer effects of the metabolic products of the resveratrol analogue, DMU-212: structural requirements for potency. Eur J Med Chem 2011; 46(6): 2586–2595. [DOI] [PubMed] [Google Scholar]
- 6.Ciolino HP, Daschner PJ and Yeh GC. Resveratrol inhibits transcription of CYP1A1 in vitro by preventing activation of the aryl hydrocarbon receptor. Cancer Res 1998; 58(24): 5707–5712. [PubMed] [Google Scholar]
- 7.Haworth RS and Avkiran M. Inhibition of protein kinase D by resveratrol. Biochem Pharmacol 2001; 62(12): 1647–1651. [DOI] [PubMed] [Google Scholar]
- 8.Slater SJ, Seiz JL, Cook AC, et al. Inhibition of protein kinase C by resveratrol. Biochim Biophys Acta 2003; 1637(1): 59–69. [DOI] [PubMed] [Google Scholar]
- 9.Upham BL, Guzvic M, Scott J, et al. Inhibition of gap junctional intercellular communication and activation of mitogen-activated protein kinase by tumor-promoting organic peroxides and protection by resveratrol. Nutr Cancer 2007; 57(1): 38–47. [DOI] [PubMed] [Google Scholar]
- 10.Yoon SH, Kim YS, Ghim SY, et al. Inhibition of protein kinase CKII activity by resveratrol, a natural compound in red wine and grapes. Life Sci 2002; 71(18): 2145–2152. [DOI] [PubMed] [Google Scholar]
- 11.Surh YJ, Chun KS, Cha HH, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res 2001; 480–481: 243–268. [DOI] [PubMed] [Google Scholar]
- 12.Tessitore L, Davit A, Sarotto I, et al. Resveratrol depresses the growth of colorectal aberrant crypt foci by affecting bax and p21(CIP) expression. Carcinogenesis 2000; 21(8): 1619–1622. [PubMed] [Google Scholar]
- 13.Sale S, Tunstall RG, Ruparelia KC, et al. Comparison of the effects of the chemopreventive agent resveratrol and its synthetic analog trans 3,4,5,4′-tetramethoxystilbene (DMU-212) on adenoma development in the Apc(Min+) mouse and cyclooxygenase-2 in human-derived colon cancer cells. Int J Cancer 2005; 115(2): 194–201. [DOI] [PubMed] [Google Scholar]
- 14.Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res 2009; 2(5): 409–418. [DOI] [PubMed] [Google Scholar]
- 15.Fulda S. Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discov Today 2010; 15(17–18): 757–765. [DOI] [PubMed] [Google Scholar]
- 16.Bhardwaj A, Sethi G, Vadhan-Raj S, et al. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood 2007; 109(6): 2293–2302. [DOI] [PubMed] [Google Scholar]
- 17.Cai Y, Zhao L, Qin Y, et al. Resveratrol inhibits proliferation and induces apoptosis of nasopharyngeal carcinoma cell line C666–1 through AMPK activation. Pharmazie 2015; 70(6): 399–403. [PubMed] [Google Scholar]
- 18.Ding XZ and Adrian TE. Resveratrol inhibits proliferation and induces apoptosis in human pancreatic cancer cells. Pancreas 2002; 25(4): e71–e76. [DOI] [PubMed] [Google Scholar]
- 19.Estrov Z, Shishodia S, Faderl S, et al. Resveratrol blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, causes S-phase arrest, and induces apoptosis of acute myeloid leukemia cells. Blood 2003; 102(3): 987–995. [DOI] [PubMed] [Google Scholar]
- 20.Fouad MA, Agha AM, Merzabani MM, et al. Resveratrol inhibits proliferation, angiogenesis and induces apoptosis in colon cancer cells: calorie restriction is the force to the cytotoxicity. Hum Exp Toxicol 2013; 32(10): 1067–1080. [DOI] [PubMed] [Google Scholar]
- 21.Kim YA, Choi BT, Lee YT, et al. Resveratrol inhibits cell proliferation and induces apoptosis of human breast carcinoma MCF-7 cells. Oncol Rep 2004; 11(2): 441–446. [PubMed] [Google Scholar]
- 22.Mo W, Xu X, Xu L, et al. Resveratrol inhibits proliferation and induces apoptosis through the hedgehog signaling pathway in pancreatic cancer cell. Pancreatology 2011; 11(6): 601–609. [DOI] [PubMed] [Google Scholar]
