

Abstract
Recombinant adeno-associated viruses (rAAVs) produced by transfecting DNA plasmids into mammalian cells can inadvertently package host cell DNA (hcDNA) and plasmid DNA inside their capsids. Although the percentage of these DNA impurities is low compared to the rAAV genome in vector preparations, it is essential to characterize the DNA impurities in gene therapy products due to the theoretical risks associated with unwanted gene expression and potential immunogenicity and oncogenicity in treated patients. We performed long-read sequencing in rAAV vector, with a focus on analyzing residual, non-transgene DNA within the capsids. Although we detected host cell and residual plasmid DNA impurities, they were predominantly incomplete sequences without coding potential. This indicated that while DNA impurities may be present in rAAV preparations, host cell and residual plasmid genes were unlikely to be expressed. This was supported by RNA sequencing (RNA-seq) analyses that showed minimal plasmid RNA transcripts and host cell RNA transcripts in the livers of mice dosed with rAAV. Overall, the results from these studies enable data-based risk assessment of co-packaged DNA impurities and a better understanding of potential adverse effects associated with rAAV gene therapy.
Keywords: rAAV, residual DNA, host cell DNA transcription, PacBio sequencing, gene therapy, DNA impurities, AAV product quality assessment, AAV safety assessment
Graphical abstract
AAV gene therapy vectors have demonstrated clinical efficacy and safety. The current manuscript provides a detailed analysis of the nature, quantity, annotation, and transcription of residual DNA from host cells and production plasmids, thereby addressing one of the safety concerns associated with residual DNA in AAV vectors.
Introduction
Recombinant adeno-associated virus (rAAV) has emerged as a widely used vector for gene transfer in various therapeutic applications. These therapies offer promising potential for the treatment of genetic diseases, neurodegenerative disorders, and cancer.1,2 In recent years, extensive research and clinical trials have demonstrated the safety and efficacy of rAAV-based therapies.3 Currently, there are seven approved in vivo rAAV therapies with several hundred in clinical trials in the United States.4,5 Continued research and technological advancements in rAAV vector design, production, and administration will not only enhance patient care but also inform for future developments and improvements in rAAV-based gene therapies.6,7
The wild-type AAV genome is a short single-stranded DNA molecule, approximately 4.7 kb in length, containing sequences required for replication and capsid formation flanked by GC-rich palindromic inverted terminal repeats (ITRs).8,9,10 The replication and capsid genes between the two ITRs (Rep and Cap) can be removed and replaced with an expression cassette that includes the gene of interest.9,11 Production of rAAV requires host cells, such as human HEK293 cells or insect Sf9 cells, and is achieved through either transient transfection, viral infection, or stable cell line production.12,13,14 Ideally, encapsidated DNA would consist solely of full-length vector genome sequences, including the ITRs and the expression cassette. However, rAAV products typically contain varying degrees of nucleic acid impurities,15,16 with the DNA impurities arising from the plasmid DNA or helper virus used in production, and the host cell DNA from the production cell line.12,17 Depending on the analytical methods and purification techniques, the level of these DNA impurities can range from 0.5% to 13% of the vector genome.18
The presence of DNA impurities in rAAV therapy raises concerns about potential safety risks to patients.18,19,20 For example, a study in non-human primates (NHP) showed that small amounts of DNA impurities in rAAV therapies could trigger inflammation in the NHP brain.21 Also, a major concern regarding residual host cell DNA (hcDNA) is the possibility of expression of harmful genetic elements, like oncogenes.22,23 Current World Health Organization (WHO) recommendations for residual hcDNA in products produced from cell substrates include a maximum of 10 ng/dose and <200 bp in size.24,25 However, these thresholds were based on guidance for biologic therapies, such as antibodies and viral vaccines. Since the production of rAAV involves extensive cellular materials and rAAV occasionally encapsidates unwanted DNA fragments at varying rates, current rAAV gene therapy products cannot meet these stringent criteria.26
Although the co-packaged plasmid DNA in rAAV vector preparations is not expected to possess functional mammalian promoters and therefore should not undergo transcription, evidence suggests that DNA impurities in rAAV preparations may undergo nonhomologous recombination27,28 with vector genomes, leading to the presence of ITR-containing plasmid DNA sequences.29 AAV ITRs can function as a weak mammalian promoter30,31 and could potentially enable transcription of DNA impurities. These ITR-containing impurities with potential for transcription and expression raise concerns about unintended production of non-therapeutic proteins and other unwanted RNA or protein expression. It is therefore crucial to further investigate and understand the presence and behavior of DNA impurities in rAAV preparations and evaluate the extent of potential implications for patients treated with rAAV therapies.32
Results
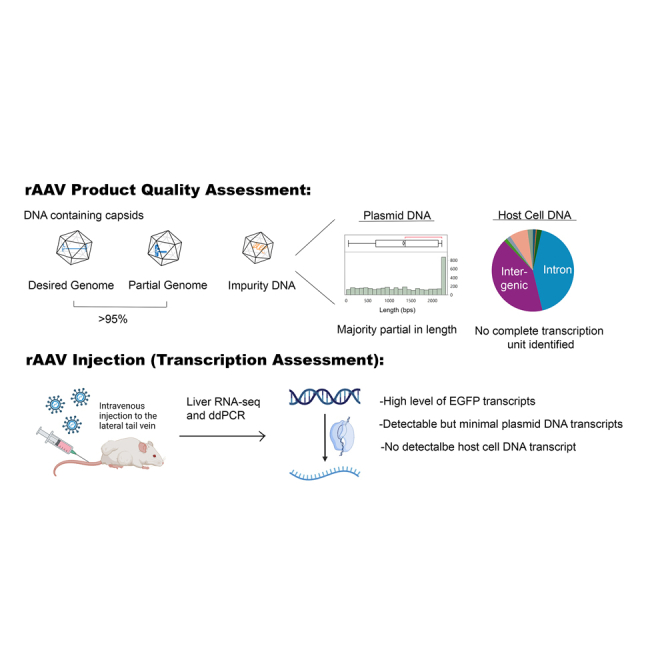
Quality measurements of rAAV materials
Recombinant AAVhu37 containing the transgene enhanced green fluorescent protein (rAAVhu37-EGFP) was produced using triple transfection of HEK293 cells (Figure 1A) to a genomic titer of 1.7E13 GC/mL, as measured by droplet digital PCR (ddPCR) (Figure 1). rAAV vector purification was performed as described in the materials and methods. The amount of residual hcDNA was measured by qPCR using either the 18S or Alu amplicon with host cell genomic DNA as standard. The concentration of residual hcDNA was approximately 95 ng per 1E+12 viral genome copies based on 18S amplicon and approximately 15 ng per 1E+12 viral genome copies based on an Alu amplicon, which suggested hcDNA quantification in rAAV vectors is dependent on target of choice. We then measured residual plasmid DNA impurities, including AAV2 Rep, AAVhu37 Cap, and Kanamycin resistance (KanR) genes by a dPCR assay. Results are summarized in Figure 1C. Overall, the amount of DNA impurities was similar to previous findings in vectors made with transfection-based processes.18
Figure 1.

