

ABSTRACT
The oxygen reduction reaction (ORR) to either H2O2 or H2O generation is important to meet diverse application demands. The product selectivity of the ORR is strongly correlated with the nature of the catalyst. We report herein that alkali-metal cations (AM+) can steer the product selectivity of the ORR catalysed on a molecular model catalyst with Co–N4 sites. The electron-transfer number of the ORR increases with Li+ ≈ Na+ < K+ < Rb+ < Cs+. A series of electrochemical measurements reveal the 2e−+2e− ORR pathway in large AM+ electrolytes at neutral pH. In situ electrochemical scanning tunneling microscopy resolves the formation of high-contrast species in the cobalt octaethylporphine (CoOEP) monolayer on Au(111) in large AM+ electrolytes when the ORR occurs. The high-contrast species is assigned to the HO2−, as the 2e− ORR product, adsorbed on CoOEP. Combined electrochemical scanning tunneling microscopy, electrochemical measurements and theoretical calculations reveal that large AM+ can stabilize HO2− on CoOEP and promote its further reduction, which accounts for the AM+-dependent selectivity of the ORR. Revealing the unrecognized effect of AM+ on ORR selectivity opens up new avenues for modulating the distribution of ORR products by adjusting the electrolyte composition.
Keywords: electrochemical scanning tunneling microscopy, oxygen reduction reaction, single-atom catalysts, alkali-metal cations
This study reveals that alkali metal cations stabilize reaction intermediates and steer oxygen reduction pathways, enabling selective hydrogen peroxide or water production through simple electrolyte tuning.
INTRODUCTION
The oxygen reduction reaction (ORR) is one of the most widely investigated cathodic processes owing to its important role in energy conversion [1–4]. The ORR in neutral environments has received widespread attention due to its mild reaction conditions and promising applications in biological systems and seawater electrolysis [5,6]. Both 2e− reduction producing H2O2 and 4e− reduction producing H2O by using the ORR are valuable and highly pursued [7–10]. Generally, the product selectivity of the ORR is largely determined by the nature of the electrocatalytically active centers. For instance, 4e− reduction is promoted on Pt, Fe–N4 and Mn–N4 catalytic sites [11–13] and 2e− reduction dominates on Co–N4 sites [14,15]. To meet different practical demands, tremendous efforts have been devoted to developing specialized ORR catalysts with excellent activity and high selectivity through elaborately designed synthesis [7–10]. Recently, the influence of electrolyte components on the selectivity of small-molecule electrocatalysis has received increasing attention [16–18]. A versatile and cost-effective solution to steer the product selectivity of the ORR and to fit different application requirements is highly desirable.
Alkali-metal cations (AM+) are one of the most widely used supporting electrolytes. AM+ significantly affect the performance of many reduction reactions including the ORR [19–21], hydrogen evolution reaction [22,23] and CO2 reduction reaction [24–26]. The mechanism of the AM+ effect is a fundamental issue in electrochemistry that is currently receiving great attention. AM+ have been reported to modulate the potential profile in the electric double layer (EDL) and the surface charge density [24,27–32], buffer the interfacial pH and affect proton diffusion [33–36] and interact directly with reaction intermediates [37]. For example, it has been reported that hydrated AM+, especially Li+ and Na+, interact with the surface-adsorbed OH− on Pt(111) and inhibit O2 from reaching the Pt surface, resulting in the lowered ORR activity [20]. M–N4 electrocatalysts are highly active towards the ORR and are promising alternatives to noble-metal materials [12–15]. The effect of AM+ on ORRs catalysed by M–N4 sites such as product selectivity has not yet been investigated. Metal porphyrins (MPors) and phthalocyanines are molecular models for practical M–N4 catalysts, which have been widely used in mechanistic studies of electrocatalysis. In this work, we used MPors as molecular models and explored the effect of AM+ on the ORR catalysed by M–N4 sites. Moreover, in situ observation of catalytic processes on surface active sites is valuable to provide visual evidence of the catalytic mechanism. Electrochemical scanning tunneling microscopy (EC–STM) allows imaging of surface catalysis at atomic and molecular levels under electrochemical conditions [38–44]. For instance, in situ EC–STM resolves the adsorption of O2 on FePc and reveals the catalytic conversion of O2 on FePc when the ORR occurs [45]. Moreover, it is reported that the ORR activity of Co–N4 sites is positively correlated with the Co–O2 binding strength, which provides in situ evidence of the scaling relationship [46].
Herein, we report that the product selectivity of ORRs catalysed by cobalt octaethylporphine (CoOEP) depends on the species of AM+ in the electrolyte. The electron-transfer number increases in the order of Li+ ≈ Na+ < K+ < Rb+ < Cs+. The O2 is reduced to H2O2 in small AM+ electrolytes and more completely to H2O in large AM+ electrolytes. The 2e−+2e− ORR mechanism in large AM+ is revealed based on Damjanović kinetics. EC–STM is employed to in situ investigate the catalytic conversion of O2 on the CoOEP. High-contrast species are observed in large AM+ electrolytes when the ORR occurs and assigned to the 2e− ORR product adsorbed on the CoOEP. Moreover, the electron-transfer number of the ORR is positively correlated with the surface coverage of the adsorbed 2e− ORR product. The result shows that AM+ modulate the product selectivity of the ORR by regulating the stability of the adsorbed 2e− ORR product. In addition to the previously established dependence of ORR selectivity on the nature of the catalyst, this study opens up a new avenue to steer ORR selectivity by modifying the composition of the electrolyte.
RESULTS AND DISCUSSION
Selectivity of ORR catalysed by the CoOEP in various AM+ electrolytes
Rotating ring-disk voltammetry is employed to study the effect of AM+ on the ORR performance catalysed by using CoOEP. The ORR is measured on the CoOEP-modified Au disk and the produced hydrogen peroxide is detected on the Pt ring. Figure 1a shows the linear-sweep voltammograms (LSVs) in O2-saturated AMClO4 electrolytes (AM = Li, Na, K, Rb and Cs). The reduction current is ascribed to the ORR and is not detected in Ar-saturated electrolytes. The onset potential for the ORR is more positive and the reduction current density is higher in larger AM+ electrolytes. Figure 1b shows the product selectivity of the ORR in different AM+ electrolytes. The electron-transfer number increases following the trend of Li+ ≈ Na+ < K+ < Rb+ < Cs+. It has been reported that cobalt porphyrins and phthalocyanines predominantly promote the 2e− ORR [47]. The influence of AM+ on the product selectivity of ORRs catalysed by CoOEP suggests the noticeable role of large AM+ in facilitating the 4e− ORR process.