- 23.Poussier B, Cordova AC, Becquemin JP, et al. Resveratrol inhibits vascular smooth muscle cell proliferation and induces apoptosis. J Vasc Surg 2005; 42(6): 1190–1197. [DOI] [PubMed] [Google Scholar]
- 24.Zhai XX, Ding JC and Tang ZM. Resveratrol inhibits proliferation and induces apoptosis of pathological scar fibroblasts through the mechanism involving TGF-β1/Smads signaling pathway. Cell Biochem Biophy 2015; 71: 1267–1272. [DOI] [PubMed] [Google Scholar]
- 25.Charytoniuk T, Drygalski K, Konstantynowicz-Nowicka K, et al. Alternative treatment methods attenuate the development of NAFLD: a review of resveratrol molecular mechanisms and clinical trials. Nutrition 2017; 34: 108–117. [DOI] [PubMed] [Google Scholar]
- 26.Patel KR, Scott E, Brown VA, et al. Clinical trials of resveratrol. Ann N Y Acad Sci 2011; 1215: 161–169. [DOI] [PubMed] [Google Scholar]
- 27.Tome-Carneiro J, Larrosa M, Gonzalez-Sarrias A, et al. Resveratrol and clinical trials: the crossroad from in vitro studies to human evidence. Curr Pharm Des 2013; 19(34): 6064–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosca L and Hortelano S. Mechanisms of nitric oxide-dependent apoptosis: involvement of mitochondrial mediators. Cell Signal 1999; 11(4): 239–244. [DOI] [PubMed] [Google Scholar]
- 29.Chan SH, Wu KL, Wang LL, et al. Nitric oxide- and superoxide-dependent mitochondrial signaling in endotoxin-induced apoptosis in the rostral ventrolateral medulla of rats. Free Radic Biol Med 2005; 39(5): 603–618. [DOI] [PubMed] [Google Scholar]
- 30.Babini G, Bellinzona VE, Morini J, et al. Mechanisms of the induction of apoptosis mediated by radiation-induced cytokine release. Radiat Prot Dosimetry 2015; 166: 165–169. [DOI] [PubMed] [Google Scholar]
- 31.Chang J, Wang H, Wang X, et al. Molecular mechanisms of Polyphyllin I-induced apoptosis and reversal of the epithelial-mesenchymal transition in human osteosarcoma cells. J Ethnopharmacol 2015; 170: 117–127. [DOI] [PubMed] [Google Scholar]
- 32.Chung WH. Mechanisms of a novel anticancer therapeutic strategy involving atmospheric pressure plasma-mediated apoptosis and DNA strand break formation. Arch Pharm Res 2015; 39: 1–9. [DOI] [PubMed] [Google Scholar]
- 33.Han YX, You LS, Liu H, et al. Apoptosis of acute myeloid leukemia HL-60 cells induced by CDK inhibitor SNS-032 and its molecular mechanisms. Zhejiang Da Xue Xue Bao Yi Xue Ban 2015; 44(2): 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan YS, Ooi KK, Ang KP, et al. Molecular mechanisms of apoptosis and cell selectivity of zinc dithiocarbamates functionalized with hydroxyethyl substituents. J Inorg Biochem 2015; 150: 48–62. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Tomar MS and Acharya A. HSF1-mediated regulation of tumor cell apoptosis: a novel target for cancer therapeutics. Future Oncol 2013; 9(10): 1573–1586. [DOI] [PubMed] [Google Scholar]
- 36.Delogu G, Moretti S, Famularo G, et al. Circulating neutrophils exhibit enhanced apoptosis associated with mitochondrial dysfunctions after surgery under general anaesthesia. Acta Anaesthesiol Scand 2001; 45(1): 87–94. [DOI] [PubMed] [Google Scholar]
- 37.Inoue T and Suzuki-Karasaki Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic Biol Med 2013; 61: 273–284. [DOI] [PubMed] [Google Scholar]
- 38.Viola G, Fortunato E, Cecconet L, et al. Induction of apoptosis in Jurkat cells by photoexcited psoralen derivatives: implication of mitochondrial dysfunctions and caspases activation. Toxicol Vitro 2007; 21(2): 211–216. [DOI] [PubMed] [Google Scholar]
- 39.Brown GC and Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 2007; 75(2): 283–290. [DOI] [PubMed] [Google Scholar]