rAAVhu37-EGFP vector diagram and analytics
(A) Plasmids used in triple transfection for viral production. (B) rAAV products and capsid variants that contain different DNA species. (C) Analytical data for rAAVhu37-EGFP product.
Assessment of transcription potential for residual plasmid DNA
PacBio sequencing was performed to understand the genome composition of the packaged rAAVhu37-EGFP vector. PacBio sequencing provides high-quality, single DNA molecule sequencing of the entire genome and assesses genome quality and impurity size/quantity. Circular consensus sequencing (CCS) reads were generated from rAAVhu37-EGFP vector DNA, and the results are summarized in Table 1. The majority (95.38%) of the sequencing reads mapped to the expression cassette. Out of all reads mapped to the expression cassette, 92.86% of the reads contain the full expression cassette (Figure S1). Approximately 4% of the reads mapped to either the plasmid backbone (2.26%) or the human genome (2.01%). Of all the plasmid DNA sequences mapped to the plasmid backbone, 32.05% contained at least one ITR sequence, whereas only 1.15% of the reads mapped to the human genome contained any ITR sequences. ITRs can form a hairpin structure that serves as a primer for DNA polymerase, enabling the synthesis of the second strand of DNA without the need for additional primers.33,34
Table 1.
Mapping statistics for PacBio sequencing
| Total CCS reads (>200 bps) | CCS reads mapped to expression cassette | CCS reads mapped to all plasmid backbone |
CCS reads with ITR mapped to human genome |
|||
|---|---|---|---|---|---|---|
| All | With ITR | All | With ITR | |||
| rAAVhu37-EGFP | 574,836 | 548,260 | 12,976 | 4,159 | 11,547 | 133 |
| Percent of Reads | 100.00% | 95.38% | 2.26% | 32.05%a | 2.01% | 1.15%b |
Percentage of CCS reads mapped to plasmid backbone containing ITR sequences.
Percentage of CCS reads mapped to human genome containing ITR sequences.
Reads that mapped to plasmid DNA genes were analyzed for transcription potential. The abundance of each impurity gene was calculated based on coverage of the impurity gene relative to the coverage of the expression cassette. The levels measured for the impurity genes were: AAV.hu37 Capsid (0.68%), AAV2 Rep (0.46%), KanR (0.28%), E4 (0.13%), E2A (0.10%), and VARNA (0.10%) (Figure 2; Table S1). No detectable adenovirus E1 (E1A and E1B) sequences were in the rAAV materials. Analysis of all open reading frames (ORFs) indicates that most of the reads mapped to the plasmid genes were partial and lacked a complete ORF. The percentages of reads containing complete ORFs for each impurity were calculated: AAV.hu37 Cap (22%), AAV2 Rep (12%), KanR (63%), E4 (2%), E2A (14%), and VARNA (25%) (Figure 2; Table S1). These findings suggested that the encapsidated plasmid DNA molecules are mostly fragmented and are thus unlikely to be transcribed and translated into functional protein.
Figure 2.

Quantity and size distribution of residual plasmid DNA
(A) Size distribution for residual plasmid DNA impurities. (B) Number of residual plasmid DNA genes and their complete open reading frames.
Genomic feature of residual hcDNA
Genome annotation was performed on the 2.01% of reads that mapped to the human genome. These reads were evenly distributed across the genome (Figure 3A), with most reads annotated as either intronic (41.3%) or intergenic (42.7%; Figure 3B). Approximately 2% of loci were annotated as exons (Figure 3B), but only 10 were single-exon genes. Upon manual analysis using the UCSC genome browser, two reads contained whole exons (Table S2); however, none had a complete transcription unit, including both the upstream untranslated region (5′ UTR) and downstream untranslated region (3′UTR). This indicates that the hcDNA impurities are mostly fragmented and lack promoters or gene bodies to produce a functional transcript. Interestingly, >7% of the residual hcDNA mapped to promoter regions in the human genome (Figure 3B). Given that only 1%–2% of the human genome contains protein-coding genes,35,36 it appears that hcDNA impurities are enriched with promoter regions.
Figure 3.

Distribution of residual HcDNA
(A) Genome distribution of hcDNA sequences. (B) Distribution of residual hcDNA in different chromosomes. (C) Motif enrichment analysis of the hcDNA.
The AAV Rep protein binds the Rep binding element (RBE) within the ITR and promotes genome replication, packaging, and AAV genome integration into the host genome.37,38 To determine if Rep binding promotes the packaging of hcDNA, we screened PacBio CCS reads mapped to the host genome for the AAV2 ITR RBE motif, GAGCGAGCGAGCGCGC.39 Simple enrichment analysis revealed significant enrichment of the RBE motif in the mapped regions (p value = 1.03E−146) (Figure 3C; Table S3). RBE motif are significantly enriched in the promoter (p value = 2.76E−17, n = 1823), intronic (p value = 5.33E−66, n = 10238), intergenic regions (p value = 2.03E−72, n = 10475), and transcription termination site (TTS) (p value = 1.49E−13, n = 558). Analysis with partial ITR sequence (AAGGTCGCCCGACGCC) showed slight enrichment (p-value = 5.8E−12), which aligns with the previous result that there are 1.15% of ITR-containing hcDNA (Table 1). These results suggested that hcDNA containing RBE sequences is more likely to be packaged. In our analysis, we found that 32% of hcDNA molecules have RBE sequences, whereas 68% of hcDNA molecules do not, suggesting that other mechanisms may be involved in residual hcDNA packaging.