Figure 1.

Electrochemical measurements of ORR in different AM+ electrolytes. (a and b) ORR catalysed by CoOEP in various AMClO4 electrolytes. (a) LSVs of ORR catalysed by CoOEP in O2-saturated 80 mM AMClO4 electrolytes (AM = Li, Na, K, Rb, Cs). (b) Electron-transfer number of ORR measured in (a). (c) Tafel plots of ORR measured in (a). (d) Analysis of Damjanović kinetics of ORR catalysed by CoOEP in O2-saturated 80 mM KClO4, RbClO4 and CsClO4 electrolytes.
Furthermore, the Tafel slope values of the ORR in different AM+ electrolytes are similar (∼120 mV/dec, Fig. 1c), suggesting that the rate-determining step (RDS) of the O2 reduction in all AM+ electrolytes is the first electron transfer. M–N4 sites can catalyse the reduction of O2 to H2O via either the direct 4e− or the 2e−+2e− ORR pathway. The cleavage of the M–O bond occurs after the four-electron transfer in the direct 4e− pathway. In the 2e−+2e− pathway, O2 is reduced via the 2e− ORR to hydrogen peroxide, which is further reduced to achieve the four-electron transfer. To study the pathway of the nearly 4e− O2 reduction in large AM+ electrolytes, we analysed the Damjanović kinetics of the ORR (Fig. S1) [48–50]. The rotating ring-disk voltammograms are measured on the CoOEP in O2-saturated CsClO4 electrolyte at various rates of electrode rotation (ω). As shown in Fig. 1d, ID/(IR/N0) (ID, IR and N0 are the disk current, ring current and collection efficiency, respectively) measured at −0.2 V (vs. a saturated calomel electrode (SCE)) is plotted versus ω−1/2. The intercept is fitted to be 0.96 and represents 1+(2k1/k2), where k1 and k2 are the rate constants of the direct 4e− ORR and 2e− ORR, respectively. According to the Damjanović kinetics of the ORR [48–50], an intercept of ∼1 indicates that k1 is close to 0, showing that the direct 4e− process barely occurs. Here, the intercept of 0.96 suggests that the ORR catalysed by CoOEP in the Cs+ electrolyte follows the 2e−+2e− reduction. Similarly, the intercept is fitted to be 0.98 and 1.03 in K+ and Rb+ electrolytes, respectively (Fig. 1d). The electron-transfer number of the ORR positively correlates with the size of the AM+, showing that the reduction of the 2e− ORR product is facilitated in large AM+ electrolytes, which is summarized as:
 |
(1) |
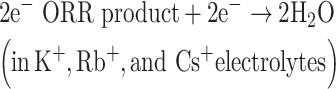 |
(2) |
EC–STM study of the ORR in large AM+ electrolytes
EC–STM was employed to investigate the surface species in the self-assembled CoOEP monolayer on Au(111) prior to and during the ORR. As shown in Fig. 2a, in the Ar-saturated CsClO4 electrolyte, the CoOEP molecules (as marked by the solid square in Fig. 2) appear as bright spots with an apparent height of ∼0.1 nm and they barely undergo any morphological transformation in the potential range of 0.2 to −0.2 V (Fig. S2). In the O2-saturated CsClO4 electrolyte, species with an apparent height of ∼0.15 nm (as marked by the solid circle in Fig. 2b) appears in the CoOEP monolayer at 0.2 V, which is assigned to the O2 adsorption on the CoOEP [46]. At −0.2 V, the high-contrast species (i.e. surface species with relatively high apparent height) [51,52] with an apparent height of ∼0.2 nm (as marked by the dotted circle in Fig. 2c) is observed in the CoOEP monolayer. The high-contrast species is not observed in the Ar-saturated electrolyte and at 0.2 V (i.e. when the ORR does not occur) in the O2-saturated electrolyte. Moreover, the high-contrast species is not observed in the O2-saturated AM+-free and small AM+ electrolytes when the ORR occurs (see above) [46]. These results suggest that the high-contrast species is related to the O2 reduction and indicate the essential role of large AM+ in promoting the formation of high-contrast species. It is reported that the 2e− ORR product readily desorbs from Co–N4 sites due to the relatively weak Co–O bond, resulting in the 2e− ORR pathway [47]. Enhancing the stability of the 2e− ORR product adsorbed on the Co–N4 sites is important for the 4e− ORR pathway. Here, the high-contrast species generated via the ORR is more stable in large AM+ electrolytes. Additionally, electrochemical measurements (Fig. 1) have shown that, in large AM+ electrolytes, the reduction of the 2e− ORR product is facilitated and the 4e− ORR pathway is favored. The previous studies and our results inspire us to further investigate the correlation between the high-contrast species and the 2e− ORR product [24,47].
Figure 2.

EC–STM investigation of ORR in large AM+ electrolytes. (a) EC–STM image of CoOEP monolayer in Ar-saturated 80 mM CsClO4 electrolyte at 0.2 V. (b and c) EC–STM images of CoOEP monolayer in O2-saturated 80 mM CsClO4 electrolyte at (b) 0.2 V and (c) −0.2 V. (d–f) EC–STM images of CoOEP monolayer in electrolyte containing 3 mM of hydrogen peroxide at −0.2 V. (d) CoOEP monolayer in Ar-saturated 80 mM CsClO4 electrolyte. (e) CoOEP monolayer in Ar-saturated 80 mM Cs+ electrolyte with pH 12.5. (f) CoOEP monolayer in Ar-saturated 80 mM Na+ electrolyte with pH 12.5. Electrolytes used in (e) and (f) are prepared with AMClO4 and AMOH. Cross section corresponding to the dashed box. Scale bar represents 5 nm. (g and h) Calculated molecular structures and simulated EC–STM images of (g) CoOEP and (h) HO2−–CoOEP.
