

Abstract
Background
The outgrowth of new vessels – angiogenesis – in the tumour mass is considered to be a limiting factor of tumour growth. To inhibit the matrix lysis that is part of the tumour angiogenesis, we employed the chimeric protein mhATF-BPTI, composed of the receptor binding part of the urokinase (ATF) linked to an inhibitor of plasmin (BPTI).
Methods
For delivery, recombinant adenovirus encoding the transgene of interest was injected intravenously or locally into the tumour. The anti tumour effect of this compound was compared to that of human endostatin and of mhATF alone in two different rat bronchial carcinomas growing either as subcutaneous implants or as metastases.
Results
Significant inhibition of the tumour growth and decrease of the number of lung metastasis was achieved when the concentration of mhATF-BPTI at the tumour site was above 400 of ng / g tissue. This concentration could be achieved via production by the liver, only if permissive to the recombinant adenovirus. When the tumour cells could be transduced, local delivery of the vector was enough to obtain a response. In the case of metastasis, the capacity of the lung tissue to concentrate the encoded protein was essential to reach the required therapeutic levels. Further, endostatin or mhATF could not reproduce the effects of mhATF-BPTI, at similar concentrations (mhATF) and even at 10-fold higher concentration (endostatin).
Conclusion
The ATF-BPTI was shown to inhibit tumour growth of different rat lung tumours when critical concentration was reached. In these tumour models, endostatin or ATF induce almost no tumour response.
Background
It has been suggested that solid tumours can not grow beyond 60–80 cells without inducing the formation of new blood vessels: tumour angiogenesis [1]. This blood vessel formation requires proliferation of endothelial cells, cell migration and the lysis of the different components of the matrix (collagen, elastin, fibronectin) by several proteases. One of great importance, urokinase (uPA) and its high affinity receptor, the GPI-anchored membrane CD87 (uPAR), is critical in tumour progression and its level of expression is correlated with poor prognosis of various cancers such as prostate [2], breast [3], colon [4], ovarian [5], gastric [6], brain [7], and lung cancer [8]. Urokinase (uPA) and its main product plasmin belong to the very early proteases [9-11] that activate the metallo-protease (MMP) cascade and therefore control cell motility, tissue remodelling and the activation of various angiogenic factors (VEGF, TGFβ). Most of these proteases (e.g. uPA) are localised at the tumour site through receptors that are highly expressed on the surface of activated endothelial cells and tumour cells. A high level of uPAR at the invasive edge of the tumour concentrates uPA where neoangiogenesis occurs [12], generating the full activation of the MMP cascade [13]. Furthermore, various cellular migrations proceed upon the transient attachment of the uPAR to matrix components (e.g. vitronectin) [14], and uPA as well as plasmin activate angiogenic factors such as VEGF, HGF, FGFs, and TGFβ [15-17].
The inhibitors of the matrix lysis can be divided in two categories: the synthetic inhibitors and the endogenous ones. Most of the synthetic inhibitors are competitive inhibitors of the metallo-proteases, mimicking the substrate of the enzymes (marismatat and derivatives). The endogenous inhibitors (e.g. endostatin, angiostatin, thrombospondin) differ in their angiostatic activity from synthetic inhibitors, since they appear to also block endothelial cell proliferation and/or migration. In this paper we described the antitumour effects of a hybrid protein, consisting of the Amino Terminal Fragment linked to Bovine Pancreatic Trypsin Inhibitor (ATF-BPTI), delivered by gene therapy with adenoviral vector.
ATF is the N-terminal part of the uPA (residues 1–135) that binds to the uPA receptor (uPAR) and thus inhibits the formation of the uPA-uPAR complex. Systemic and local delivery of an adenovirus encoding ATF have been shown to inhibit the growth of primary tumour implants as well as of tumour metastasis in different animal models [18,19]. In the ATF-BPTI chimeric protein, ATF has two functions (1) the anti uPA effect with its known angiostatic activity and (2) delivery of BPTI at the tumour site due to over-expression of uPAR on tumour and endothelial cells. The ATF is species specific between the human and the mouse [20] and is not specific between the mouse and the rat. The murinised version of ATF-BPTI differs by four amino acids from the human version It was shown to bind to the rat receptor and to inhibit plasmin activity in a rat model [21]. BPTI (commercialised as aprotinin) is a serine protease inhibitor prepared from bovine lung tissue which dose dependently inhibits human trypsin, plasmin and kallikrein. Aprotinin acts in a number of interrelated ways to provide an anti-fibrinolytic effect. Thus, aprotinin is mainly used in the clinic as anti-haemorrhagic agent in extracorporal circulation (valve replacement, coronary by-pass), for preventing postoperative haemorrhage, and as anti protease in acute pancreatitis. BPTI has been used systemically in a few animal tumour models, and showed anti-tumour effects in a mouse hepatoma, lung carcinoma, and mammary carcinoma [22-24], and in a fibrosarcoma of the hamster [24]. However, BPTI was not further developed as a systemic anti tumour agent probably because of its anti fibrinolytic property that might preclude its use in cancer patients. Its linkage to ATF is expected to ameliorate or reduce that side effect.
In this study, the tumour growth inhibiting effect of ATF-BPTI gene transfer was investigated and compared to endostatin and ATF. The adenoviral vector was administered intravenously, which leads to the ATF-BPTI production and secretion by the liver, or by intra tumoral injection. In the latter case, the protein is mostly produced by transduced tumour cells. Two transplantable rat bronchial carcinomas, growing either as subcutaneous implants or as lung metastasis were employed as targets. Levels of ATF-BPTI in the plasma and in tissues were determined and the association with tumour responses is described.
Methods
Animals
Pathogen-free inbred male Wag/Rij and Brown Norway rats, weighing 300 to 350 gr were purchased from Harlan, The Netherlands. All animals were fed ad libitum with laboratory chow and water and were kept under standard laboratory conditions. For assay of plasma hEndostatin, and mhATF-BPTI, rats were anaesthetised with isoflurane and bled by tail vein puncture and the blood was collected in EDTA containing tubes. All animal procedures were performed in accordance with the official guidelines after obtaining permission of the animal welfare committee.
ATF-BPTI cloning
The plasmid encoding the ATF-BPTI (pAB) was provided by P. Quax (TNO-PG, Leiden). The ATF plasmid was constructed by deleting the DNA sequences encoding amino acids 139 to 401 of the urokinase cDNA plasmid. Then a DNA fragment encoding the amino acids residues 36–96 of the bovine pancreatic trypsin inhibitor (BPTI) was cloned into the ATF plasmid resulting in a plasmid encoding the ATF, the BPTI and the 11 C-terminal amino acids of urokinase including the stop codon. After sequence confirmation, the ATF-BPTI cDNA was cloned into the recombinant adenovirus (Ad) shuttle vector Adapt. The murinised ATF-BPTI (mhAB) plasmid was constructed by an identical procedure, using the (Tyr22, Arg27,29,30) uPA mutant cDNA as starting sequence [33].
Adenoviral vectors
Recombinant adenovirus vectors were generated in PER.C6™ cells by homologous recombination between an Adapter plasmid (pAdapt) and the E1 deleted Ad 5 DNA plasmid as described elsewhere [34]. The expression cassette contains a CMV promoter and SV40 poly A signal. As a result of the absence of sequence overlap between the Adapt plasmid and the Ad5 E1 sequences integrated into the genome of PER.C6, the vector stocks used in this study does not contain replicative competent adenovirus (RCA) [35].
The Ad Adapt mhAB encodes for a murinised form of the human ATF-BPTI [21]. In this construct the mhAB, is preceded by the native secretion signal peptide of the human urokinase. The human endostatin coding sequence (InvivoGen, CA, USA) was cloned in the Ad Adapt shuttle vector. The encoded endostatin corresponds to the 183 residue of the human endostatin described by O'Reilly et al (1997) with an intact N-terminus (HSHRDFQ...), preceded by the secretion signal peptide of the human IL-2. The Ad.Adapt.Luc and the Ad.Adapt.LacZ are recombinant adenoviral vectors in which the E3 region of Ad5 is retained and the gene of interest (luciferase or LacZ) replaces the E1 region. The Ad Adapt empty is identical to Ad Adapt mhAB except that it does not encode any transgene.
All vectors were produced on PER.C6™ using standard procedures [35]. Infectious units (iu) were determined by end point cytopathogenic effect (CPE) assay on 911 cells [36]. Viral particles were determined by HPLC [37]. The particle to infectious unit ratio was always lower than 10.
Elisa and activity analysis
An mhATF-BPTI ELISA was developed by P. Quax et al [21], using a monoclonal antibody specific for the ATF as the capture antibody and a polyclonal antibody directed against BPTI as the detector antibody. As a standard we used urokinase or mhATF-BPTI. The biologic activity of the mhATF-BPTI was tested in vitro in a plasmin inhibition assay using the chromogenic substrate S2251 (Chromogenix, Mölndahl, Sweden). Diluted samples of Ad Adapt mhAB infected A549 cells medium were incubated for 15 min at room temperature with 200 pmol/l plasmin. S251 was then added and after 24 hours incubation at 37 C the A405 was measured to determine the conversion of the chromogenic substrate. Controls consisted of plasmin solution incubated with buffer or with 1.0 KIU /ml aprotinin.
The human endostatin activity was tested in the human umbilical vein endothelial cells HUVEC proliferation assay. In short, HeLa cells were incubated for 2 days with the Ad Adapt human endostatin and the medium was collected. Fibroblast growth factor (bFGF)-stimulated HUVEC in 96 wells plate were incubated for 2 days with hEndostatin infection medium. The proliferation of the HUVEC cell was determined by measuring the incorporation of BrdU. Levels of human endostatin in rat plasma was determined with a human enzyme-linked immunoabsorbent assay (ELISA) kit (InvivoGen, CA USA) according to the manufacturers procedure.