Rep protein can bind to transcription factors directly, thereby promoting rAAV genome replication40,41,42,43 without binding to DNA. Since there is an enrichment of promoter regions in residual hcDNA sequences, we wanted to determine if there was enrichment of any transcription factors. A known motif analysis of transcription factors was performed on reads mapped to hcDNA. Results showed an enrichment of certain motifs compared with the background (Table S4), suggesting that transcription factor binding might be involved in hcDNA encapsidation in rAAV vector.
rAAVhu37-EGFP successfully transduces mouse cells and induces EGFP expression
As a first step to understand whether rAAV-encapsidated residual plasmid DNA would enter mammalian cells and undergo RNA transcription, we analyzed the expression of the transgene EGFP and DNA impurities in animals dosed with rAAVhu37-EGFP. A single dose of rAAVhu37-EGFP was administered to adult mice at 2.0E+14 GC/kg, and two or three weeks later the mice were sacrificed. As expected, we found high protein expression of EGFP in the dosed mice livers, and samples taken 3 weeks post-dosing had approximately twice the level of EGFP protein compared with samples taken at 2 weeks (Figure 4A). Given that GFP protein has a half-life of approximately 24–48 h,44 this result showed sustained viral vector EGFP production and expression beyond the first 2 weeks of viral injection.
Figure 4.

Expression of EGFP in the rAAVhu37-EGFP infected mouse liver
(A) EGFP protein measured by ELISA (n = 3 controls, n = 5 per experimental group). (B) EGFP mRNA calculated from RNA-seq (n = 3). Data are shown as means (SD), p values were determined by non-paired student t test. (C) Mapping results of RNA-seq reads from livers of control (n = 3) and rAAVhu37-EGFP-dosed mice (n = 3 per group).
We then performed RNA sequencing (RNA-seq) on the livers of the dosed mice to assess the transcription levels of the EGFP transgene and residual plasmid DNA impurities. To distinguish endogenous mouse RNA from viral DNA-derived RNA, the total reads were first mapped to the mouse genome (mm10). As expected, the majority of RNA reads (95%–98%) were mapped to the mouse genome. The reads that failed to align concordantly or non-concordantly were extracted and mapped to the EGFP plasmid references. A significant number of reads were mapped to the EGFP gene of interest (GOI) region in the livers of mice dosed with rAAVhu37-EGFP, as expected, compared to the control group (100× GPF expression compared to control; Figures 4B and 4C). The two peaks observed in the reads mapped to EGFP in Figure 4C are likely due to fragmentation bias introduced during library preparation. Similar to the protein expression, RNA transcripts in samples taken 3 weeks post-treatment with rAAVhu37-EGFP showed an approximately 2-fold increase in the percentage of reads mapping to the EGFP expression cassette compared with samples taken 2 weeks post-treatment (Figure 4B). This indicates that rAAVhu37-EGFP effectively induces EGFP RNA transcription and protein translation in the mouse liver.
Residual plasmid DNA expression assessment in mice livers
To determine whether residual plasmid DNA undergoes transcription, we analyzed the RNA-seq reads that mapped to plasmid references. The average read depth for each impurity is summarized in Table S5. As expected, there were almost no detectable reads mapped to the plasmid DNA in the control samples (Figures 5A and 5B). On the contrary, detectable levels of some transcripts from DNA impurities were observed in samples obtained 2 and 3 weeks post-rAAVhu37-EGFP injection (Figures 5A and 5B).
Figure 5.

RNA expression of genes from residual plasmid DNA in mice dosed with rAAVhu37-EGFP
(A) Abundance of plasmid DNA-derived mRNA in control and rAAVhu37-EGFP-dosed mice by RNA-seq (n = 3). (B) Average read depth for the residual plasmid DNA impurities. Data are shown as means (SD). (C) Normalized quantification of plasmid DNA impurities by dPCR (n = 3). (D) Correlation between percentage of residual DNA transcripts (in mice livers) and percentage of residual DNA (in AAV vectors) with or without open reading frame.
Statistical analysis revealed a significant enrichment of AAVhu37 Capsid, AAV2 Rep, and KanR transcripts in samples taken 2 and 3 weeks post-rAAVhu37-EGFP dosing compared with controls (Figure 5B). There was no statistical difference in transcripts from samples taken 2 weeks post-injection versus those taken 3 weeks post-injection (Figure 5B).
Further analysis by reverse transcription (RT) dPCR was performed to confirm the RNA-seq results. For the RNA samples from control animals, no amplification was detected for EGFP or the plasmid DNA impurities. In contrast, RNA from the rAAVhu37-EGFP-transduced samples showed high levels of EGFP expression, consistent with previous RNA-seq findings (Figure 4). Additionally, detectable levels of transcripts from DNA impurities were observed in samples obtained 2 and 3 weeks post-rAAVhu37-EGFP injection. The residual plasmid-DNA-derived transcripts were normalized to the EGFP transcripts. As shown in Figure 5C, the relative transcript levels for all DNA impurities in mice livers were less than 0.01% of EGFP, indicating a significantly lower relative transcript level after entering animal cells compared to the relative DNA level observed in the rAAVhu37-EGFP vector (relative transcript level is 10–100 times lower than the DNA level). There was no correlation between the percentage of impurity reads detected by PacBio sequencing and their transcription level (Figure 5D); however, there was a positive correlation between the percentage of impurity reads containing a complete ORF and their transcription level (Figure 5D). These results suggest that a complete open reading frame is necessary for transcription from co-packaged plasmid-derived DNA impurities.
Residual hcDNA expression assessment in mice livers
To better understand the transcription potential of residual hcDNA, RNA-seq reads from mouse liver samples were analyzed for hcDNA (human genome)-derived transcripts. Reads that failed to map to the mouse genome or to the three production plasmids were extracted and mapped to the human genome. While control samples showed minimal reads mapped to plasmid references, 1%–3% of reads from livers of dosed mice mapped to production plasmids (Table S6), mainly to the EGFP region (Table S5), and 2%–3% of RNA-seq reads from all samples, including control samples, mapped to the human genome. When comparing the percentage of reads mapped to the human genome, no difference was observed between the control and rAAV-treated samples. The 2%–3% RNA-seq reads aligned to the human genome could result from environmental contamination during library preparation or sequencing45 or from mouse transcript variants that did not align with the mouse genome.46 Additionally, principal component analysis of the transcriptomic data mapped to the human genome showed no distinct clustering by sample type (Figure S2). These results indicate that no human-DNA-derived transcripts were detected in rAAV-treated mice livers by RNA-seq and support the absence of complete coding sequences of human genes in the PacBio long-read sequencing analysis of vector DNA (Table S2).
Discussion
Gene therapies have the potential to be curative treatments for patients by delivering genetic materials encapsidated within viruses to their target tissues. rAAV is one of the main viral vectors for gene therapy, with several rAAV-based gene therapy products being approved in recent years.4,5
Current Food and Drug Administration (FDA) guidance for human gene therapy new drug applications24,25 recommends that the residual DNA impurity from the host cell be less than 10 ng per dose and for the DNA size to be below 200 bps. This guidance was based on previous guidance for biologics such as antibodies and viral vaccines,24 which was based on the recommendation of the WHO in 1998. Recent studies have shown that rAAV gene therapy products far exceed these limits (1–210 pg/1E9GC).47,48 Thus, it is critical to understand further the risk associated with these DNA impurities, including hcDNA and plasmid-derived DNA sequences. Unlike other biologic therapies, the DNA moiety in the viral-vector-based gene therapy product is an intrinsic component. As a result, vectors containing DNA impurities introduced through a cellular production platform can be difficult to separate from the desired product.