To assign the high-contrast species, EC–STM is conducted to observe the CoOEP monolayer with H2O2 in the electrolyte. As shown in Fig. 2d and Fig. S3, in the Ar-saturated CsClO4 electrolyte with 3 mM H2O2, the surface is fully covered by CoOEP molecules with an apparent height of ∼0.1 nm. The high-contrast species barely appears in the monolayer, demonstrating that the high-contrast species observed during the ORR in the Cs+ electrolyte does not originate from the H2O2 adsorption. It is reported that the O2 reduction leads to the elevated pH value in the vicinity of the electrode surface [53]. The elevated pH value affects the acid–base equilibrium of the hydrogen peroxide and favors the presence of the HO2− form. These results inspire us to investigate the binding of HO2− on CoOEP and its effect on the apparent height of the surface species [54–56]. As shown in Fig. 2e, the CoOEP monolayer is observed in the Ar-saturated Cs+ electrolyte with pH 12.5 containing 3 mM of hydrogen peroxide. The pKa for H2O2 is 11.6 and the hydrogen peroxide in the electrolyte exists mainly in the form of HO2− with pH 12.5 [55]. Surface species with an apparent height of ∼0.2 nm is observed in the monolayer (Fig. 2e), which is assigned to the adsorbed HO2− on the CoOEP. The assignment is further confirmed by the absence of the 0.2-nm-height species in the HO2−-free Cs+ electrolyte with pH 12.5 (Fig. S4). Moreover, the adsorption of HO2− on the CoOEP is barely observed in the Na+ electrolyte with pH 12.5 containing 3 mM of hydrogen peroxide (Fig. 2f), which suggests that the adsorbed HO2− is stabilized by large AM+. The adsorbed HO2− and the high-contrast species observed in the O2-saturated CsClO4 electrolyte when the ORR occurs not only exhibit the same contrast, but their formation also shares consistent AM+ dependence. Thus, the high-contrast species is assigned to the adsorbed HO2− on the CoOEP. Furthermore, EC–STM is employed to observe the CoOEP monolayer in O2-saturated various AM⁺ electrolytes at pH 1.5 (Figs S5 and S6). The 2e⁻ ORR product exists in the form of H2O2 due to the acid–base equilibrium of hydrogen peroxide in acidic environments. Under ORR conditions, the high-contrast species is not observed in all AM⁺ electrolytes. These results are consistent with the assignment of the high-contrast species as the adsorbed HO2− on the CoOEP. Overall, EC–STM results demonstrate that adsorbed HO2− as the 2e− ORR product is stabilized by large AM+ in the electrolyte.
The assignment of surface species is further confirmed by using theoretical calculations based on density functional theory (DFT). The calculated molecular structures of CoOEP and HO2−–CoOEP are shown in Fig. 2 and Fig. S7. In the simulated EC–STM images (Fig. 2g and h), HO2−–CoOEP shows significantly more enhanced contrast than CoOEP, which is in good agreement with the experimental observation.
Furthermore, EC–STM is conducted to investigate the stabilizing effect of all AM+ on HO2− binding to CoOEP when the ORR occurs. As shown in Fig. 3a and b, the adsorbed HO2− is barely observed in the Li+ and Na+ electrolytes. In the K+ electrolyte (Fig. 3c), a noticeable amount of adsorbed HO2− is observed with a surface coverage of ∼4%. The HO2− coverage increases to ∼10% in the Rb+ electrolyte (Fig. 3d) and ∼12% in the Cs+ electrolyte (Fig. 3e). The statistical results quantitatively demonstrate that the surface HO2− coverage is positively correlated with the size of the AM+ in the electrolyte (Fig. 3f). Additionally, this result is in good agreement with the presence and absence of adsorbed HO2− in the Cs+ and Na+ electrolytes (with HO2− in the electrolyte), respectively (Fig. 2e and f). It is known that the surface coverage of adsorbates is positively correlated with the equilibrium constant or adsorption energy [37,46]. The EC–STM results suggest that the stabilizing effect of AM+ on the adsorbed HO2− increases in the order of Li+ ≈ Na+ < K+ < Rb+ < Cs+.
Figure 3.

AM+ effect on HO2− binding to CoOEP. (a–e) EC–STM images of CoOEP monolayer in O2-saturated 80 mM AMClO4 electrolytes at −0.2 V. Scale bar represents 10 nm. (f) Statistics of surface HO2−–CoOEP coverage measured in (a–e).
It is reported that AM+ accumulates in the EDL during cathodic polarization [57,58] in the order of Li+ < Na+ < K+ < Rb+ < Cs+. The AM+ in EDL undergo partial dehydration [59] and can stabilize surface-adsorbed species such as reactants, intermediates and products [24,37]. For instance, it has been proposed that the dipole field generated by AM+ in the outer Helmholtz plane can stabilize CO during CO2 reduction [24]. Here, we investigated the effect of the electric field on the stability of HO2−–CoOEP. As the electric field strength increases, the free-energy change of HO2−–CoOEP (compared with that without the electric field) gradually increases (Figs S8 and S9). We propose that a similar dipole field effect may account for the stabilization of HO2−–CoOEP in large AM+ electrolytes.
Stabilization of the 2e− ORR product results in an increased electron-transfer number
Furthermore, we investigated the correlation between the surface HO2− coverage and the electron-transfer number of the ORR in O2-saturated electrolytes with Cs+ concentrations ([Cs+]) of 10, 30 and 80 mM. As the effect of Li+ on ORR selectivity (Fig. 1) and HO2− adsorption (Fig. 3) is negligible, the electrolytes are prepared here by using CsClO4 and LiClO4 to keep [AMClO4] at 80 mM. As shown in Fig. 4a, the surface coverage of HO2− at −0.2 V is 1.2% ([Cs+] = 10 mM) ˂ 6.3% ([Cs+] = 30 mM) ˂ 11.9% ([Cs+] = 80 mM). In addition, the electron-transfer number of the ORR at −0.2 V is 2.5 ([Cs+] = 10 mM) ˂ 3.1 ([Cs+] = 30 mM) ˂ 3.7 ([Cs+] = 80 mM) (Fig. 4b). The statistical result suggests the positive correlation between the electron-transfer number of the ORR and the surface HO2− coverage (Fig. 4b).
Figure 4.