Tumour models
The L42 tumour is a bronchial squamous carcinoma that originated in a Wag/Rij rat after intrapulmonary implantation of an iodine-125 seed [38]. L44 adenocarcinoma was induced in Brown Norway rats by local irradiation of the thorax [39]. Both tumours are also adapted to cell culture and give rise to tumours with the same histological characteristics as the original tumours. These two carcinomas were serially passaged in syngeneic rats for more than 50 passages. The CC531 tumour is a dimethylhydrazine-induced, moderately differentiated colon adenocarcinoma exhibiting weak immunogenicity [40]. The tumour is transplantable in syngeneic Wag/Rij rats. The subcutaneously growing tumour has a low microvascular density of 1.4% ± 0.6 (n = 10) and a volume doubling time of 10 days ± 0.8 (n = 10). For the experiments, the tumours were established in the flank of rats by implanting small pieces (3 × 3 × 3 mm) of tumour tissue sub-cutaneously as described before [41].
The size of the subcutaneous tumours was measured in three dimensions at regular intervals, using a Vernier calliper. The volume of the tumours was calculated, from the three tumour dimensions measures (a, b, c), with the formula V = πabc/6. The doubling time of the different tumours was calculated between volume of 1000 and 2000 mm3.
Microvascular density (MVD) in the L44, L42, and CC531 tumours was determined as follows: When the tumours reached the size of 10 × 10 × 10 mm, the animals were sacrificed and frozen sections through the middle of the tumours were prepared. We used an anti-mouse CD31 antibody (American Diagnostica), that cross reacts with rat CD31, to stain specifically the endothelial cells that can then be easily selected by the image analyser software Qwin™. The software allowed us to quantify the surface of the selected endothelial cells and to relate it to the surface of non-necrotic tumour tissue. The end value is the surface of all endothelial cells as percentage of the surface of non-necrotic tissue: the microvascular density.
The global process including cell infection and protein production is commonly called in the literature adenoviral permissiveness. The in vitro permissiveness of the different cell lines to adenovirus was determined by infection of the cells by graded MOI of Ad5 Adapt Luciferase. The Luciferase activity was measured in the supernatant of the cell culture lysate after 2 days of culture and expressed in RLU / milligram of protein. The in vivo permissiveness of the tumours was assessed by analysing the lysate of the whole tumour (without any surrounding tissue) 2 days after injection of the Ad Adapt Luc. Adenoviral vector was administered in a volume of 100 μL to 8 × 8 × 8 mm tumours.
For the metastases model the L44 and L42 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. When the cells reached 80% of confluence, they were trypsinised, washed and resuspended in PBS. 104 L44 tumour cells or 5.104 L42 tumour cells (0.5 ml) were injected into the tail vein of the syngeneic rat. The animals were sacrificed after 22 days (L44 bearing Brown Norway) and 53 days (L42 bearing Wag/Rij), the lungs fixed in Bouin solution and the nodules on the surface were counted. The first experiments, in which animals were not treated, showed that all animals developed multiple metastases in the lungs. The average number of nodules counted on the lung surface was 58 in Wag/Rij and 93 in Brown Norway rats. The volume doubling time of the metastases in the lung was extrapolated from the volumes determined at two time points. The volume at 11 days (for the L44) and at 20 days (for the L42) was determined by measuring the size of microscopic metastasis on lung sections (10 metastases per rat, 4 rats per tumour model). The final volume of the nodules on the lung surface was assessed, on day 22 for the L44 and day 53 for the L42, using a Vernier calliper (10 nodules per rat, 10 rats per tumour model).
Luciferase assays in tissues
Rats were sacrificed by an overdose of isoflurane and whole organs were dissected out, frozen in liquid nitrogen and stored at -80°. Organs were homogenised in phosphate buffered saline pH 7.8 using a blender. To lyse the cells, DTT (SIGMA, The Netherlands) (1 mM) and Triton x-100 (0.1%) (Merck, The Netherlands) were added. After centrifugation at 10,000 rpm for 10 min, 20 μl of the supernatant was added to 100 μl of luciferase assay substrate (Promega, The Netherlands). Relative light units (RLU) were determined for 30 s using a luminometer (Lumat 951, Wallac, Belgium). The amount of protein in the extracts was determined using a commercial kit (Bio-Rad laboratories, The Netherlands) based on the Coomasie brilliant blue G250 binding assay developed by Bradford [42]. The level of luciferase activity in the tissue homogenates was expressed in RLU / mg protein. The background level of this assay is <1000 RLU/mg protein and was defined by measuring luciferase activity in organs of non treated rats.
mhATF-BPTI determination in tissues
Rats were sacrificed by an overdose of isoflurane and the blood was flushed out by cutting the vena cava and injecting 100 ml PBS into the right ventricle. The whole organs or tumours were dissected out and weighted, frozen in liquid nitrogen and stored at -80 C. Tissue and organs were homogenised in phosphate buffer pH 7.8 using a blender. To prepare a cell lysate, DTT (SIGMA, The Netherlands) (1 mM) and Triton x-100 (0.1%) (Merck, The Netherlands) were added. After centrifugation at 10,000 rpm for 10 min, the ELISA was performed on 1:100 dilution of the supernatant. Total protein in the extracts was determined as described above. The concentration of mhATF-BPTI was expressed in ng / g tissue. The background level of this assay is < 20 ng / g tissue and was defined by measuring the mhATF-BPTI in organs of rats treated with the empty vector.
LacZ expression assay
Forty-eight hours after Ad.Adapt.LacZ administration rats were sacrificed and organs were removed and cut in 2 mm sections. Sections were fixed in 10% phosphate buffered formalin (pH 7.0) for 60 min at room temperature and incubated overnight in 0.5 M sucrose at 4°C. The samples were subsequently frozen in liquid nitrogen. Ten micron thick frozen sections were prepared and stained with 5-bromo-4-chloro-3-indolyl-β-galactopyranoside (X-gal) solution (Molecular Probes, The Netherlands) overnight at 37 C. Finally, sections were counterstained with Hematoxylin-Phloxin-Safran.
Statistical analysis
Survival time until tumour size of 20 ml was analysed by log range analysis of Kaplan-Meier curves. Analysis of variance (ANOVA) was used to analyse the differences in tumour growth delay. Scheffe's test was employed to analyse the difference between the numbers of metastases, between plasma concentrations, and between MVDs.
Results
Tumour characteristics
Carcinomas of the lung were chosen, as it is the most frequent tumour in the cancer population. Several vascularisation phenotypes can be observed in the different histological type of lung cancer. Therefore a bronchial squamous cell carcinoma and an adenocarcinoma with different microvascular density (MVD) were selected to assess the inhibition effect of the ATF-BPTI. The growth characteristics of the tumours are listed in Table 1. The L44 adenocarcinoma grows roughly 2.5-fold faster than the squamous cell carcinoma L42. This difference of growth rate holds for in vitro culture of the cells as well as for subcutaneous tumours and lung metastasis. Probably related to the growth rate, the vascularisation of the L44 tumours was significantly higher (p < 0.001, Scheffe'test) than that of the L42 tumours. The distribution of endothelial cells in each of the tumour is homogeneous in the viable part of the tumour. However, the L42 tumour develops a necrotic area with no vessels in the centre of the tumour mass as it grows above 8 × 8 × 8 mm.
Table 1.
Doubling time and vascularisation of the tumours
| N | L44 adenocarcinoma | L42 squamous cell carcinoma | |
| Rat strain | Brown Norway | Wag/Rij | |
| Volume Doubling time | |||
| • in vitro [hours] | 4 | 19.3 ± 1.9 | 46.2 ± 5.7 |
| • subcutaneous tumour [days] | 10 | 3.5 ± 0.5 | 8 ± 0.7 |
| • lung metastasis [days] | 10 | 3.9 ± 0.3 | 9.9 ± 0.7 |
| MVD [%] ± SD | |||
| • subcutaneous tumour | 5 | 3.5 ± 0.1 | 2 ± 0.1 |
The in vitro doubling time is determined after two days of culture of 106 cells / well in a 6-wells plate. The Volume Doubling Time of subcutaneous tumours is assessed between 1000 and 2000 mm3. For the lung metastasis the doubling time was determined between day 11 and 22 for the L44 and between day 20 and 53 for the L42 carcinoma. The micro-vascular density (MVD) represents the surface of all endothelial cells as percentage of the surface of non-necrotic tissue. This was measured in 10 × 10 × 10 mm subcutaneous tumours. The values indicate means ± SD.
The in vitro permissiveness to Ad5 of the cultured cell line showed that the L42 carcinoma had a 2 log higher permissiveness than the L44 tumour (Fig 1A). These characteristics are preserved in vivo when using the luciferase as well as the LacZ marker gene delivered in 10 × 10 × 10 mm tumours. Luciferase activity, 2 days after intra tumoral injection of 1010iu Ad Adapt Luc, was 2 log higher in the L42 tumours than in the L44 adenocarcinoma (Fig 1B). The luciferase activity in various organs of the Wag/Rij rats is about the background level, and 1 to 2 log higher in those of the Brown Norway rats. Our interpretation is that a significant proportion of the vector escapes into the circulation and reaches the liver and spleen, because the L44 tumour cells are poorly infected by the adenovirus. These observations were corroborated by the results of the intratumoral injection of the β-Galactosidase encoding adenovirus. Histological examination, 2 days after administration of 1010iu Ad Adapt LacZ, showed that the L42 tumour preparations were almost completely blue whereas the L44 tumours showed few blue cells around the needle track (Fig 2). This inhomogeneous distribution is not specific for the L44 tumour, as it was also observed in the L42 tumours when injecting a lower dose of virus (data not shown).
Figure 1.