Transcription potential of residual plasmid DNA
The transcription potential of residual plasmid DNA within gene therapy vectors is a critical concern due to the risk of unintended gene expression. A previous study suggested undetectable transcription of an AAV cap gene in an rAAV product48; however, a recent study showed detectable RNA transcripts for residual plasmid DNA.18 The discrepancy in data from the abovementioned studies might come from differences in plasmid design, purification methods, or sensitivity of the quantification methods. In our analysis of rAAVhu37-EGFP, PacBio sequencing was employed to map sequencing reads to the expression cassette, plasmid backbone, and hcDNA. Results indicated that although the majority of reads mapped to the intended expression cassette, approximately 4% of total reads mapped to either the plasmid backbone or human genome sequences (hcDNA). These levels of residual plasmid DNA are at the lower end of what is expected from an rAAV product.18 In addition, a significant portion of these reads, except for KanR, contained fragmented sequences lacking complete ORFs, which reduced their protein expression potential. The low transcription potential of these impurities is aligned with findings from earlier research.18,19
A subset of reads contains complete ORFs, suggesting potential transcription of these DNA impurities. We found that the level of residual plasmid DNA transcripts correlates with the number of residual plasmid DNA sequences containing complete ORFs identified in the PacBio sequencing using rAAVhu37-EGFP. KanR DNA, with the most number of reads with complete ORF, expressed at the highest level, whereas AAVhu37 Cap and AAV2 Rep DNA reads have a smaller number of complete ORF, expressed at a lower level compared to KanR. These data suggest that it is possible to use PacBio long-read sequencing results to predict the likelihood of residual plasmid DNA transcription in rAAV vectors.
While EGFP expression increased in mice livers dosed with rAAVhuh37-EGFP for 3 weeks compared to those for 2 weeks, the expression level of residual plasmid DNA remained the same. It is known that rAAV DNA in animal tissues may persist in the nucleus as monomer episomes or concatemeric circles,49 as these structures stabilize the rAAV DNA and promote sustained expression from the transgene. The expression of impurity DNA suggests that unlike rAAV DNA, the impurity DNA may not be as stable as the transgene in animal tissues. This is supported by the low percentage (∼32.05%) of plasmid sequences that contain ITRs (Table 1) and even lower percentage (1.15%) of hcDNA that contain ITRs. It is expected that residual hcDNA and plasmid DNA are less stable due to a lack of ITR structures in their sequences.
Several studies have shown that in addition to second strand synthesis, double-stranded rAAV vector genomes may be formed through complementary strand annealing.50,51,52 In this case, a plus or minus single-strand vector genome anneals to another single-strand vector genome with a complement sequence to form a double-stranded vector genome.50,51,52 The importance of this mechanism to transduction was demonstrated in a study where infecting cells with a mixture of plus and minus single-strand rAAV vectors led to increased transduction compared to preparations consisting of only a single polarity.50 In our study, the EGFP expression cassette was the predominant vector genome, whereas the DNA impurities were present at a much lower level in the liver tissue. Since the EGFP rAAV preparation contained equimolar quantities of plus and minus strand vectors, the probability of single-strand vectors containing EGFP forming stable double-stranded vectors through complementary strand annealing was much higher than single-strand vectors containing DNA impurities. We predict this would contribute to a reduced number of DNA impurity vectors forming stable double-stranded DNA and result in lower and less efficient expression of DNA impurities compared to EGFP.
It is also possible that impurity RNA transcripts are less stable due to the absence of a strong mammalian promoter and the polyadenylation signal sequences.53,54 Samples from more extended pre-clinical studies in the future could answer whether residual plasmid DNA transcription is temporary or permanent.
Mechanism of residual plasmid DNA transcription
The exact mechanism underlying co-packaged residual plasmid DNA transcription remains unclear. Theoretically, DNA lacking a mammalian promoter would not undergo transcription. In our trans plasmid, the P5 promoter is at the 3′ end of the AAVhu37 Capsid and AAV2 Rep genes. The P5 promoter consists of several transcription factor binding sites, including a YY1 initiator and TATA domain, both of which are known to initiate transcription in mammalian cells.55,56,57,58 This could explain the RNA expression observed for AAVhu37 Cap and AAV2 Rep genes. To minimize residual Cap/Rep expression, a recent study used a modified P5-HS promoter, which incorporates a spacer sequence between the P5 Rep binding sequences and the P5 YY1 initiator; this design greatly reduced Cap/Rep expression.59
Unlike the Cap and Rep genes, the KanR gene has no functional mammalian promoter driving its expression in the production plasmids. However, it has been shown that the ITR sequence could function as a weak promoter.21,30,31 The function of ITR in transcription was demonstrated by the decreased transcription when ITRs are mutated.31 In our study, about 32% of the residual plasmid DNA contains ITR sequences, which may drive the expression of KanR genes.
Residual hcDNA transcription potential
The transcription potential of residual hcDNA in gene therapy vectors is a safety concern that has been given increased attention. Therefore, understanding the genomic constitution of hcDNA in each rAAV product and the potential risks associated with the hcDNA is important. Since the median size of protein-coding genes in the human genome is 26,288 bp,60 it is expected that the hcDNA in AAV products contains a very small amount, if any, of short coding sequences, which are unlikely to be fully transcribed to proteins. In our rAAVhu37-EGFP vector, PacBio sequencing revealed that approximately 2% of the reads mapped to human genomic sequences, mostly to intronic (41.3%) or intergenic (42.7%) regions. Only a tiny fraction of reads mapped to the exonic areas and even fewer contained the complete exon. Manual analysis of the exonic fragments confirmed that none contained complete ORFs (including untranslated regions), consistent with previous assumptions that fragmented hcDNA is unlikely to produce functional proteins.19 Furthermore, the RNA-seq results suggested no human DNA transcripts are enriched in livers of the mice treated with rAAV.