The 2e−+2e− ORR process in large AM+ electrolytes. (a) EC–STM images of CoOEP monolayer in O2-saturated 80 mM AMClO4 electrolytes with [Cs+] of 10, 30 and 80 mM at −0.2 V. Electrolytes were prepared by using LiClO4 and CsClO4. Scale bars represent 10 nm. (b) Correlation between the surface HO2−–CoOEP coverage measured in (a) and the electron-transfer number of the ORR at −0.2 V in electrolytes with various [Cs+]. (c) Surface HO2− coverage in Ar-saturated electrolytes containing 3 mM of hydrogen peroxide and various [Cs+]. (d) LSVs of HO2− reduction on CoOEP in Ar-saturated electrolytes with 3 mM of hydrogen peroxide and various Cs+ concentrations. Electrolytes for measurements in (c) and (d) were prepared by using AMClO4 and AMOH to give a pH value of 12.5. Li+ was used to maintain the AM+ concentration at 80 mM. (e) Scheme illustrating the 2e− ORR process in small AM+ (Li+ and Na+) electrolytes and the 2e−+2e− ORR process in large AM+ (K+, Rb+ and Cs+) electrolytes.
Moreover, to explore the effect of stabilized HO2− adsorption on HO2− reduction, we measured the HO2− reduction current in Ar-saturated electrolytes containing 3 mM of hydrogen peroxide and various [Cs+]. The electrolyte is prepared by using AMClO4 and AMOH to allow pH 12.5. Li+ is used to maintain the AM+ concentration at 80 mM. Electrochemical measurements suggest the positive correlation between the onset potential of the HO2− reduction and [Cs⁺], indicating the involvement of [Cs⁺] in HO2− reduction. We then investigated the influence of Cs⁺ on the surface coverage of HO2−, as well as the relationship between the surface HO2− coverage and the onset potential of the HO2− reduction. In EC–STM images (Fig. S10), the surface HO2− coverage in the CoOEP monolayer is ∼0.7% ([Cs+] = 1 mM) ˂ 3.1% ([Cs+] = 10 mM) ˂ 7.9% ([Cs+] = 30 mM) ˂ 18.2% ([Cs+] = 80 mM) (Fig. 4c). Correlative EC–STM and LSVs (Fig. S11) demonstrate that the HO2− reduction current is positively correlated with the surface HO2− coverage. To further probe the interfacial dynamics, we plotted the LSVs on the HO2−-coverage-corrected electrode (CCE) scale [60], which is defined as:
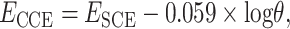 |
(3) |
where θ represents the surface coverage of adsorbed HO2− on the CoOEP. When the surface coverage of HO2− is varied, the onset potentials for HO2− reduction vs SCE are distinct (Fig. S11) yet they become unified when referenced to the CCE (Fig. 4d), suggesting that the adsorbed HO2− acts as the reactant in the RDS of the HO2− reduction (Fig. S11). The HO2− reduction is facilitated by the increased coverage of adsorbed HO2− under the stabilization effect of large AM+. In addition, the high-contrast species was shown previously to be the 2e− ORR product and herein to be the reaction substrate for HO2− reduction, which further confirms that the high-contrast species is adsorbed HO2− on the CoOEP. Briefly, the large AM+ stabilize the adsorbed HO2− and promote the reduction of HO2− to H2O, thereby facilitating the 2e−+2e− ORR.
It is reported that the Co–O bond is relatively weak compared with Fe–O and Mn–O bonds, leading to the facile desorption of the 2e− ORR product (Fig. S9) and consequently favoring the 2e− ORR (in contrast to FeN4 and MnN4 sites, which promote the 4e− ORR) [47]. Here, we show that the surface coverage of adsorbed HO2− is positively correlated with the size of AM+ and the concentration of large AM+. These results suggest that, in small AM+ electrolytes, the 2e− ORR product generated on CoN4 sites is barely stabilized by AM+ and readily desorbs, resulting in predominant 2e− ORR selectivity. In contrast, large AM+ stabilize the adsorbed 2e− ORR product on CoN4 sites and facilitate its further reduction, thereby enabling the nearly 4e− O2 reduction (Fig. 4e).
Beyond model electrocatalysts
Furthermore, the effect of AM+ on ORR selectivity was measured on the practical Co–N4 electrocatalyst. Here, we examined the ORR catalysed by COF-366-Co, which is an accessible and widely investigated covalent organic framework (COF) constructed from CoPor (Fig. S12) [61,62], in different AM+ electrolytes. The O2 reduction current commences at ∼0 V in LSVs measured on the COF-366-Co-coated electrode (Fig. S13). The electron-transfer number of the ORR in different AM+ electrolytes at −0.35 V increases in the order of Li+ (2.52) < Na+ (2.57) < K+ (3.16) < Rb+ (3.43) < Cs+ (3.58). On practical electrocatalysts with Co–N4 sites as active centers, the electron-transfer number of the ORR is positively correlated with the size of the AM+ in the electrolyte, suggesting that the AM+ effect revealed on model catalytic sites can be extended to practical electrocatalysts. Moreover, COF-366-Co represents a series of electrocatalysts with high structural designability [63–65]. The electronic properties of active sites can be tuned by modifying the molecular building blocks [66–69]. Structural engineering and functional group modification endow these catalysts with synergistic effects [70,71]. Notably, the active site structures are similar to metalloporphyrin, ensuring the generality of the strategy revealed in molecular models for modulating the product distribution by adjusting the electrolyte composition.
CONCLUSION
In summary, through correlative electrochemical measurements and in situ EC–STM, we found that AM+ steer the product selectivity of ORRs catalysed by Co–N4 sites. Rotating ring-disk voltammetry reveals that the electron-transfer number of the ORR increases in the order of Li+ ≈ Na+ < K+ < Rb+ < Cs+. Analysis of the Damjanović kinetics of the ORR in large AM+ electrolytes reveals the 2e−+2e− ORR pathway. In situ EC–STM resolves HO2− binding on CoOEP in large AM+ electrolytes when the ORR occurs. The surface CoOEP–HO2− coverage is higher in larger AM+ electrolytes. Moreover, the electron-transfer number of the ORR is positively correlated with the surface coverage of the adsorbed HO2−. A series of voltammograms suggest that the HO2− reduction is promoted in large AM+ electrolytes, facilitating the 2e−+2e− ORR process. Conclusively, the stabilization and further reduction of adsorbed HO2− on CoOEP is promoted by large AM+, resulting in the increased electron-transfer number of the ORR. This work reveals unrecognized effects of AM+ on the product selectivity of ORRs catalysed by Co–N4 sites and the mechanisms involved. The demonstration of the AM+ effect provides a promising approach to modulate ORR selectivity to meet diverse application demands by adjusting the electrolyte composition rather than replacing the catalyst.