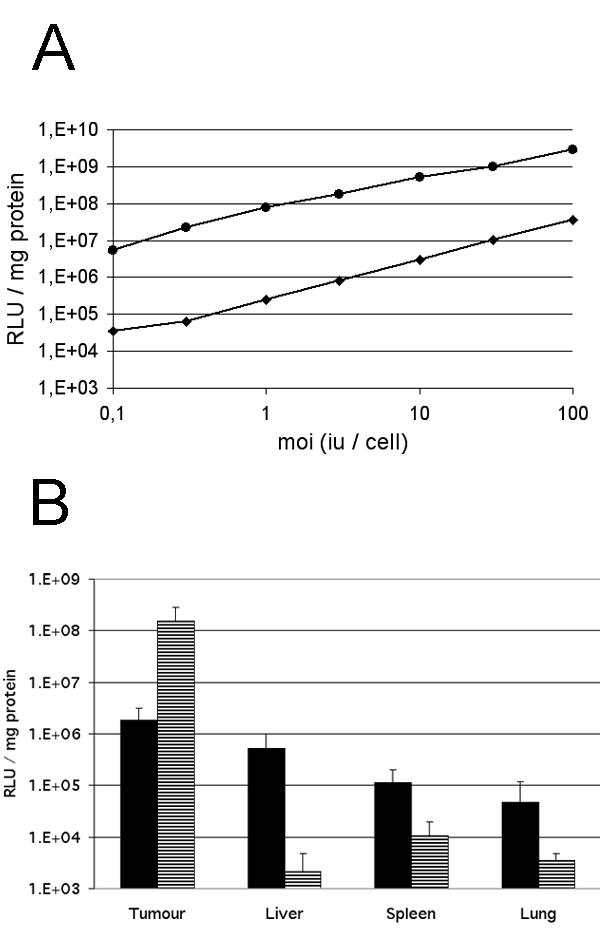
Tumour permissiveness to recombinant adenovirus. A: Luciferase activity in the L42 (circles) and L44 (diamonds) cell lines infected with increasing amounts of Ad Adapt Luc. Values are the mean of duplicate experiments. B: Luciferase expression in subcutaneous L44 tumours (black bars) and in L42 tumours (striped bars) after intratumoral injection of 1010iu Ad Adapt Luc. The luciferase was determined 2 days after virus injection. Four tumours per group, vertical lines indicate SD.
Figure 2.
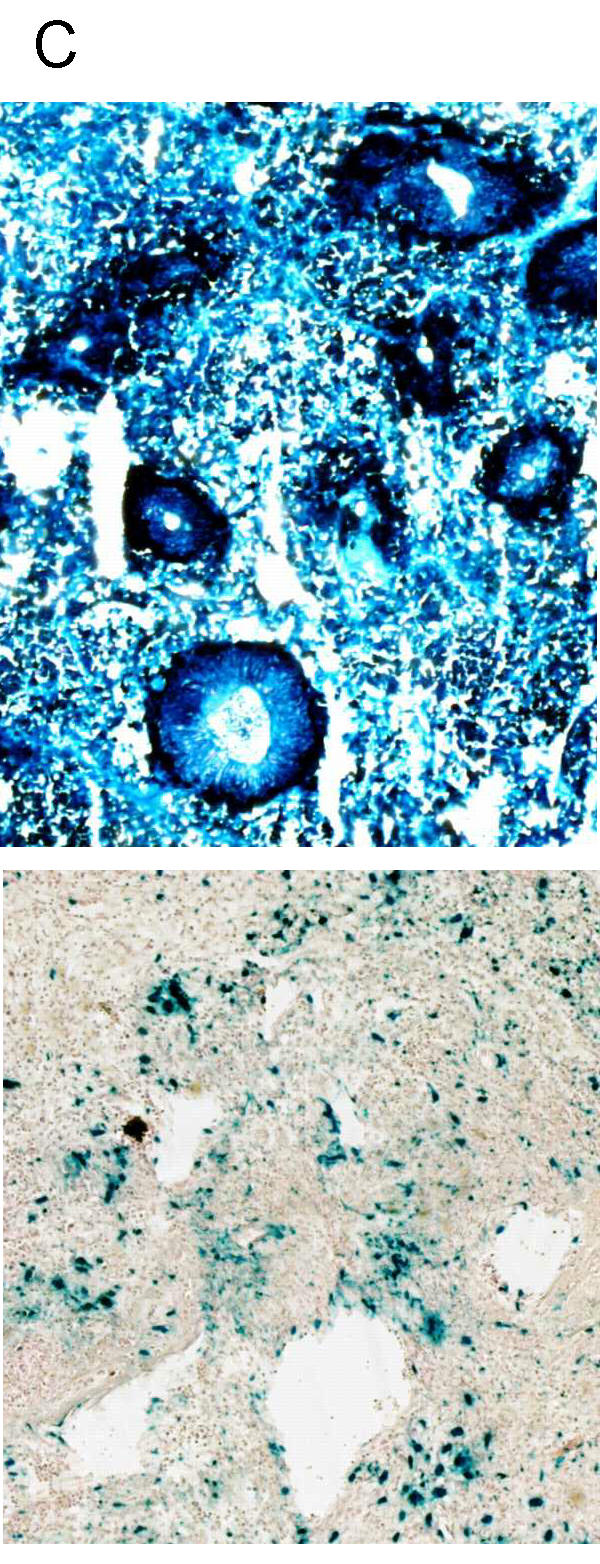
Distribution of adenoviral infection in the tumours. Histological sections (magnification × 50) of L42 (above) and L44 (below) tumours stained for β-galactosidase (blue). The necropsy was performed 2 days after intra tumoral injection of 1010iu Ad Adapt LacZ, when the tumours were about 10 × 10 × 10 mm.
In vitro activity of mhATF-BPTI and hEndostatin
To assess the functionality of the vectors, the biological activities of the encoded transgenes were tested in vitro. As depicted in Fig 3A, the mhATF-BPTI expressed in the supernatant of Ad Adapt mhAB infected A459 cells was able to inhibit 70% of the plasmin activity at a picomolar concentration (mol wt: 25 kD). Furthermore, mhATF-BPTI was shown to inhibit plasmin activity at the cell surface of rat aortic smooth muscle cells (RASMC) expressing rat urokinase receptor [21].
Figure 3.
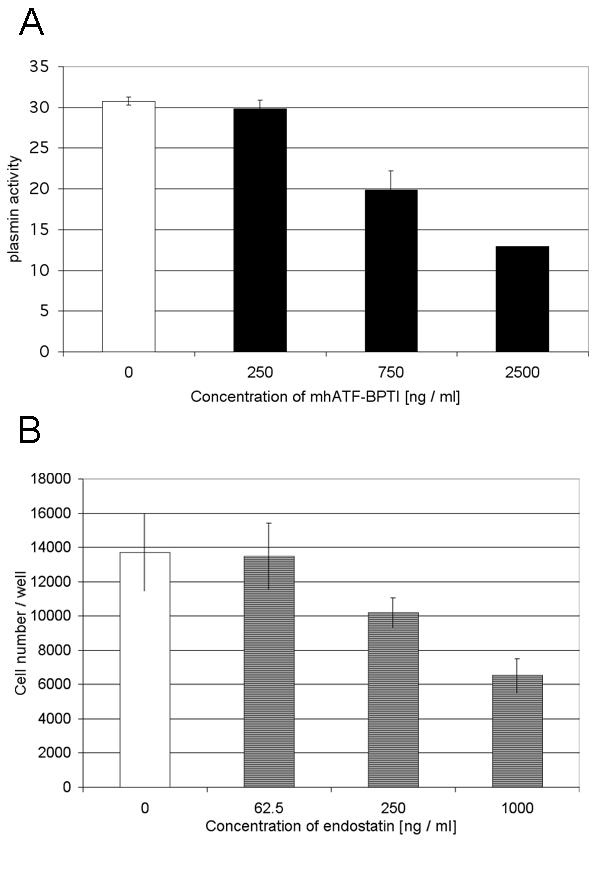
Biological activity of the encoded proteins. A: Inhibition of the plasmin activity (arbitrary units) by increasing amount of mhATF-BPTI in supernatant of A459 cells infected by Ad Adapt mhAB (black bars) or Ad Adapt Empty (white bar). Values are means of triplicate experiments. Vertical lines indicate SD. B: In vitro assay for endostatin activity. Inhibition of bFGF (1 ng / ml) induced proliferation of HUVEC cells by increasing amounts of hEndostatin in supernatant of A459 cells infected by Ad Adapt hEndo (stripped bars). Ad Adapt Empty infection medium was used as positive control (open bar). Values are means of quadruplicate experiments. Vertical lines indicate SD.
The biologic activity of the endostatin encoded in the adenoviral vector was determined through the bFGF simulated HUVEC proliferation assay. A linear dose response relationship between endostatin concentration and percentage of inhibition of HUVEC proliferation could be demonstrated (Fig. 3B). The ED50 of endostatin (1000 ng / ml) was in the range of published data [25]. Further, the angiostatic and antitumoral activities of the human endostatin has been demonstrated in different rat models [26-28].
Secretion of mhATF-BPTI and hEndostatin in animals
Brown Norway rats were injected intravenously with different doses of Ad Adapt mhAB (from 108 to1010iu) and the concentration of mhATF-BPTI was assessed in the plasma at various time points post-infection. The detection limits of the ELISA in plasma is around 5 ng / ml. The results, depicted in Fig 4, show a linear dose response with a maximum level of 500–800 ng / ml obtained when 1010iu Ad Adapt mhAB was injected intravenously. A linear dose response relationship was also observed in both rat strains after iv injection of graded doses (109iu to 1010iu) of vectors encoding the marker genes luciferase and LacZ (data not shown). In the Wag/Rij rats the plasma levels of mhATF-BPTI were 10 times lower (Fig. 5A) after the same vector dose. The plateau concentration after intravenous injection of 1010iu Ad Adapt mhAB was around 50 ng /ml plasma. In both rat strains the plasma levels reached a plateau after 3 days and remained constant for at least 1 month. The levels of ATF in the plasma were similar to those of the ATF-BPTI in the Brown Norway as well as in the Wag/Rij rats (Fig. 5B).
Figure 4.