In our rAAVhu37-EGFP vector, a relatively higher percentage of transcription start sites from the hcDNA was packaged into rAAV, compared to the transcription start site composition in human genome. The actual percentage of DNA containing transcription start sites might be higher since more than 1,000,000 potential promoter sequences are still unannotated.61 Promoter regions may be more accessible to protein binding,62,63,64 allowing Rep proteins to bind to transcription factors to initiate DNA replication and promote encapsidation of hcDNA. The enrichment of active transcription sites could explain the quantification difference between hcDNA measured by qPCR using the 18S amplicon versus the Alu amplicon (Figure 1C), since 18S ribosomal RNA is actively transcribed in cells, whereas Alu repeats are not.
Our study found enrichment of many transcription factor binding sites and RBE in the residual hcDNA, suggesting hcDNA containing RBE sequences are more likely to be packaged into rAAV capsids. A recent study with comprehensive mutagenesis of Rep protein provided a detailed sequence-to-function map and showed single codon mutation can improve rAAV production.65 In the AAV field, it is still unclear whether mutated Rep protein with different binding capacities could alter residual hcDNA packaging.
Additional safety risks associated with DNA impurities in rAAV vectors
This paper assessed sequences of residual hcDNA and residual plasmid DNA in purified rAAV vector. We provided evidence of negligible transcription of plasmid-derived genes and no transcription of hcDNA in mice livers after rAAV transduction. The DNA impurity profile in rAAV vectors can vary between different production and purification systems. Other studies may therefore have different results if rAAV vectors were generated from an alternative manufacturing process.
Additional safety concerns are not assessed in this paper such as (1) carcinoma potential triggered by oncogenes in the hcDNA, (2) immune response triggered by AAV genome and AAV capsids,66,67 and (3) rAAV genome integration into the host genome.68,50,51,52 The broad safety aspect of rAAV products can be better understood by continuously assessing the host genome and host responses to rAAV treatments in research labs and AAV gene therapy industry in long-term pre-clinical, non-clinical, and clinical studies.
In summary, this study showed that the majority of packaged hcDNA is intergenic and intronic, with no complete ORF. RNA-seq analysis found no difference of human-DNA-derived transcripts in livers of mice treated with or without rAAV. Vector genome and RNA-seq analysis of in vivo samples suggest a relatively low expression risk of residual hcDNA. Low-level transcription of some plasmid DNA impurities was detected in mice dosed with high levels of AAVhu37-EGFP at 2.0E14 GC/kg. Although there are currently no reports of clinical manifestations of safety risks specific to DNA impurities in AAV gene therapy products, with therapeutic dose levels exceeding 1.0E14 GC per kg, it is prudent to reduce these impurities through improvements in vector design and process development. Continuous long-term follow-up of patients treated with rAAV gene therapy vector is also essential to assess the overall safety of these products.
Materials and methods
Recombinant adeno-associated virus generation and purification
rAAV generation was performed by triple transfection of HEK293 cells as described in previous studies.69 The AAV vectors were produced in 250L bioreactors and subjected to a full-scale downstream purification process, with high purity and low DNA and protein impurities. Briefly, three plasmids—the transgene plasmid containing EGFP expression cassette (1,610 bps) and ITRs, the AAV helper plasmid containing AAV2 Rep and Hu37 Cap, and the Adenovirus helper plasmid (Figure 1A)—were transfected into HEK293 cells. Four days post-transfection, an endonuclease, Benzonase, was added to the bioreactors to digest non-encapsidated hcDNA. The cultured cells were harvested 5 days post-transfection. Harvested materials were loaded onto a chromatography column packed with Poros AAVX affinity resin. The elution pool was further purified by anion exchange chromatography using a CIM-Q monolith from BIA separations. The elution peak containing the full capsids was concentrated to a target concentration and diafiltered into the final formulation buffer to yield the final rAAV vector material.
Quantification of vector genomes and residual DNA by (droplet) digital PCR
For quantification of EGFP and DNA impurities, ddPCR and dPCR were performed as previously described70,71,72,73 using primer probes specific for EGFP sequence as follows: (1) forward primer, 5′- GCA TCG ACT TCA AGG AGG AC -3’; (2) reverse primer, 5′- TCA CCT TGA TGC CGT TCT TC- 3’; and (3) probe, 5’/56 FAM/AGC CAC AAC/ZEN/GTC TAT ATC ATG GCC G/3IABkFQ-3’. Residual plasmid DNA was quantified by multiplex dPCR using primers and probes specifically for the KanR, AAV2 Rep, and Hu37 Cap. The primer probe sequences for KanR are as follows: (1) forward primer, 5′- GGC CTG TTG AAC AAG TCT GG-3’; (2) reverse primer, 5′- TCC GAC TCG TCC AAC ATC AA-3’; and (3) probe, 56-ROXN/AC GAC TGA ATC CGG TGA GAA TGG C/3IAbRQSp/-3’. The primer probe sequences for AAV2 Rep are as follows: (1) forward primer, 5′-TCA CCA AGC AGG AAG TCA AAG-3’; (2) Reverse primer, 5′-CCC GTT TGG GCT CAC TTA TAT C-3’; and (3) probe, 5’/56-TAMN/AC GTG GTT GAG GTG GAG CAT GAA T/3IAbRQSp-3’. The primer probe sequences for Hu37 Cap are as follows: (1) forward primer, 5′-GCA GCG ACT CAT CAA CAA -3’; (2) reverse primer, 5′- GTC TTG GTG CCT TCA TTC -3’; and (3) probe, 5’/56 FAM/AAC ATC CAG/ZEN/GTC AAG GA GGT CAC G/3IABkFQ-3’. Residual HEK293 hcDNA was quantified using TaqMan real-time PCR assay targeting 18s rRNA (Thermo Fisher, Cat: 4333760T) or Alu repeats ([1] forward primer, 5′- GAG GCG GGC GGA TCA -3’; [2] reverse primer, 5′- CCC GGC TAA TTT TTG TAT TTT TAG TAG -3’; [3] probe, 5’/56-FAM/CAG CCT GGC/ZEN/CAA CAT GGT GAA ACC) with SgrI-digested genome HEK293 DNA standard.
PacBio AAV sequencing
To prepare viral DNA for long-read sequencing, 100 μL of rAAV material (at 1.7E13 GC/mL) was digested in proteinase K reaction at 55°C for 30 min followed by deactivation of proteinase K at 80°C for 15 min while gradually dropping the temperature to 4°C to allow single-stranded DNA to anneal to double-stranded DNA. After this, DNA was purified by Monarch DNA and PCR Purification Kit (NEB, Cat: #T1130) following the manufacturer-recommended protocol starting with a mixture of one volume of sample buffer and three volumes of binding buffer.