METHODS
Chemicals and materials
The CoOEP was from Sigma-Aldrich (98%). 5,10,15,20-Tetrakis(4-aminophenyl)porphyrin cobalt (CoTAPP) was from J&K (95%). CoOEP and CoTAPP were utilized without further purification. 1,4-Benzenedicarboxaldehyde (BDA) was from J&K (99%). N,N-Dimethylformamide (DMF) was from Sigma-Aldrich. H2O2 was from Sigma-Aldrich (70%). HClO4 was from ALDRICH (70%, purity > 99.999%). LiClO4 was from Sigma-Aldrich (purity > 99.99%). LiOH was from J&K (purity > 99%). NaClO4 was from ACROS (purity > 99%). NaOH was from MACKLIN (purity > 99.9%). KClO4 was from ALDRICH (purity > 99.99%). KOH was from J&K (purity > 90%). Rb2CO3 was from MACKLIN (purity > 99.9%). RbClO4 electrolyte was prepared by using Rb2CO3 and HClO4. CsClO4 was from Thermo Scientific (reagent grade). CsOH was from MACKLIN (purity > 99.9%). Milli-Q water (18.2 MΩ·cm, TOC < 4 ppb) was used throughout the investigation.
The COF-366-Co was prepared according to previous reports [61,62]. CoTAPP (18 mg, 0.025 mmol), BDA (6.7 mg, 0.05 mmol), 1,2-dichlorobenzene (1 mL), n-butanol (1 mL) and 6 M aqueous acetic acid (0.2 mL) were added in a 5-mL Pyrex tube. The tube was sonicated for 15 minutes and then flash-frozen at 77 K (liquid N2 bath). After three freeze–pump–thaw cycles, the tube was evacuated to an internal pressure of 50 mTorr and flame-sealed. After heating at 120°C for 72 h, a dark purple precipitate was produced at the bottom of the tube. The precipitate, separated by filtration, was transferred to a Soxhlet extractor and washed thoroughly with tetrahydrofuran (THF) (24 h) and acetone (24 h). The material was then dried in a vacuum oven at 80°C for 12 h. Then, 1 mg of COF-366-Co was dispersed in 1 mL of solution (water:isopropanol:5 wt% Nafion solution in the ratio of 10 : 10 : 0.1 by volume) and sonicated to form the mixture. Subsequently, 1 mg of XC-72 carbon black was added to the mixture. The mixture was sonicated for 1 h to yield the homogeneous ink, after which 25 μL of catalyst ink was dropwise coated onto the Au disk of the rotating ring-disk electrode as the working electrode (loading: 0.2 mg cm−2).
Powder X-ray diffraction patterns were recorded on a PANalytical Empyrean Diffractometer operating at 40 kV and 40 mA using Cu Kα radiation (λ = 1.5416 Å) at ambient temperature in the range of 2°–30° at 3.5°/min.
EC–STM measurements
All EC–STM images were collected by using the NanoScope E scanning tunneling microscope (Bruker, Inc.). Tungsten wire (Alfa Aesar, 0.25 mm in diameter) was electrochemically etched (0.6 M KOH, 20 V DC) and coated with nail polish to prepare the EC–STM tips. The Au(111) single crystal prepared by using the Clavilier method was used as working electrode [72]. The Au(111) electrode was annealed in a hydrogen–oxygen flame before each experiment. The self-assembled CoOEP monolayer was prepared by immersing the Au(111) electrode in the DMF solution of the CoOEP. The working electrode was transferred into the electrochemical cell with two platinum wires as the reference electrode and counter electrode.
Theoretical calculations
All DFT calculations were performed by using the projector augmented wave method. The electronic exchange and correlation were described by using the Perdew–Burke–Ernzerhof functional with the generalized gradient approximation [73]. The cut-off of the plane wave was set as 600 eV. The criteria of force convergence were <0.02 eV/Å for geometry optimization [74].
Electrochemical measurements
The electrochemical measurements were conducted on an RRDE-3A (ALS, Japan) and an Autolab PGSTAT302N (Metrohm, Netherlands) electrochemical workstation. The LSVs were measured on the CoOEP-modified (as well as COF-366-Co-coated) Au disk electrode (4 mm in diameter). The ring electrode of the rotating ring-disk electrode was Pt. Prior to electrochemical measurements, the rotating-disk electrode and the rotating ring-disk electrode were polished with 0.3 and 0.05 μm of alumina slurries and then cleaned and subsequently sonicated in Milli-Q water and 2-propanol to a mirror-finish state. The Pt ring was then electrochemically cleaned. An SCE and a Pt wire were used as the reference and counter electrodes, respectively. All electrode potentials were reported with respect to the SCE. The LSVs were recorded at a scan rate of 10 mV s−1 without iR-compensation. Except for specific indications, the rate of electrode rotation was 100 rpm. The electron-transfer number of the ORR was calculated as n = 4ID/[(IR/N0)+ID].
Supplementary Material
ACKNOWLEDGEMENTS
The Supercomputing Environment of the Chinese Academy of Sciences is acknowledged for providing computational resources.
Contributor Information
Yue Feng, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
Yu-Qi Wang, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China.
Zi-Cong Wang, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
Hong Li, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
Liang Ding, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
Jin-Song Hu, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
Li-Jun Wan, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
Dong Wang, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 101408, China.
FUNDING
This work was supported by the National Key R&D Program of China (2021YFA1501002) and the National Natural Science Foundation of China (22132007 and 22421001).
AUTHOR CONTRIBUTIONS
Y.-Q.W., Y.F. and D.W. conceived the project. Y.F. and Y.-Q.W. conducted EC–STM experiment. Y.F., Y.-Q.W., Z.-C.W., H.L. and L.D. conducted material preparation and characterization, electrochemical measurements and theoretical calculation. Y.-Q.W., Y.F. and D. W. wrote the paper. All authors contributed to the analysis and discussion.
Conflict of interest statement. None declared.