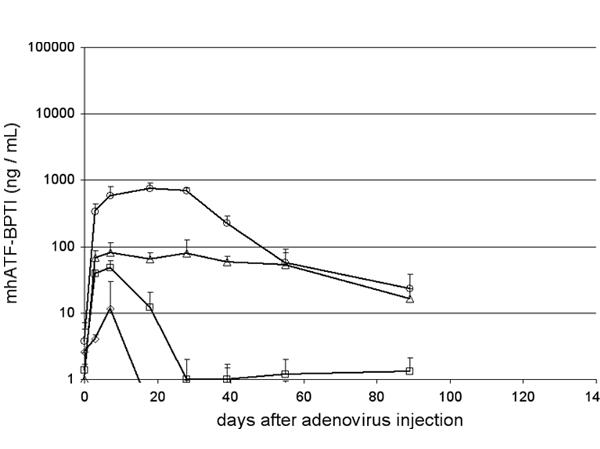
Plasma levels of mhATF-BPTI in Brown Norway rats. The rats received an iv injection of 1010iu (circles), 2.5109iu (triangles), 109iu (squares), and 108iu (diamonds). Five animals per group, vertical lines indicate SD.
Figure 5.
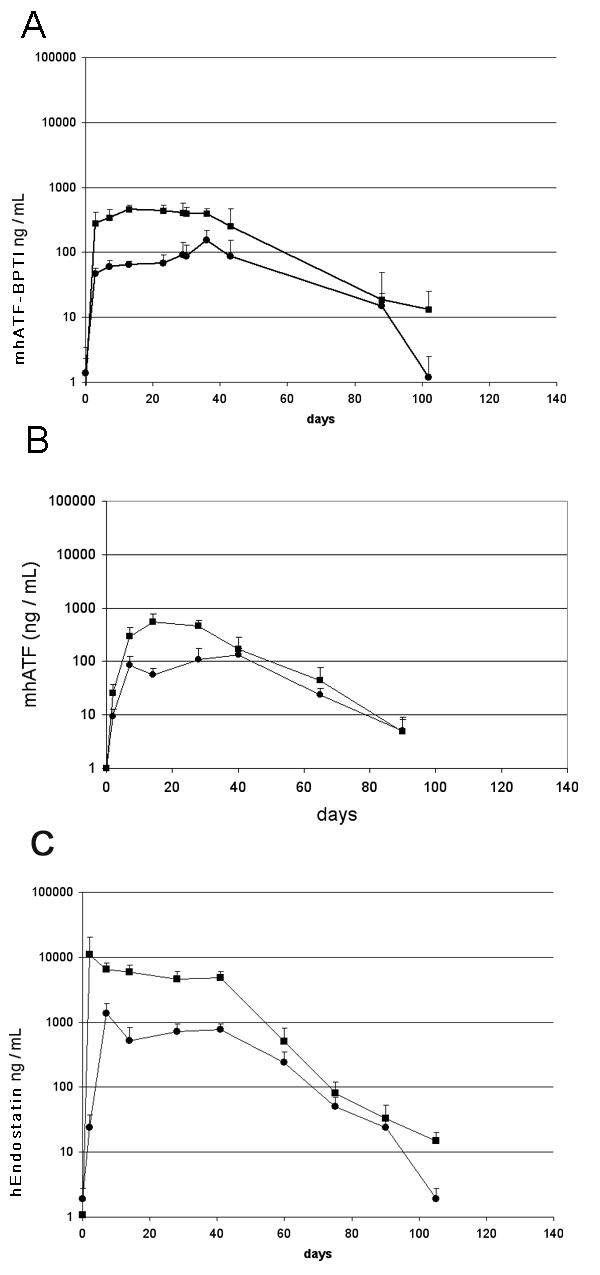
Plasma levels of the encoded proteins in Brown Norway (squares) and Wag/Rij (circles) rats. The rats received an iv injection of 1010iu of the respective vector on day 0. Ten animals per group, vertical lines indicate SD. A: mhATF-BPTI. B: mhATF. C: human Endostatin. These concentrations were reproducible in other experiments.
The human Endostatin was measured in the plasma of animals that received 1010iu Ad Adapt hEndostatin intravenously (Fig. 5C). The concentrations of endostatin were much higher than those of mhATF-BPTI. The levels reached a plateau around 10 μg / ml in the Brown Norway rats and around 1 μg / ml in the Wag/Rij rats. Otherwise the kinetics were roughly similar to those of mhATF-BPTI, with a plateau lasting for a month and one log lower levels in the Wag/Rij than in the Brown Norway rats.
Effects of systemic administration of Ad mhAB on tumour growth
The purpose of these experiments was to investigate the response to angiostatic gene therapy of the two selected lung tumours with different characteristics. Firstly, we investigated the effects of systemic delivery of mhATF-BPTI on the growth of subcutaneous tumour implants.
Brown Norway rats bearing L44 tumours were treated with intravenous injections of Ad Adapt mhATF-BPTI daily for 4 consecutive days. As depicted in Fig 6, three different doses of vector were compared and a significant inhibition of the tumour growth (p < 0.001, ANOVA) was observed with the 2 highest doses (109iu and 2.5 109iu). Seven days after the viral injection, the plasma concentrations of mhATF-BPTI were about 120 ng / ml and 250 ng / ml respectively, which is slightly higher than the levels obtained after a single injection. In contrast, treatment of such tumours with a low dose of adenoviral vector (108iu), that induced lower plasma levels of mhATF-BPTI of 10 ng / ml, did not significantly inhibit tumour growth. Thus it appears that the anti tumour activity of mhATF-BPTI is dependant on its systemic concentration. Higher doses could not be achieved without inducing unacceptable liver toxicity, as manifested by a severe inflammatory infiltrate and vacuolisation of the hepatocytes. Therefore 1010iu was the maximal accepted dose for these experiments.
Figure 6.
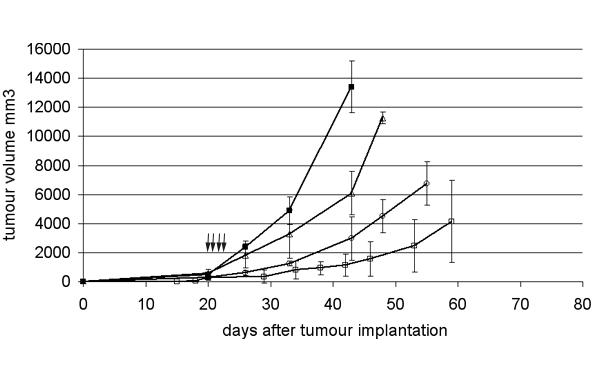
L44 tumour responses to a dose escalation of Ad Adapt mhATF-BPTI. The animals were treated with daily iv injections of the vector for 4 consecutive days. The treatment began 20 days after the tumour implantation when the tumours were 8 × 8 × 8 mm. Arrows indicate days of injection. The doses employed are 108iu (open triangles, n = 4), 109iu (open circles, n = 4), and 2.5 109iu (open squares, n = 6) Ad Adapt mhATF-BPTI and 1010iu (closed squares, n = 6) Ad Adapt empty. Vertical lines indicate SD.
In a second experiment Ad Adapt mhAB and Ad Adapt hEndostatin were compared (Fig. 7). L44 tumour pieces were implanted subcutaneously on day 0 and 1010iu of each vector were injected iv in a single dose on day 20. The treatment with Ad Adapt mhAB caused a significant inhibition (p < 0.0001, ANOVA) of the growth of the L44 tumours as compared to the Ad Adapt empty or Ad Adapt mhATF treated animals (Fig. 7A). The human endostatin, which plasma level was around 10 μg / ml 2 days after vector injection, showed a slight but not significant effect (p = 0.007, ANOVA) on tumour growth compared to the empty vector. The mhATF-BPTI effect was achieved with a plasma level of mhATF-BPTI of about 800 ng / ml between day 5 and 30 after the virus injection. It is required by Dutch law to sacrifice animals when the tumour volume has surpassed 20 ml. That intervention precluded the continuation of the growth curves after the time point at which the first animal is sacrificed. Therefore the animals with a tumours volume under 20 ml were plotted on a Kaplan Meier analysis graph. The result showed a significant (p < 0,0001, rank test) improvement in time to sacrifice showing an Ad Adapt mhAB specific protective effect. In the control group, the rats (n = 9) reached the limiting size after 38 to 50 days, whereas in the Ad AB treated group (n = 9), the rats had tumours above 20 ml after 55 to108 days. Mean time to sacrifice was prolonged by 30 days (Fig. 7B).
Figure 7.