DNA samples were quantified using a Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). Sample purity and integrity was checked using Nanodrop and Agilent TapeStation. Following quality control, PacBio SMRTbell amplicon libraries for PacBio Sequel were constructed using the SMRTbell prep kit 3.0 (PacBio, Menlo Park, CA, USA) using the manufacturer-recommended protocol. Primer annealing, polymerase binding, and complex cleanup were performed using the Sequel binding kit 3.1 & cleanup beads (PacBio) and loaded onto the PacBio Sequel IIe. Sequencing was performed on the PacBio Sequel IIe SMRT cell with AAV mode enabled. Sequel IIe sequencing generated subread files, as well as HiFi reads via on-board circular consensus sequencing (CCS) analysis. Demultiplexing was performed using PacBio Liam. Sequencing data was deposited in GEO under accession code: GSE280789.
PacBio sequencing data analysis
CCS reads with length >200 bp and average quality score of 99% were used for mapping and alignment. Sequences were uploaded to DNAnexus, an Amazon-based cloud server for bioinformatic analysis. Reference sequences of the expression cassette and residual plasmid DNA were also uploaded to DNAnexus.
To determine the size of residual plasmid DNA and residual hcDNA, CCS reads were first mapped to the expression cassette (excluding ITR sequences) by MiniMap2, and unmapped reads were extracted by Samtools and mapped to the plasmid backbone sequence including AAV2 Rep, Hu37 Cap, KanR, and the rest of the Ad genes from helper plasmids. The unmapped reads were then mapped to human genomic DNA sequences. The reads mapped to different impurities and human genome DNA sequences were remapped to the 5′ITR sequences via MiniMap2 to quantify the percentage of reads containing ITRs.
To identify the genomic loci for the impurity hcDNA, the PacBio reads mapped to the human genome (hg38) were annotated using Homer annotatePeaks.pl. The results were then further analyzed using JMP.
MEME suite version 5.5 was used for motif enrichment discovery and motif analysis of hcDNA. PacBio CCS reads mapped to hcDNA were further trimmed with Cutadapt to remove any sequences with the PacBio primer sequences: 5′-AAA AAA AAA AAA AAA AAA TTA ACG GAG GAG GAG GA-3′ and PacBio Blunt Adapter sequences: 5′-ATC TCT CTC TTT TCC TCC TCC TCC GTT GTT GTT GTT GAG AGA GAT-3′ as an input file. The AAV2 REP binding motifs were determined based on a previous publication.39 The Atoh1 binding motif was extracted from the JASPAR database as the motif input. SEA (simple enrichment analysis) was used to identify the occurrence of REP motifs on our input sequences. De novo and known motif enrichment analysis was conducted with Homer.
In vivo sample preparation
C57BL/6 adult male mice between 7 and 8 weeks old were used for this study. Control mice were untreated, whereas the treated mice were dosed with 2.0E14 GC/kg of rAAVhu37-EGFP via intravenous tail vein injection. Following dosing, mice were monitored daily and weighed every other day. No adverse effects were observed through the course of the study. Two and three weeks after dosing, mice were perfused, and the liver was isolated. Small 3–4 mm punches from the median lobe were collected and placed in separate cryotubes, snap frozen in liquid nitrogen, and stored at −80°C. The remaining median lobe was placed in a cryotube, snap frozen in liquid nitrogen, and stored at −80°C. All animal experiments were performed in accordance with national guidelines and regulations.
EGFP quantification with the ELISA kit
A single 3–4 mm liver punch was placed in cell extraction buffer PTR containing cell extraction enhancer solution from the GFP ELISA Kit (Abcam, Cat: ab171581). Halt protease inhibitor cocktail (Thermo Fisher Scientific Cat:78430) was also added to the solution. The tissue was homogenized in a chilled Bullet Blender Gold (Next Advance), left on ice for 20 min, centrifuged at 18,000 g for 20 min, and the supernatant was collected. The protein concentration of the sample was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Cat: 23225). The samples were then diluted to a stock solution of 12 picogram protein/μL. Each well received 50 μL of the protein stock concentration, was incubated with the capture and detector antibodies, and developed according to the manufacturer’s instructions (Abcam Cat: ab171581). A single absorbance value at 450 nm was recorded upon addition of the Stop Solution, and a standard curve was used to calculate the total EGFP per milligram of protein for each sample.
RNA sequencing
Total RNA was extracted from fresh frozen tissue samples using the Qiagen RNeasy Plus Universal mini kit following manufacturer instructions (Qiagen, Cat: 74881). RNA samples were quantified using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA), and RNA integrity was checked with the Agilent Tapestation 4200 (Agilent Technologies, Palo Alto, CA, USA). ERCC Ex-fold RNA reagent (Cat: 4456739) from Thermo Fisher Scientific was added to normalized total RNA prior to library preparation following the manufacturer protocol. Samples were treated with TURBO DNase (Thermo Fisher Scientific, Cat: AM2238) to remove DNA impurities. The next steps included performing rRNA depletion using the QIAseq FastSelect−rRNA HMR kit (Qiagen, Cat: 334386), which was conducted following the manufacturer’s protocol. RNA-seq libraries were constructed with the NEBNext Ultra II RNA Library Preparation Kit for Illumina (NEB, Cat: E7770) by following the manufacturer’s recommendations. Briefly, enriched RNAs were fragmented for 15 min at 94°C. First-strand and second-strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3′ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment with limited cycle PCR. Sequencing libraries were validated using the Agilent Tapestation 4200 (Agilent Technologies, Palo Alto, CA, USA) and quantified using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA).
Sequencing libraries were multiplexed and clustered onto two lanes of a Flowcell. After clustering, the Flowcell was loaded onto the Illumina HiSeq instrument (Illumina, San Diego, CA, USA) according to manufacturer’s instructions. Samples were sequenced using a 2 × 150 bp paired-end (PE) configuration. Image analysis and base calling were conducted by the Illumina Control Software. Raw sequence data (.bcl files) were converted into fastq files and de-multiplexed using Illumina bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification. Each sample was sequenced to approximately 100 million reads. The sequencing data were deposited in GEO under accession code: GSE280788.
Bioinformatic analysis of RNA-seq results
Total pair-end FASTQ reads for each sample were uploaded to DNAnexus. All analysis was performed in DNAnexus unless otherwise stated. Adapters and short reads (<50 bps) and reads with more than 4 “Ns” were removed using Flexbar FASTQ Read Trimmer. The quality and size of the reads were checked with FastQC. The processed reads were mapped to the mouse genome (mm10) using Bowtie2 FASTQ Read Mapping. Unmapped reads were extracted using Samtools and Bedtools and were then mapped to the EGFP reference genome using the Bowtie2 FASTQ Read Mapper. The same Samtools and Bedtools commands were then used to extract reads not mapped to either mouse genome mm10 or the EGFP reference genome. The FASTQ files for the unmapped reads were then mapped to pAAV2/8 and pAdF6 using the Bowtie2 FASTQ Read Mapper.