REFERENCES
- 1. Xiao M, Chen Y, Zhu J et al. Climbing the apex of the ORR volcano plot via binuclear site construction: electronic and geometric engineering. J Am Chem Soc 2019; 141: 17763–70. 10.1021/jacs.9b08362 [DOI] [PubMed] [Google Scholar]
- 2. Li M, Zhao Z, Cheng T et al. Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction. Science 2016; 354: 1414–9. 10.1126/science.aaf9050 [DOI] [PubMed] [Google Scholar]
- 3. Guo D, Shibuya R, Akiba C et al. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016; 351: 361–5. 10.1126/science.aad0832 [DOI] [PubMed] [Google Scholar]
- 4. Tian X, Zhao X, Su Y-Q et al. Engineering bunched Pt-Ni alloy nanocages for efficient oxygen reduction in practical fuel cells. Science 2019; 366: 850–6. 10.1126/science.aaw7493 [DOI] [PubMed] [Google Scholar]
- 5. Cao B, Zhao Z, Peng L et al. Silver nanoparticles boost charge-extraction efficiency in Shewanella microbial fuel cells. Science 2021; 373: 1336–40. 10.1126/science.abf3427 [DOI] [PubMed] [Google Scholar]
- 6. Santoro C, Bollella P, Erable B et al. Oxygen reduction reaction electrocatalysis in neutral media for bioelectrochemical systems. Nat Catal 2022; 5: 473–84. 10.1038/s41929-022-00787-2 [DOI] [Google Scholar]
- 7. Lu Z, Chen G, Siahrostami S et al. High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials. Nat Catal 2018; 1: 156–62. 10.1038/s41929-017-0017-x [DOI] [Google Scholar]
- 8. Kim HW, Ross MB, Kornienko N et al. Efficient hydrogen peroxide generation using reduced graphene oxide-based oxygen reduction electrocatalysts. Nat Catal 2018; 1: 282–90. 10.1038/s41929-018-0044-2 [DOI] [Google Scholar]
- 9. Mehmood A, Gong M, Jaouen F et al. High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat Catal 2022; 5: 311–23. 10.1038/s41929-022-00772-9 [DOI] [Google Scholar]
- 10. Li J, Chen M, Cullen DA et al. Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells. Nat Catal 2018; 1: 935–45. 10.1038/s41929-018-0164-8 [DOI] [Google Scholar]
- 11. Wang Y, Wang D, Li Y. A fundamental comprehension and recent progress in advanced Pt-based ORR nanocatalysts. SmartMat 2021; 2: 56–75. 10.1002/smm2.1023 [DOI] [Google Scholar]
- 12. Lee B-H, Shin H, Rasouli AS et al. Supramolecular tuning of supported metal phthalocyanine catalysts for hydrogen peroxide electrosynthesis. Nat Catal 2023; 6: 234–43. 10.1038/s41929-023-00924-5 [DOI] [Google Scholar]
- 13. Fan W, Duan Z, Liu W et al. Rational design of heterogenized molecular phthalocyanine hybrid single-atom electro catalyst towards two-electron oxygen reduction. Nat Commun 2023; 14: 1426. 10.1038/s41467-023-37066-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen S, Luo T, Li X et al. Identification of the highly active Co–N4 coordination motif for selective oxygen reduction to hydrogen peroxide. J Am Chem Soc 2022; 144: 14505–16. 10.1021/jacs.2c01194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tian Y, Li M, Wu Z et al. Edge-hosted atomic Co-N4 sites on hierarchical porous carbon for highly selective two-electron oxygen reduction reaction. Angew Chem Int Ed 2022; 134: e202213296. 10.1002/ange.202213296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu S, Yamauchi H, Wang S et al. CO2-to-methanol electroconversion on a molecular cobalt catalyst facilitated by acidic cations. Nat Catal 2024; 7: 1000–9. 10.1038/s41929-024-01197-2 [DOI] [Google Scholar]
- 17. Zhu Q, Rooney CL, Shema H et al. The solvation environment of molecularly dispersed cobalt phthalocyanine determines methanol selectivity during electrocatalytic CO2 reduction. Nat Catal 2024; 7: 987–99. 10.1038/s41929-024-01190-9 [DOI] [Google Scholar]
- 18. Zhang Z-M, Wang T, Cai Y-C et al. Probing electrolyte effects on cation-enhanced CO2 reduction on copper in acidic media. Nat Catal 2024; 7: 807–17. 10.1038/s41929-024-01179-4 [DOI] [Google Scholar]
- 19. Luo M, Koper MTM. A kinetic descriptor for the electrolyte effect on the oxygen reduction kinetics on Pt(111). Nat Catal 2022; 5: 615–23. 10.1038/s41929-022-00810-6 [DOI] [Google Scholar]
- 20. Strmcnik D, Kodama K, van der Vliet D et al. The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum. Nat Chem 2009; 1: 466–72. 10.1038/nchem.330 [DOI] [PubMed] [Google Scholar]
- 21. Kumeda T, Laverdure L, Honkala K et al. Cations determine the mechanism and selectivity of alkaline oxygen reduction reaction on Pt(111). Angew Chem Int Ed 2023; 135: e202312841. 10.1002/ange.202312841 [DOI] [PubMed] [Google Scholar]
- 22. Shah AH, Zhang Z, Huang Z et al. The role of alkali metal cations and platinum-surface hydroxyl in the alkaline hydrogen evolution reaction. Nat Catal 2022; 5: 923–33. 10.1038/s41929-022-00851-x [DOI] [Google Scholar]
- 23. Goyal A, Koper MTM. The interrelated effect of cations and electrolyte pH on the hydrogen evolution reaction on gold electrodes in alkaline media. Angew Chem Int Ed 2021; 60: 13452–62. 10.1002/anie.202102803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Resasco J, Chen LD, Clark E et al. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J Am Chem Soc 2017; 139: 11277–87. 10.1021/jacs.7b06765 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Z, Li H, Shao Y et al. Molecular understanding of the critical role of alkali metal cations in initiating CO2 electroreduction on Cu(100) surface. Nat Commun 2024; 15: 612. 10.1038/s41467-024-44896-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan B, Wang Y, Li Y. Understanding and leveraging the effect of cations in the electrical double layer for electrochemical CO2 reduction. Chem Catal 2022; 2: 1–10. 10.1016/j.checat.2022.03.012 [DOI] [Google Scholar]