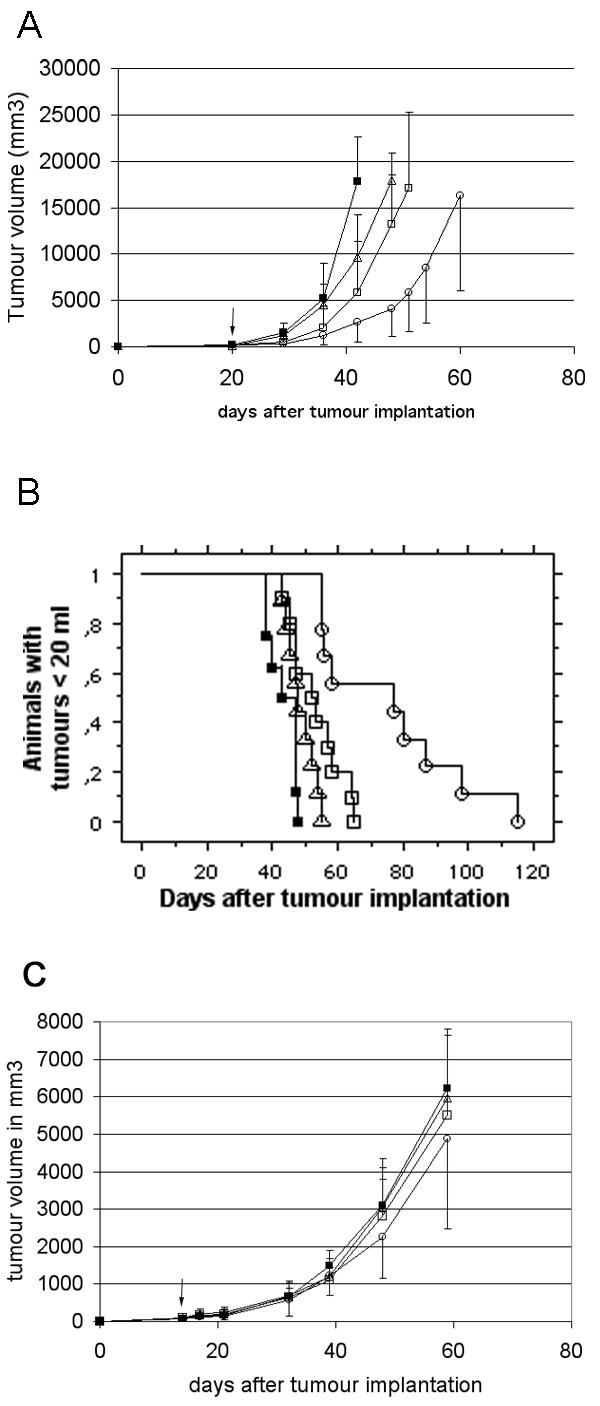
Tumour response after systemic treatment with a single iv dose of 1010iu of the various vectors. The vectors employed are: Ad Adapt mhATF-BPTI (open circles), Ad Adapt hEndostatin (open squares), Ad Adapt mhATF (open triangles), and Ad Adapt empty (closed squares). 10 animals per group, vertical lines indicate SD. The arrow indicates the time of treatment. A: L44 tumours treated by Ad Adapt mhAB (p < 0.0001, ANOVA), Ad Adapt mhATF (p = 0.03, ANOVA), and Ad Adapt hEndo treatment (p = 0.007, ANOVA). The treatment began at day 20 when the tumours were 8 × 8 × 8 × m. B: Kaplan-Meyer plots of treated rats: end point tumour volume 20,000 mm3 (for the Ad Adapt mhAB, p < 0.0001, rank test). C: L42 tumour growth curves, the viruses were injected on day 14 when the tumours were 4 × 4 × 4 mm.
To complete the study, the same treatment protocol was applied to Wag/Rij rats bearing the slowly growing and poorly vascularised L42 adenocarcinoma. A single dose of 1010iu of the different vectors was injected iv on day 14 (Fig. 7C). The plateau concentration of mhATF, mhATF-BPTI and endostatin were low, around 50 ng/ml for the 2 first and around 0.5 μg/ml for the last respectively. No significant inhibition of the tumour growth was observed in either the mhATF-BPTI, the mhATF, or the endostatin groups as compared to the empty vector group. We concluded that the plasma levels of our therapeutic agents were too low to induce any response in this model. In an attempt to improve this issue we studied direct injection of our vectors into the tumour.
Intra-tumoral Ad Adapt mhAB administration delays tumour growth in L42
Adenoviral-mediated delivery of the mhATF-BPTI directly at the tumour site was studied in the L42 lung carcinoma implanted subcutaneously into Wag/Rij rats. 19 days after implantation, one intratumoral virus injection (1010iu in 0.1 ml PBS) per day for 3 consecutive days was performed and tumour growth monitored. Seven days after the beginning of the treatment, a significant (p < 0.0001, ANOVA) delay of tumour growth was seen in the Ad Adapt mhAB injected group (Fig. 8A). The mhATF-BPTI plasma levels were under the detection limit of the ELISA.
Figure 8.
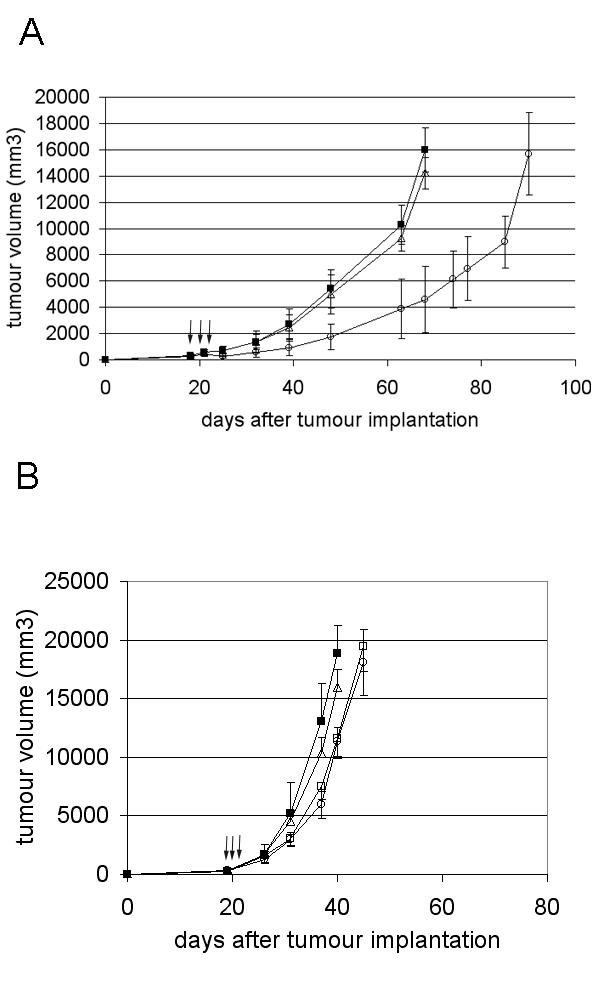
Tumour response after local injection of 3 consecutive doses of 1010iu. A: L42 tumour treated with Ad Adapt mhATF-BPTI (open circles, p < 0.0001 ANOVA), Ad Adapt mhATF (open triangles), and Ad Adapt empty (closed squares). Arrows indicate the days of injection (19 – 21) and the tumours were 8 × 8 × 8 mm at the first day of injection. 10 animals per group, vertical lines indicate SD. B: L44 tumour after treatment with 1010iu Ad Adapt mhATF-BPTI (open circles), Ad Adapt mhATF (open squares), Ad Adapt hEndostatin (open squares), and Ad Adapt empty (closed squares). Arrows indicate the days of injection (19 – 21) and the tumours were 8 × 8 × 8 mm at the first day of injection. 5 animals per group, vertical lines indicate SD.
In contrast, local delivery of the vector to the very low permissive L44 tumour did not lead to any significant tumour growth inhibition by Ad Adapt mhAB (Fig. 8B).
Local concentration of mhATF-BPTI
The results of the systemic administration in the Brown Norway suggested a correlation between the plasma concentration of mhATF-BPTI and the tumour response. The concentrations of mhATF-BPTI were measured in the plasma and in the tumour at 7 and 14 days after intravenous or intra-tumoral injection of the mhATF-BPTI vector (Table 2). The systemic delivery of 1010iu Ad Adapt mhAB in L44 bearing Brown Norway rats resulted in a concentration of mhATF-BPTI in the tumour of about 1.2–1.8 μg / g tumour. This intra-tumoral level was maintained for at least 2 weeks and is 2–3 times higher than the plasma level. This suggests that mhATF-BPTI concentrates in the tumour. The concentration of mhATF-BPTI in the L42 tumours after intravenous delivery of the vector was much lower around 150 – 160 ng / g tumour, but as in the Brown Norway rats 2–3 time higher than the plasma concentration.
Table 2.
Concentration of mhATF-BPTI in the subcutaneous tumours
| L44 | L42 | ||||
| Route of administration | n | Day 7 | Day 14 | Day 7 | Day 14 |
| Intravenous [ng / g tumour] | 6 | 1791 ± 274 | 1256 ± 187 | 162 ± 98 | 152 ± 34 |
| [ng / ml plasma] | (658 ± 56) | (506 ± 62) | (52 ± 23) | (61 ± 21) | |
| Intratumoral [ng / g tumour] | 6 | 59 ± 35 | 88 ± 34 | 1415 ± 147 | 1048 ± 485 |
| [ng / ml plasma] | - | - | - | - | |
The concentrations were determined after a single iv injection of 1010iu Ad Adapt mhAB and after intratumoral injection of Ad Adapt mhAB (1010iu / day for 3 days). The concentration of mhATF-BPTI [ng / g of fresh tissue] was determined in the tumour lysate at 7 and 14 days after the vector delivery. The plasma concentration [ng / ml] is indicated under the intra tumoral values. In case of the intra tumoral administration the plasma levels were under the detection limit.
The local delivery of the transgene in the tumour resulted in a low concentration of ATF-BPTI in the L44 tumour (60 – 90 ng / g tumour) and high concentration in the L42 tumours (1 – 1.4 μg / g tumour). This was expected from the 2 log higher permissiveness to adenoviral infection of the L42 compared to the L44 tumours.
Effect of mhATF-BPTI on lung metastasis
In the subcutaneous tumour model, the majority of the tumour cells implanted die, and the surviving cells grow in the fibrous interstitial tissue of the skin. The subsequent angiogenesis give rise to one small artery that will vascularise the whole tumour. This probably does not reflect accurately the clinical situation, where a primary carcinoma cell grows in a highly vascularised epithelium. The angiogenesis in non-small-cell lung carcinomas in patients shows four different patterns of vascularisation: basal, diffuse, papillary and alveolar [29]. The basal and the diffuse are the most frequent patterns found in squamous carcinomas and adenocarcinomas respectively. Both are accompanied by a destruction of the normal lung parenchyma and the production of newly formed vessels. Our lung metastasis models in the rat also represent these two main patterns: the L44 (adenocarcinoma) shows a diffuse architecture and the L42 (squamous cell carcinoma) grows with a basal histological architecture. Further characterisation showed that the growth rate of the lung metastases is roughly similar to that of subcutaneous implants, the difference between L42 and L44 being maintained at both locations (Table 1).