Read count for the plasmid DNA and hcDNA
Library preparation for RNA sequencing had a PCR amplification step; therefore, duplicate reads could be generated. To ensure proper read count, duplicate reads were removed in the mapped reads using Picard MarkDuplicates Mapping Deduplicator. The depth of the reads in specific genomic sites was then counted using the Samtool depth-d. The counted depth was downloaded from DNAnexus, and the average depth across each gene was analyzed. Reads that did not map to the mouse genome or the plasmids were extracted and mapped to the human genome (hg38). Mapped read files were transferred to UseGalaxy, where each alignment file was individually annotated and counted using the FeatureCounts program. The FeatureCounts output was then used for differential expression analysis with DESeq2. Additionally, individual genes were annotated using the annotateMyIDs tool.
Quantification of RNA levels by QIAcuity dPCR
The concentration of RNA from each sample was measured using a NanoDrop spectrophotometer. For cDNA synthesis, 2.5 ng of RNA was used with the Superscript IV VILO Reverse Transcriptase Kit (Thermo Fisher Scientific Cat:11766500), following the manufacturer’s protocol with minor modifications. Briefly, various volumes of the 2.5 ng RNA samples were treated with ezDNase mix to remove residual DNA. Subsequently, 4 μL of Superscript IV VILO master mix and 6 μL of water were added to the reaction mixture to perform cDNA synthesis. Simultaneously, no RT controls were prepared using 4 μL of “no RT” master mix added to the same samples.
After RT, the reaction mixture was diluted 1:3 with nuclease-free water. The samples were either used directly for dPCR or aliquoted and stored at −80°C. Digital PCR was performed according to the manufacturer’s protocol. Different dilutions were used for different targets due to differences in abundance and quantification limits of dPCR: for plasmid DNA, no additional dilution was performed before amplification to increase sensitivity; for EGFP, dilutions of 1:10,000 and 1:100,000 were performed to achieve measurable concentrations. The absolute concentration of each target gene was calculated based on the dilution level and the detected concentration. The “no RT” control showed no amplification of any DNA impurities targets, indicating no DNA contamination.
Data availability
All sequencing data generated during this study are available in the Gene Expression Omnibus (GEO) database, under accession codes: GSE280788 and GSE280789.
Acknowledgments
The authors thank the following members in Ultragenyx’s Pharmaceutical Development, Analytical Development team for assay support: Lauren Panolini for generating the genome titer data for rAAVhu37-EGFP using ddPCR and Dennis G Chen for generating the residual DNA qPCR and dPCR measurement targeting residual plasmid and hcDNA for rAAVhu37-EGFP drug substance. The authors are grateful to Ultragenyx’s Pharmaceutical Development, Pilot Plant team for their support in generating the AAVhu37-EGFP samples used in this study; to Ultragenyx’s Gene Therapy Research team members: Irena Ignatova for helping to design the mouse study, Mayra Senices and Karima Zahiri for dosing the mice and tissue collection, Mattewhew Fuller for his scientific review and comments to improve the manuscript; to Ultragenyx’s Bioinformatics Team: Julien Couthouis and Sean Daugherty for their support on DNAnexus. The authors are grateful for all the scientific and communication input provided by Lorelei Stoica and James Warren.
Author contributions
W.Z., H.-I.J., and X.L. contributed to the conceptualization of the study. H.-I.J. and P.W. performed the data curation and formal analysis. H.-I.J. conducted the investigation, developed the methodology, and wrote the original draft of the manuscript. W.Z. provided supervision. W.Z., H.-I.J., P.W., and X.L. contributed to the review and editing of the manuscript.
Declaration of interests
This work was supported by Ultragenyx Pharmaceutical Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2025.101503.
Supplemental information
References
- 1.Bower J.J., Song L., Bastola P., Hirsch M.L. Harnessing the Natural Biology of Adeno-Associated Virus to Enhance the Efficacy of Cancer Gene Therapy. Viruses. 2021;13 doi: 10.3390/v13071205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mijanovic O., Brankovic A., Borovjagin A., Butnaru D.V., Bezrukov E.A., Sukhanov R.B., Shpichka A., Timashev P., Ulasov I. Battling Neurodegenerative Diseases with Adeno-Associated Virus-Based Approaches. Viruses. 2020;12 doi: 10.3390/v12040460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J.H., Gessler D.J., Zhan W., Gallagher T.L., Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct. Targeted Ther. 2024;9:78. doi: 10.1038/s41392-024-01780-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Approved Cellular and Gene Therapy Products. 2024. https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products
- 5.Clinical Trials - Gene Therapy. https://clinicaltrials.gov/search?term=%22gene%20therapy%22
- 6.Meng J.S., He Y., Yang H.B., Zhou L.P., Wang S.Y., Feng X.L., Yahya Al-Shargi O., Yu X.M., Zhu L.Q., Ling C.Q. Melittin analog p5RHH enhances recombinant adeno-associated virus transduction efficiency. J. Integr. Med. 2024;22:72–82. doi: 10.1016/j.joim.2024.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Ling C., Yu C., Wang C., Yang M., Yang H., Yang K., He Y., Shen Y., Tang S., Yu X., et al. rAAV capsid mutants eliminate leaky expression from DNA donor template for homologous recombination. Nucleic Acids Res. 2024;52:6518–6531. doi: 10.1093/nar/gkae401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atchison R.W., Casto B.C., Hammon W.M. Adenovirus-Associated Defective Virus Particles. Science. 1965;149:754–756. doi: 10.1126/science.149.3685.754. [DOI] [PubMed] [Google Scholar]
- 9.Samulski R.J., Srivastava A., Berns K.I., Muzyczka N. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell. 1983;33:135–143. doi: 10.1016/0092-8674(83)90342-2. [DOI] [PubMed] [Google Scholar]
- 10.Lusby E., Fife K.H., Berns K.I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. J. Virol. 1980;34:402–409. doi: 10.1128/JVI.34.2.402-409.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hermonat P.L., Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA. 1984;81:6466–6470. doi: 10.1073/pnas.81.20.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grieger J.C., Samulski R.J. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol. 2012;507:229–254. doi: 10.1016/B978-0-12-386509-0.00012-0. [DOI] [PubMed] [Google Scholar]