- 27. Murata A, Hori Y. Product selectivity affected by cationic species in electrochemical reduction of CO2 and CO at a Cu electrode. Bull Chem Soc Jpn 1991; 64: 123–7. 10.1246/bcsj.64.123 [DOI] [Google Scholar]
- 28. Hori Y, Suzuki S. Electrolytic reduction of carbon dioxide at mercury electrode in aqueous solution. Bull Chem Soc Jpn 1982; 55: 660–5. [Google Scholar]
- 29. Frumkin AN. Influence of cation adsorption on the kinetics of electrode processes. Trans Faraday Soc 1959; 55: 156–67. 10.1039/tf9595500156 [DOI] [Google Scholar]
- 30. Ringe S, Clark EL, Resasco J et al. Understanding cation effects in electrochemical CO2 reduction. Energy Environ Sci 2019; 12: 3001–14. 10.1039/C9EE01341E [DOI] [Google Scholar]
- 31. Chen LD, Urushihara M, Chan K et al. Electric field effects in electrochemical CO2 reduction. ACS Catal 2016; 6: 7133–9. [Google Scholar]
- 32. Hussain G, Pérez-Martínez L, Le J-B et al. How cations determine the interfacial potential profile: relevance for the CO2 reduction reaction. Electrochim Acta 2019; 327: 135055. 10.1016/j.electacta.2019.135055 [DOI] [Google Scholar]
- 33. Singh MR, Kwon Y, Lum Y et al. Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu. J Am Chem Soc 2016; 138: 13006–12. 10.1021/jacs.6b07612 [DOI] [PubMed] [Google Scholar]
- 34. Ayemoba O, Cuesta A. Spectroscopic evidence of size-dependent buffering of interfacial pH by cation hydrolysis during CO2 electroreduction. ACS Appl Mater Interfaces 2017; 9: 27377–82. 10.1021/acsami.7b07351 [DOI] [PubMed] [Google Scholar]
- 35. Zhang F, Co AC. Direct evidence of local pH change and the role of alkali cation during CO2 electroreduction in aqueous media. Angew Chem Int Ed 2020; 59: 1674–81. 10.1002/anie.201912637 [DOI] [PubMed] [Google Scholar]
- 36. Qin H-G, Li F-Z, Du Y-F et al. Quantitative understanding of cation effects on the electrochemical reduction of CO2 and H+ in acidic solution. ACS Catal 2023; 13: 916–26. 10.1021/acscatal.2c04875 [DOI] [Google Scholar]
- 37. Wang Y-Q, Dan X-H, Wang X et al. Probing the synergistic effects of Mg2+ on CO2 reduction reaction on CoPc by in situ electrochemical scanning tunneling microscopy. J Am Chem Soc 2022; 144: 20126–33. 10.1021/jacs.2c09862 [DOI] [PubMed] [Google Scholar]
- 38. Jacobse L, Huang Y-F, Koper MTM et al. Correlation of surface site formation to nanoisland growth in the electrochemical roughening of Pt(111). Nat Mater 2018; 17: 277–82. 10.1038/s41563-017-0015-z [DOI] [PubMed] [Google Scholar]
- 39. He Y, Borguet E. Metastable phase of the Au(111) surface in electrolyte revealed by STM and asymmetric potential pulse perturbation. J Phys Chem C 2011; 115: 5726–31. 10.1021/jp110484w [DOI] [Google Scholar]
- 40. Kwon S, Kim Y-G, Baricuatro JH et al. Dramatic change in the step edges of the Cu(100) electrocatalyst upon exposure to CO: operando observations by electrochemical STM and explanation using quantum mechanical calculations. ACS Catal 2021; 11: 12068–74. 10.1021/acscatal.1c02844 [DOI] [Google Scholar]
- 41. Amirbeigiarab R, Tian J, Herzog A et al. Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions. Nat Catal 2023; 6: 837–46. 10.1038/s41929-023-01009-z [DOI] [Google Scholar]
- 42. Pfisterer JHK, Liang Y, Schneider O et al. Direct instrumental identification of catalytically active surface sites. Nature 2017; 549: 74–7. 10.1038/nature23661 [DOI] [PubMed] [Google Scholar]
- 43. Kosmala T, Baby A, Lunardon M et al. Operando visualization of the hydrogen evolution reaction with atomic-scale precision at different metal-graphene interfaces. Nat Catal 2021; 4: 850–9. 10.1038/s41929-021-00682-2 [DOI] [Google Scholar]
- 44. Auer A, Andersen M, Wernig E-M et al. Self-activation of copper electrodes during CO electro-oxidation in alkaline electrolyte. Nat Catal 2020; 3: 797–803. 10.1038/s41929-020-00505-w [DOI] [Google Scholar]
- 45. Gu J-Y, Cai Z-F, Wang D et al. Single-molecule imaging of iron-phthalocyanine-catalyzed oxygen reduction reaction by in situ scanning tunneling microscopy. ACS Nano 2016; 10: 8746–50. 10.1021/acsnano.6b04281 [DOI] [PubMed] [Google Scholar]
- 46. Wang Y-Q, Dan X-H, Yi Z-Y et al. Single-molecule study on the catalytic role of Co–O2 binding in ORR by in situ ECSTM. J Phys Chem C 2023; 127: 2929–35. 10.1021/acs.jpcc.2c07891 [DOI] [Google Scholar]
- 47. Zagal JH, Koper MTM. Reactivity descriptors for the activity of molecular MN4 catalysts for the oxygen reduction reaction. Angew Chem Int Ed 2016; 55: 14510–21. 10.1002/anie.201604311 [DOI] [PubMed] [Google Scholar]
- 48. Damjanovic A, Genshaw MA, Bockris JO.'M. Distinction between intermediates produced in main and side electrodic reactions. J Chem Phys 1966; 45: 4057–9. 10.1063/1.1727457 [DOI] [Google Scholar]
- 49. Jin S, Van Neste A, Ghali E et al. New cathode materials for chlorate electrolysis. J Electrochem Soc 1997; 144: 4272. 10.1149/1.1838177 [DOI] [Google Scholar]
- 50. Wroblowa HS, Yen-Chi-Pan RG. Electroreduction of oxygen: a new mechanistic criterion. J Electroanal Chem Interfacial Electrochem 1976; 69: 195–201. 10.1016/S0022-0728(76)80250-1 [DOI] [Google Scholar]