The protective effect of systemic administration of Ad Adapt mhAB was assessed by treating the animals with a single iv dose of Ad Adapt mhAB, or Ad Adapt hEndostatin, or with Ad Adapt Empty as negative control virus. The treatment was administered when the micro metastasis were a few hundreds cells large as observed in not-treated rats. This size was reached at 4 days after injection of the L44 tumour cells and after 13 days in the L42 model. The animals were sacrificed when the lung nodules were expected to be 2 mm diameter in the control group. As depicted in Fig. 9A the L42 metastasis-bearing rats responded to treatment with Ad Adapt mhAB with a more than 50% reduction in the number of metastases. Macroscopic inspection of the lung surface and microscopic examination of lung sections gave similar responses. It is also noteworthy that treatment with the endostatin vector, which results in a much higher level of the angiostatic protein (1–3 μg / ml), does not significantly reduce the number of metastases. In contrast, the L44 lung metastasis were not significantly influenced by either the Ad Adapt mhATF-BPTI or the Ad Adapt mhATF treatments (Fig 9B).
Figure 9.
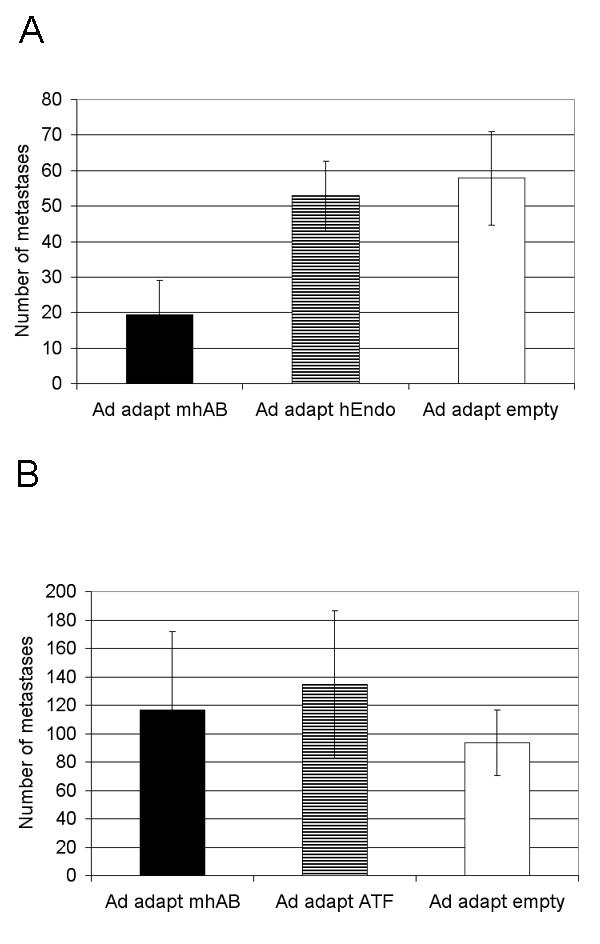
Effect of systemic treatment with 1010iu adenovirus on lung metastasis. A: L42 metastases. The lungs were analysed 53 days after the tumour cell injection (p < 0.0001, Scheffe's test for difference between mhAB and empty group). 10 animals per group, vertical lines indicate SD. B: L44 metastases. The lungs were analysed 22 days after the tumour cell injection.10 animals per group, vertical lines indicate SD.
Surprisingly, the systemic administration of Ad Adapt mhATF-BPTI does inhibit the growth of L42 lung metastases, but not of the L44 metastases. In view of the high permissiveness of the L42 tumour cells, it seemed possible that the lung metastases were transfected with the mhATF-BPTI gene. Therefore the permissiveness of the lung micrometastasis was estimated, by determining the β-galactosidase and luciferase activity 2 and 7 days after the iv injection of 1010iu Ad Adapt LacZ or Ad Adapt Luc into animals bearing L44 or L42 micrometastasis in the lungs. The results were however essentially negative. The luciferase activity was under the background level in all cases and the sections of the frozen lungs showed no tumour cell stained for the β-galactosidase in either L44 or L42 metastasis bearing animals (data not shown, 6 animals per group). Thus, it seems that the metastases are not actively contributing to the levels of mhATF-BPTI in the lungs and the concentration in the metastases should be similar to the concentration in the lung parenchyma (all the blood being washed out). Therefore, the concentration of mhATF-BPTI in the lung of the two rat strains was determined. The results depicted on Table 3 show that the mhATF-BPTI concentration is significantly higher (p < 0.001, Scheffe's test) in the Wag/Rij rats than in the Brown Norway rats and is not influenced by the presence of metastasis. The Wag/Rij rats concentrate the mhATF-BPTI in the lung parenchyma about 10 fold, reaching therapeutic levels. In contrast, the same systemic treatment has no effect on the L42 tumour growing subcutaneously, and this can be explained by the fact that tumour does not concentrate the mhATF-BPTI sufficiently.
Table 3.
Concentration of mhATF-BPTI in the lung
| N | Brown Norway (L44) | Wag/Rij (L42) | |
| mhATF-BPTI [ng / g] animals with metastasis | 7 | 164 ± 39 | 547 ± 90 |
| mhATF-BPTI [ng / g] healthy animals | 3 | 142 ± 51 | 479 ± 64 |
The concentration of mhATF-BPTI [ng / g of tissue] was determined at 7 days after a single intravenous injection of 1010iu Ad Adapt mhAB in healthy animals and in rats with lung metastasis. The values indicate means ± SD.
Influence of the tumour mass on the anti tumour activity of mhATF-BPTI
Radio- and chemotherapy are usually expected to be less effective on large tumour mass than on small tumours. In contrast, it was shown that certain anti-angiogenic compounds (e.g. Combrestatin) act the other way around, namely that they are more active on large tumours (>10 cm3) than on small tumours (<1 cm3). The supposed explanation of this peculiar volume – response relationship is based on the concept that the requirement for new vessels increases with the size of the tumour [30]. Nevertheless, this is not a general rule for angiostatics. Various compounds such as endostatin, metallo proteinase (MMP) inhibitors, and TNP-470 do not inhibit the tumour growth when its size is too large. Thus, large tumours that are richly vascularised might be more sensitive to angiostatic therapy. In order to investigate this issue in the case of ATF-BPTI, two subcutaneous tumours with difference vascular density were treated at different time points (figure 10 and 11). In the highly vascularised L44 as well as in the poorly vascularised CC531 tumours, ATF-BPTI could significantly inhibit the growth of the tumours when they were around 100 mm3 at the time of treatment. In contrast, when the tumours reached a volume of 1000 mm3, the treatment had no more influence on the tumour growth. The receptor of ATF-BPTI (uPAR) is known to be principally present at the outer edge of the tumour mass and as the volume increases, the proportion of uPAR in the tumour will decrease. Thus, it was expected that large tumour mass were insensitive to the treatment. In this extent, ATF-BPTI is similar to the MMPs inhibitors, in that their best indication would probably be for treatment of residual disease or small metastases.
Figure 10.
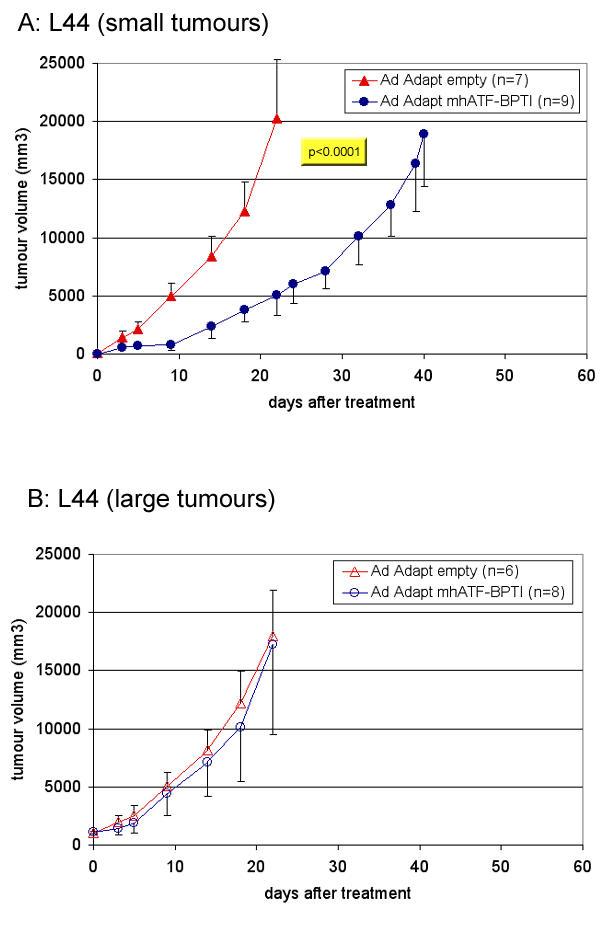
Influence of the tumour mass on the anti tumour effect of ATF-BPTI in the L44 model. The animals were treated when the tumours were about 100 mm3 (A) or about 1000 mm3 (B). The lung adenocarcinoma L44 was implanted subcutaneously in Brown Norway rats and in order to achieve therapeutic levels, the animals were treated by intravenous injection of the vector (1010 iu Ad Adapt mhAB or Ad Adapt empty). The p value between the treated group and the control is indicated in the figure when the difference was significant according to the ANOVA test. The number of animals per group is indicated in the figures. Vertical lines indicate SD.
Figure 11.