- 40.Benamar A, Rolletschek H, Borisjuk L, et al. Nitrite-nitric oxide control of mitochondrial respiration at the frontier of anoxia. Biochim Biophys Acta 2008; 1777(10): 1268–1275. [DOI] [PubMed] [Google Scholar]
- 41.Shin SM, Cho IJ and Kim SG. Resveratrol protects mitochondria against oxidative stress through AMP-activated protein kinase-mediated glycogen synthase kinase-3beta inhibition downstream of poly(ADP-ribose)polymerase-LKB1 pathway. Mol Pharmacol 2009; 76(4): 884–895. [DOI] [PubMed] [Google Scholar]
- 42.Zhou X, Chen M, Zeng X, et al. Resveratrol regulates mitochondrial reactive oxygen species homeostasis through Sirt3 signaling pathway in human vascular endothelial cells. Cell Death Dis 2014; 5:e1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crek C, Mutlu Altundag E, Karademir B, et al. The effect of resveratrol on signal transduction pathways and the role of proapoptotic Bax protein on apoptosis in HCT- 116 colon carcinoma cell lines. Free Radic Biol Med 2014; 75(Suppl. 1): S27. [DOI] [PubMed] [Google Scholar]
- 44.Garvin S, Ollinger K and Dabrosin C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett 2006; 231(1): 113–122. [DOI] [PubMed] [Google Scholar]
- 45.Kumar S, Eroglu E, Stokes JA 3rd, et al. Resveratrol induces mitochondria-mediated, caspase-independent apoptosis in murine prostate cancer cells. Oncotarget 2017; 8(13): 20895–20908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen WM, Shaw LH, Chang PJ, et al. Hepatoprotective effect of resveratrol against ethanol-induced oxidative stress through induction of superoxide dismutase in vivo and in vitro. Exp Ther Med 2016; 11(4): 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SK, Joe Y, Zheng M, et al. Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production. Antioxid Redox Signal 2014; 20(16): 2589–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juan ME, Wenzel U, Daniel H, et al. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem 2008; 56(12): 4813–4818. [DOI] [PubMed] [Google Scholar]
- 49.Ko CH, Shen SC, Yang LY, et al. Gossypol reduction of tumor growth through ROS-dependent mitochondria pathway in human colorectal carcinoma cells. Int J Cancer 2007; 121(8): 1670–1679. [DOI] [PubMed] [Google Scholar]
- 50.Shibayama-Imazu T, Sonoda I, Sakairi S, et al. Production of superoxide and dissipation of mitochondrial transmembrane potential by vitamin K2 trigger apoptosis in human ovarian cancer TYK-nu cells. Apoptosis 2006; 11(9): 1535–1543. [DOI] [PubMed] [Google Scholar]
- 51.Agarwal MK, Agarwal ML, Athar M, et al. Tocotrienol-rich fraction of palm oil activates p53, modulates Bax/Bcl2 ratio and induces apoptosis independent of cell cycle association. Cell Cycle 2004; 3(2): 205–211. [DOI] [PubMed] [Google Scholar]
- 52.Tang HL, Le AH and Lung HL. The increase in mitochondrial association with actin precedes Bax translocation in apoptosis. Biochem J 2006; 396(1): 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johansson P, Fasth A, Ek T, et al. Validation of a flow cytometry-based detection of γ-H2AX, to measure DNA damage for clinical applications. Cytometry B Clin Cytom. Epub ahead of print 24 May 2016. DOI: 10.1002/cyto.b.21374. [DOI] [PubMed] [Google Scholar]
- 54.Sedelnikova OA, Pilch DR, Redon C, et al. Histone H2AX in DNA damage and repair. Cancer Biol Ther 2003; 2(3): 233–235. [DOI] [PubMed] [Google Scholar]
- 55.Lu C, Zhu F, Cho YY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell 2006; 23(1): 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rogakou EP, Nieves-Neira W, Boon C, et al. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 2000; 275(13): 9390–9395. [DOI] [PubMed] [Google Scholar]