- 51. Sautet P. Images of adsorbates with the scanning tunneling microscope: theoretical approaches to the contrast mechanism. Chem Rev 1997; 97: 1097–116. 10.1021/cr9600823 [DOI] [PubMed] [Google Scholar]
- 52. Chiang S. Scanning tunneling microscopy imaging of small adsorbed molecules on metal surfaces in an ultrahigh vacuum environment. Chem Rev 1997; 97: 1083–96. [DOI] [PubMed] [Google Scholar]
- 53. Botz A, Clausmeyer J, Öhl D et al. Local activities of hydroxide and water determine the operation of silver-based oxygen depolarized cathodes. Angew Chem Int Ed 2018; 57: 12285–9. [DOI] [PubMed] [Google Scholar]
- 54. Ruggiero BN, Gutierrez KMS, George JD et al. Probing the relationship between bulk and local environments to understand impacts on electrocatalytic oxygen reduction reaction. J Catal 2022; 414: 33–43. [Google Scholar]
- 55. Blech DM, Borders CL Jr.. Hydroperoxide anion, HO2−, is an affinity reagent for the inactivation of yeast Cu, Zn superoxide dismutase: modification of one histidine per subunit. Arch Biochem Biophys 1983; 224: 579–86. 10.1016/0003-9861(83)90245-X [DOI] [PubMed] [Google Scholar]
- 56. Zheng YL, Mei D, Chen Y-X et al. The redox reaction of hydrogen peroxide at an Au(100) electrode: implications for oxygen reduction kinetics. Electrochem Commun 2014; 39: 19–21. 10.1016/j.elecom.2013.12.005 [DOI] [Google Scholar]
- 57. Waegele MM, Gunathunge CM, Li J-Y et al. How cations affect the electric double layer and the rates and selectivity of electrocatalytic processes. J Chem Phys 2019; 151: 160902–27. 10.1063/1.5124878 [DOI] [PubMed] [Google Scholar]
- 58. Li J-Y, Li X, Gunathunge CM et al. Hydrogen bonding steers the product selectivity of electrocatalytic CO reduction. Proc Natl Acad Sci USA 2019; 116: 9220–9. 10.1073/pnas.1900761116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ovalle VJ, Hsu Y-S, Agrawal N et al. Correlating hydration free energy and specific adsorption of alkali metal cations during CO2 electroreduction on Au. Nat Catal 2022; 5: 624–32. 10.1038/s41929-022-00816-0 [DOI] [Google Scholar]
- 60. Shin S-J, Choi H, Ringe S et al. A unifying mechanism for cation effect modulating C1 and C2 productions from CO2 electroreduction. Nat Commun 2022; 13: 5482. 10.1038/s41467-022-33199-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lin S, Diercks CS, Zhang Y-B et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 2015; 349: 1208–13. 10.1126/science.aac8343 [DOI] [PubMed] [Google Scholar]
- 62. Liu C, Li H, Liu F et al. Intrinsic activity of metal centers in metal–nitrogen–carbon single-atom catalysts for hydrogen peroxide synthesis. J Am Chem Soc 2020; 142: 21861–71. 10.1021/jacs.0c10636 [DOI] [PubMed] [Google Scholar]
- 63. Lyu P, Wang Z, Guo N et al. Air-stable wafer-scale ferromagnetic metallo-carbon nitride monolayer. J Am Chem Soc 2024; 146: 20604–14. 10.1021/jacs.4c02160 [DOI] [PubMed] [Google Scholar]
- 64. Hai X, Zhao X, Guo N et al. Engineering local and global structures of single Co atoms for a superior oxygen reduction reaction. ACS Catal 2020; 10: 5862–70. 10.1021/acscatal.0c00936 [DOI] [Google Scholar]
- 65. Chi K, Wang Z, Sun T et al. Simultaneously engineering the first and second coordination shells of single iron catalysts for enhanced oxygen reduction. Small 2024; 20: 2311817. 10.1002/smll.202311817 [DOI] [PubMed] [Google Scholar]
- 66. Wu Y, Jiang Z, Lu X et al. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature 2019; 575: 639–42. 10.1038/s41586-019-1760-8 [DOI] [PubMed] [Google Scholar]
- 67. Zhang X, Wu Z, Zhang X et al. Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat Commun 2017; 8: 14675. 10.1038/ncomms14675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Han N, Wang Y, Ma L et al. Supported cobalt polyphthalocyanine for high-performance electrocatalytic CO2 reduction. Chem 2017; 3: 652–64. 10.1016/j.chempr.2017.08.002 [DOI] [Google Scholar]
- 69. Morozan A, Campidelli S, Filoramo A et al. Catalytic activity of cobalt and iron phthalocyanines or porphyrins supported on different carbon nanotubes towards oxygen reduction reaction. Carbon 2011; 49: 4839–47. 10.1016/j.carbon.2011.07.004 [DOI] [Google Scholar]
- 70. Diercks CS, Lin S, Kornienko N et al. Reticular electronic tuning of porphyrin active sites in covalent organic frameworks for electrocatalytic carbon dioxide reduction. J Am Chem Soc 2018; 140: 1116–22. 10.1021/jacs.7b11940 [DOI] [PubMed] [Google Scholar]
- 71. Liang Z, Wang H-Y, Zheng H et al. Porphyrin-based frameworks for oxygen electrocatalysis and catalytic reduction of carbon dioxide. Chem Soc Rev 2021; 50: 2540–81. 10.1039/D0CS01482F [DOI] [PubMed] [Google Scholar]
- 72. Clavilier J. The role of anion on the electrochemical behaviour of a {111} platinum surface; an unusual splitting of the voltammogram in the hydrogen region. J Electroanal Chem Interfacial Electrochem 1980; 107: 211–6. 10.1016/S0022-0728(79)80023-6 [DOI] [Google Scholar]
- 73. Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett 1996; 77: 3865–8. 10.1103/PhysRevLett.77.3865 [DOI] [PubMed] [Google Scholar]
- 74. Yang Z, Wang Y, Zhu M et al. Boosting oxygen reduction catalysis with Fe–N4 sites decorated porous carbons toward fuel cells. ACS Catal 2019; 9: 2158–63. 10.1021/acscatal.8b04381 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.