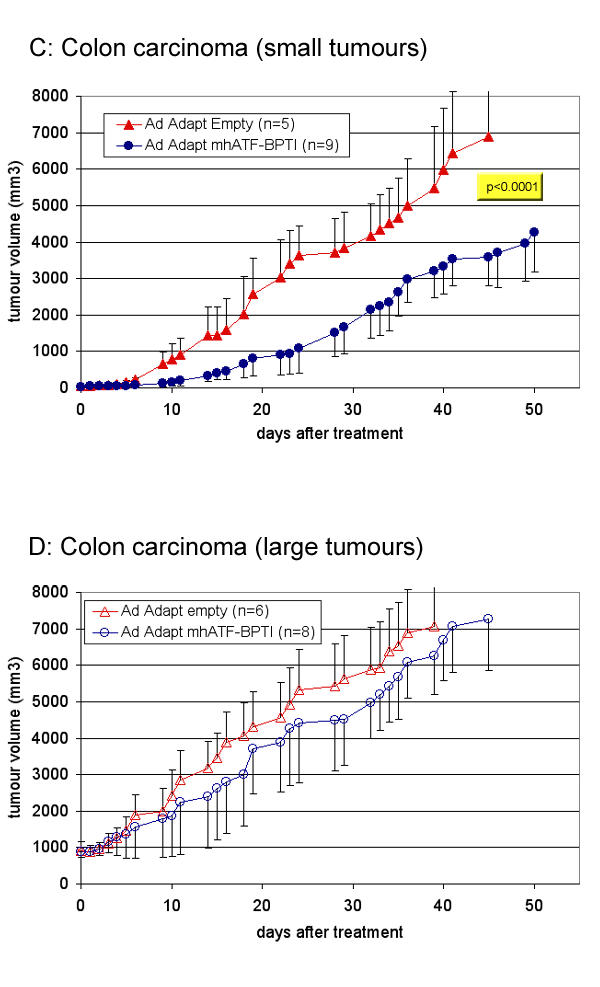
Influence of the tumour mass on the anti tumour effect of ATF-BPTI in the CC531model. The animals were treated when the tumours were about 100 mm3 (C) or about 1000 mm3 (D). The colon adenocarcinoma CC531 grows in Wag / Rij rats in which high plasma levels could not be obtain, therefore the animals were treated by one intratumoral injection of 1010 iu Ad Adapt mhAB or Ad Adapt empty. The p value between the treated group and the control is indicated in the figure when the difference was significant according to the ANOVA test. The number of animals per group is indicated in the figures. Vertical lines indicate SD.
Discussion
Agents that inhibit the growth of new vessels in the tumours include a wide range of different compounds. Most of them are not toxic for endothelial cells, but impair the dynamic process of cell proliferation, migration and matrix remodelling. Thus, these agents are mostly effective on the formation of new vessels and not on the pre-existing vasculature. Accordingly such therapy is not expected to cause a dramatic shrinkage of an established tumour. It appears from the different clinical trials with endostatin, thalidomide, the synthetic inhibitors of MMPs, and the different anti-VEGFs that angiostatics, though slightly effective in advanced diseases, are more likely indicated in combination with chemotherapy or radiotherapy and as an adjuvant therapy to treat small tumour masses, in particular local minimal residual disease or micrometastasis. In whatever situation, long term systemic delivery is considered to be the treatment of choice. Therefore gene transfer may offer an advantage. The potential of adenovirus based vectors to infect the liver cells after intravenous injection makes them suitable vectors for establishing a prolonged systemic delivery of the encoded protein. However, our findings so far show that the achievable plasma levels of mhATF-BPTI are limited by adenoviral damage to the liver. To circumvent this limitation, direct injection of the vector in the tumour was employed in an attempt to achieve higher intra tumoral levels of the therapeutic protein.
High levels of mhATF-BPTI were achieved in the permissive L42 tumour by local delivery of the vector and subsequently a significant inhibition of tumour growth was obtained. The Wag/Rij rat liver is poorly permissive to the adenoviral infection and accordingly systemic administration of the vector induces low plasma levels of mhATF-BPTI (<80 ng / ml). Consequently, the intra tumoral concentration, though 3 times higher than the plasma levels, was not high enough to inhibit its growth. In contrast, the L42 lung metastasis in the Wag/Rij did respond to the intravenous administration of the vector encoding mhATF-BPTI, though the resulting plasma level was low. It was observed that the mhATF-BPTI accumulated in the lung parenchyma, independently of the presence of metastasis, up to a concentration of about 500 ng / g. This fully explains the therapeutic response observed in this lung metastasis model.
In contrast, the L44 adenocarcinoma cells have a low permissiveness to the adenovirus. Consequently, in case of direct intra tumoral delivery of the vector, the tumour concentrations reached were low (<90 ng / g) and tumour responses were not observed. However, the Brown Norway rat liver is 10 times more permissive to the adenovirus and produced high levels of the vector-encoded protein. Additionally, the tumour tissue concentrates 2 to 3 fold the mhATF-BPTI from the plasma, resulting in high intra tumoral levels. Using graded doses of Ad Adapt mhAB, a linear relation was observed between the dose and the plasma level as well as between the dose and the tumour growth delay. It appeared that a significant tumour response could be obtained when the intra tumoral concentration of mhATF-BPTI was above 400 ng / g of tissue. In contrast, at the same plasma levels that inhibited the growth of the subcutaneous L44 could not induce any response on the metastasis in the lungs. The tissue concentration of mhATF-BPTI in the lung of the Brown Norway rats was apparently too low (<180 ng / g) for eliciting a decrease in the number of metastasis.
Conclusion
The tumour growth inhibition in both poorly and highly vascularised L44 and L42 tumours was achieved only when the concentration of mhATF-BPTI in the tumoral mass or in the surrounding tissue reached a value of at least 300 to 400 ng / g. In contrast, mhATF and the human endostatin even at higher molecular plasma concentration than the mhATF-BPTI did not achieve any significant inhibition of tumour growth as in established subcutaneous tumours as well as in micrometastasis in the lungs. Other investigators reported that at those concentrations endostatin and ATF induced responses of tumours and metastases in mice [31,32]. The absence of a response in our tumour model might be due to a lower sensitivity to angiostatic agents. In that case our results suggest that ATF-BPTI is a more potent agent than endostatin or ATF.
List of abbreviations
ATF: Amino Terminal Fragment
bFGF: Fibrobast Growth Factor b
BPTI: Bovine Pancreatic Trypsin Inhibitor
GPI: Glycophosphatidylinositol
HUVEC: Human Umbilical Vein Endothelial Cell
KIU: Kallikrein Inhibition Unit
MDV: Micro-Vascular Density
mhATF: Murinised human ATF
mhATF-BPTI: Murinised human ATF-BPTI
MMP: Matrix Metallo-Proteinase
TIMP: Tissue Inhibitor of Metallo Protease
uPA: Urokinase Plasminogen Activator
uPAR: Urokinase Receptor
VEGF: Vascular Endothelial Growth Factor
Competing interests
None declared.
Authors' Contributions
PL and JA participated in the all aspects of the study and DvB is the principal investigator.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgement
We would like to thank Margreet de Vries, Peggy Tjou Tam Sin, Chantal Pont, Pascal Merchier, Germaine Penders, and Joos Grimbergen for their excellent technical assistance. We gratefully acknowledge Dr Paul Quax for his stimulating advices. This study is supported by a BTS grant (#99107).
Contributor Information
Pierre Lefesvre, Email: p.lefesvre@crucell.com.
Joline Attema, Email: j.attema@crucell.com.
Dirk van Bekkum, Email: bekkum@crucell.com.
References
- Li CY, Shan S, Huang Q, Braun RD, Lanzen J, Hu K, Lin P, Dewhirst MW. Initial stages of tumor cell-induced angiogenesis: evaluation via skin window chambers in rodent models. J Natl Cancer Inst. 2000;92:143–147. doi: 10.1093/jnci/92.2.143. [DOI] [PubMed] [Google Scholar]
- Miyake H, Hara I, Yamanaka K, Arakawa S, Kamidono S. Elevation of urokinase-type plasminogen activator and its receptor densities as new predictors of disease progression and prognosis in men with prostate cancer. Int J Oncol. 1999;14:535–541. doi: 10.3892/ijo.14.3.535. [DOI] [PubMed] [Google Scholar]
- Costantini V, Sidoni A, Deveglia R, Cazzato OA, Bellezza G, Ferri I, Bucciarelli E, Nenci GG. Combined overexpression of urokinase, urokinase receptor, and plasminogen activator inhibitor-1 is associated with breast cancer progression: an immunohistochemical comparison of normal, benign, and malignant breast tissues. Cancer. 1996;77:1079–1088. doi: 10.1002/(SICI)1097-0142(19960315)77:6<1079::AID-CNCR12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Skelly MM, Troy A, Duffy MJ, Mulcahy HE, Duggan C, Connell TG, O'Donoghue DP, Sheahan K. Urokinase-type plasminogen activator in colorectal cancer: relationship with clinicopathological features and patient outcome. Clin Cancer Res. 1997;3:1837–1840. [PubMed] [Google Scholar]
- van der Burg ME, Henzen-Logmans SC, Berns EM, van Putten WL, Klijn JG, Foekens JA. Expression of urokinase-type plasminogen activator (uPA) and its inhibitor PAI-1 in benign, borderline, malignant primary and metastatic ovarian tumors. Int J Cancer. 1996;69:475–479. doi: 10.1002/(SICI)1097-0215(19961220)69:6<475::AID-IJC10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Ito H, Yonemura Y, Fujita H, Tsuchihara K, Kawamura T, Nojima N, Fujimura T, Nose H, Endo Y, Sasaki T. Prognostic relevance of urokinase-type plasminogen activator (uPA) and plasminogen activator inhibitors PAI-1 and PAI-2 in gastric cancer. Virchows Arch. 1996;427:487–496. doi: 10.1007/BF00199509. [DOI] [PubMed] [Google Scholar]
- Zhang X, Fei Z, Bu X, Zhen H, Zhang Z, Gu J, Chen Y. Expression and significance of urokinase type plasminogen activator gene in human brain gliomas. J Surg Oncol. 2000;74:90–94. doi: 10.1002/1096-9098(200006)74:2<90::AID-JSO2>3.3.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Takanami I, Takeuchi K, Karuke M. Expression of ETS-1 is correlated with urokinase-type plasminogen activator and poor prognosis in pulmonary adenocarcinoma. Tumour Biol. 2001;22:205–210. doi: 10.1159/000050617. [DOI] [PubMed] [Google Scholar]
- de Vries TJ, Quax PH, Denijn M, Verrijp KN, Verheijen JH, Verspaget HW, Weidle UH, Ruiter DJ, van Muijen GN. Plasminogen activators, their inhibitors, and urokinase receptor emerge in late stages of melanocytic tumor progression. Am J Pathol. 1994;144:70–81. [PMC free article] [PubMed] [Google Scholar]
- He CS, Wilhelm SM, Pentland AP, Marmer BL, Grant GA, Eisen AZ, Goldberg GI. Tissue cooperation in a proteolytic cascade activating human interstitial collagenase. Proc Natl Acad Sci U S A. 1989;86:2632–2636. doi: 10.1073/pnas.86.8.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzieri R, Masiero L, Zanetta L, Monea S, Onisto M, Garbisa S, Mignatti P. Control of type IV collagenase activity by components of the urokinase-plasmin system: a regulatory mechanism with cell-bound reactants. Embo J. 1997;16:2319–2332. doi: 10.1093/emboj/16.9.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyke C, Kristensen P, Ralfkiaer E, Grondahl-Hansen J, Eriksen J, Blasi F, Dano K. Urokinase-type plasminogen activator is expressed in stromal cells and its receptor in cancer cells at invasive foci in human colon adenocarcinomas. Am J Pathol. 1991;138:1059–1067. [PMC free article] [PubMed] [Google Scholar]
- Estreicher A, Muhlhauser J, Carpentier JL, Orci L, Vassalli JD. The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J Cell Biol. 1990;111:783–792. doi: 10.1083/jcb.111.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz DA, Natkin LR, Fujita RM, Wei Y, Chapman HA. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J Clin Invest. 1997;100:58–67. doi: 10.1172/JCI119521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin DB, Moscatelli D, Bizik J, Quarto N, Blei F, Dennis P, Flaumenhaft R, Mignatti P. Growth factor control of extracellular proteolysis. Cell Differ Dev. 1990;32:313–318. doi: 10.1016/0922-3371(90)90045-X. [DOI] [PubMed] [Google Scholar]
- Bacharach E, Itin A, Keshet E. In vivo patterns of expression of urokinase and its inhibitor PAI-1 suggest a concerted role in regulating physiological angiogenesis. Proc Natl Acad Sci U S A. 1992;89:10686–10690. doi: 10.1073/pnas.89.22.10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119:629–641. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lu H, Griscelli F, Opolon P, Sun LQ, Ragot T, Legrand Y, Belin D, Soria J, Soria C, Perricaudet M, Yeh P. Adenovirus-mediated delivery of a uPA/uPAR antagonist suppresses angiogenesis-dependent tumor growth and dissemination in mice. Gene Ther. 1998;5:1105–1113. doi: 10.1038/sj.gt.3300742. [DOI] [PubMed] [Google Scholar]
- Li H, Griscelli F, Lindenmeyer F, Opolon P, Sun LQ, Connault E, Soria J, Soria C, Perricaudet M, Yeh P, Lu H. Systemic delivery of antiangiogenic adenovirus AdmATF induces liver resistance to metastasis and prolongs survival of mice. Hum Gene Ther. 1999;10:3045–3053. doi: 10.1089/10430349950016438. [DOI] [PubMed] [Google Scholar]
- Belin D, Vassalli JD, Combepine C, Godeau F, Nagamine Y, Reich E, Kocher HP, Duvoisin RM. Cloning, nucleotide sequencing and expression of cDNAs encoding mouse urokinase-type plasminogen activator. Eur J Biochem. 1985;148:225–232. doi: 10.1111/j.1432-1033.1985.tb08829.x. [DOI] [PubMed] [Google Scholar]
- Lamfers ML, Lardenoye JH, de Vries MR, Aalders MC, Engelse MA, Grimbergen JM, van Hinsbergh VW, Quax PH. In vivo suppression of restenosis in balloon-injured rat carotid artery by adenovirus-mediated gene transfer of the cell surface-directed plasmin inhibitor ATF.BPTI. Gene Ther. 2001;8:534–541. doi: 10.1038/sj.gt.3301437. [DOI] [PubMed] [Google Scholar]
- Lage A, Diaz JW, Gonzalez I. Effect of proteinase inhibitor in experimental tumors. Neoplasma. 1978;25:257–259. [PubMed] [Google Scholar]
- Gavilondo J, Perez R, Mainardi V, Lage A. Effect of two antiproteinases on the growth of transplantable tumors and the proliferation of untransformed and transformed cells in culture. Neoplasma. 1982;29:315–322. [PubMed] [Google Scholar]
- Latner AL, Longstaff E, Turner GA. Anti-tumour activity of aprotinin. Br J Cancer. 1974;30:60–67. doi: 10.1038/bjc.1974.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanabal M, Ramchandran R, Volk R, Stillman IE, Lombardo M, Iruela-Arispe ML, Simons M, Sukhatme VP. Endostatin: yeast production, mutants, and antitumor effect in renal cell carcinoma. Cancer Res. 1999;59:189–197. [PubMed] [Google Scholar]
- Read TA, Sorensen DR, Mahesparan R, Enger PO, Timpl R, Olsen BR, Hjelstuen MH, Haraldseth O, Bjerkvig R. Local endostatin treatment of gliomas administered by microencapsulated producer cells. Nat Biotechnol. 2001;19:29–34. doi: 10.1038/83471. [DOI] [PubMed] [Google Scholar]
- Sorensen DR, Read TA, Porwol T, Olsen BR, Timpl R, Sasaki T, Iversen PO, Benestad HB, Sim BK, Bjerkvig R. Endostatin reduces vascularization, blood flow, and growth in a rat gliosarcoma. Neuro-oncol. 2002;4:1–8. doi: 10.1215/S1522851701000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroulis I, Jonas N, Saleh M. Antiangiogenic activity of endostatin inhibits c6 glioma growth. Int J Cancer. 2002;97:839–845. doi: 10.1002/ijc.10115. [DOI] [PubMed] [Google Scholar]
- Pezzella F, Pastorino U, Tagliabue E, Andreola S, Sozzi G, Gasparini G, Menard S, Gatter KC, Harris AL, Fox S, Buyse M, Pilotti S, Pierotti M, Rilke F. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am J Pathol. 1997;151:1417–1423. [PMC free article] [PubMed] [Google Scholar]
- Landuyt W, Verdoes O, Darius DO, Drijkoningen M, Nuyts S, Theys J, Stockx L, Wynendaele W, Fowler JF, Maleux G, Van den Bogaert W, Anne J, van Oosterom A, Lambin P. Vascular targeting of solid tumours: a major 'inverse' volume-response relationship following combretastatin A-4 phosphate treatment of rat rhabdomyosarcomas. Eur J Cancer. 2000;36:1833–1843. doi: 10.1016/S0959-8049(00)00173-8. [DOI] [PubMed] [Google Scholar]
- Kisker O, Becker CM, Prox D, Fannon M, D'Amato R, Flynn E, Fogler WE, Sim BK, Allred EN, Pirie-Shepherd SR, Folkman J. Continuous administration of endostatin by intraperitoneally implanted osmotic pump improves the efficacy and potency of therapy in a mouse xenograft tumor model. Cancer Res. 2001;61:7669–7674. [PubMed] [Google Scholar]
- Kim Lee Sim B, Fogler WE, Zhou XH, Liang H, Madsen JW, Luu K, O'Reilly MS, Tomaszewski JE, Fortier AH. Zinc ligand-disrupted recombinant human Endostatin: Potent inhibition of tumor growth, safety and pharmacokinetic profile. Angiogenesis. 1999;3:41–51. doi: 10.1023/A:1009058931769. [DOI] [PubMed] [Google Scholar]
- Koopman JL, Slomp J, de Bart AC, Quax PH, Verheijen JH. Mitogenic effects of urokinase on melanoma cells are independent of high affinity binding to the urokinase receptor. J Biol Chem. 1998;273:33267–33272. doi: 10.1074/jbc.273.50.33267. [DOI] [PubMed] [Google Scholar]
- Esandi MC, Verlinden SFF, Vries de MW, Pfeffer G, Bekkum van DW. Neutralizing antibodies against Adeno 5 virus open the therapeutic window for interleukin-3 gene therapy of tumors with adenovirus 5 based vectors. submitted to gene therapy. 2001.
- Fallaux FJ, Bout A, van der Velde I, van den Wollenberg DJ, Hehir KM, Keegan J, Auger C, Cramer SJ, van Ormondt H, van der Eb AJ, Valerio D, Hoeben RC. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum Gene Ther. 1998;9:1909–1917. doi: 10.1089/hum.1998.9.13-1909. [DOI] [PubMed] [Google Scholar]
- Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, Van Der Eb AJ. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther. 1996;7:215–222. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- Shabram PW, Giroux DD, Goudreau AM, Gregory RJ, Horn MT, Huyghe BG, Liu X, Nunnally MH, Sugarman BJ, Sutjipto S. Analytical anion-exchange HPLC of recombinant type-5 adenoviral particles. Hum Gene Ther. 1997;8:453–465. doi: 10.1089/hum.1997.8.4-453. [DOI] [PubMed] [Google Scholar]
- Kal HB, Zurcher C, van Bekkum DW. Induction of lung tumors by radioactive isotopes implanted in the rat lung. J Natl Cancer Inst. 1986;76:943–946. [PubMed] [Google Scholar]
- Kal HB, Meijnders PJ, Van Berkel AH, Van Bekkum DW. Response to chemotherapy of non-small cell bronchial rat tumours growing subcutaneously or in the lung. In Vivo. 1991;5:301–306. [PubMed] [Google Scholar]
- Marquet RL, Westbroek DL, Jeekel J. Interferon treatment of a transplantable rat colon adenocarcinoma: importance of tumor site. Int J Cancer. 1984;33:689–692. doi: 10.1002/ijc.2910330521. [DOI] [PubMed] [Google Scholar]
- Esandi MC, van Someren GD, Bout A, Mulder AH, van Bekkum DW, Valerio D, Noteboom JL. IL-1/IL-3 gene therapy of non-small cell lung cancer (NSCLC) in rats using 'cracked' adenoproducer cells. Gene Ther. 1998;5:778–788. doi: 10.1038/sj.gt.3300662. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]