- 13.Ayuso E., Mingozzi F., Bosch F. Production, purification and characterization of adeno-associated vectors. Curr. Gene Ther. 2010;10:423–436. doi: 10.2174/156652310793797685. [DOI] [PubMed] [Google Scholar]
- 14.Urabe M., Ding C., Kotin R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 15.Wu Z., Asokan A., Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol. Ther. 2006;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Wright J.F. Transient transfection methods for clinical adeno-associated viral vector production. Hum. Gene Ther. 2009;20:698–706. doi: 10.1089/hum.2009.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vandenberghe L.H., Wilson J.M. AAV as an immunogen. Curr. Gene Ther. 2007;7:325–333. doi: 10.2174/156652307782151416. [DOI] [PubMed] [Google Scholar]
- 18.Brimble M.A., Winston S.M., Davidoff A.M. Stowaways in the cargo: Contaminating nucleic acids in rAAV preparations for gene therapy. Mol. Ther. 2023;31:2826–2838. doi: 10.1016/j.ymthe.2023.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright J.F. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines. 2014;2:80–97. doi: 10.3390/biomedicines2010080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava A., Mallela K.M.G., Deorkar N., Brophy G. Manufacturing Challenges and Rational Formulation Development for AAV Viral Vectors. J. Pharmaceut. Sci. 2021;110:2609–2624. doi: 10.1016/j.xphs.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 21.Keiser M.S., Ranum P.T., Yrigollen C.M., Carrell E.M., Smith G.R., Muehlmatt A.L., Chen Y.H., Stein J.M., Wolf R.L., Radaelli E., et al. Toxicity after AAV delivery of RNAi expression constructs into nonhuman primate brain. Nat. Med. 2021;27:1982–1989. doi: 10.1038/s41591-021-01522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buck T.M., Wijnholds J. Recombinant Adeno-Associated Viral Vectors (rAAV)-Vector Elements in Ocular Gene Therapy Clinical Trials and Transgene Expression and Bioactivity Assays. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21124197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L., Vasan L., Kartono B., Clifford K., Attarpour A., Sharma R., Mandrozos M., Kim A., Zhao W., Belotserkovsky A., et al. Advances in Recombinant Adeno-Associated Virus Vectors for Neurodegenerative Diseases. Biomedicines. 2023;11 doi: 10.3390/biomedicines11102725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guidance for industry . Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) Guidance for Industry. FDA; 2020. [Google Scholar]
- 25.Knezevic I., Stacey G., Petricciani J., WHO Study Group on cell substrates WHO Study Group on cell substrates for production of biologicals, Geneva, Switzerland, 11-12 June 2007. Biologicals. 2008;36:203–211. doi: 10.1016/j.biologicals.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Gimpel A.L., Katsikis G., Sha S., Maloney A.J., Hong M.S., Nguyen T.N.T., Wolfrum J., Springs S.L., Sinskey A.J., Manalis S.R., et al. Analytical methods for process and product characterization of recombinant adeno-associated virus-based gene therapies. Mol. Ther. Methods Clin. Dev. 2021;20:740–754. doi: 10.1016/j.omtm.2021.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen J.M., Debelak D.J., Reynolds T.C., Miller A.D. Identification and elimination of replication-competent adeno-associated virus (AAV) that can arise by nonhomologous recombination during AAV vector production. J. Virol. 1997;71:6816–6822. doi: 10.1128/JVI.71.9.6816-6822.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X.S., Khuntirat B., Qing K., Ponnazhagan S., Kube D.M., Zhou S., Dwarki V.J., Srivastava A. Characterization of wild-type adeno-associated virus type 2-like particles generated during recombinant viral vector production and strategies for their elimination. J. Virol. 1998;72:5472–5480. doi: 10.1128/JVI.72.7.5472-5480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tai P.W.L., Xie J., Fong K., Seetin M., Heiner C., Su Q., Weiand M., Wilmot D., Zapp M.L., Gao G. Adeno-associated Virus Genome Population Sequencing Achieves Full Vector Genome Resolution and Reveals Human-Vector Chimeras. Mol. Ther. Methods Clin. Dev. 2018;9:130–141. doi: 10.1016/j.omtm.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Earley L.F., Conatser L.M., Lue V.M., Dobbins A.L., Li C., Hirsch M.L., Samulski R.J. Adeno-Associated Virus Serotype-Specific Inverted Terminal Repeat Sequence Role in Vector Transgene Expression. Hum. Gene Ther. 2020;31:151–162. doi: 10.1089/hum.2019.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor N.K., Guggenbiller M.J., Mistry P.P., King O.D., Harper S.Q. A self-complementary AAV proviral plasmid that reduces cross-packaging and ITR promoter activity in AAV vector preparations. Mol. Ther. Methods Clin. Dev. 2024;32 doi: 10.1016/j.omtm.2024.101295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorovits B., Marshall J.C., Smith J., Whiteley L.O., Neubert H. Bioanalysis of adeno-associated virus gene therapy therapeutics: regulatory expectations. Bioanalysis. 2019;11:2011–2024. doi: 10.4155/bio-2019-0135. [DOI] [PubMed] [Google Scholar]
- 33.Srivastava A. Replication of the adeno-associated virus DNA termini in vitro. Intervirology. 1987;27:138–147. doi: 10.1159/000149732. [DOI] [PubMed] [Google Scholar]
- 34.Xiao X., Xiao W., Li J., Samulski R.J. A novel 165-base-pair terminal repeat sequence is the sole cis requirement for the adeno-associated virus life cycle. J. Virol. 1997;71:941–948. doi: 10.1128/JVI.71.2.941-948.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amaral P., Carbonell-Sala S., De La Vega F.M., Faial T., Frankish A., Gingeras T., Guigo R., Harrow J.L., Hatzigeorgiou A.G., Johnson R., et al. The status of the human gene catalogue. Nature. 2023;622:41–47. doi: 10.1038/s41586-023-06490-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snyder E.E., Stormo G.D. Identification of protein coding regions in genomic DNA. J. Mol. Biol. 1995;248:1–18. doi: 10.1006/jmbi.1995.0198. [DOI] [PubMed] [Google Scholar]
- 37.McCarty D.M., Pereira D.J., Zolotukhin I., Zhou X., Ryan J.H., Muzyczka N. Identification of linear DNA sequences that specifically bind the adeno-associated virus Rep protein. J. Virol. 1994;68:4988–4997. doi: 10.1128/JVI.68.8.4988-4997.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Z.X., Chen J.Z., Yue Y.B., Zhang J.Q., Li Z.H., Feng D.M., Ruan Z.C., Tian L., Xue J.L., Wang Q.J., Jia W. A 16-bp RBE element mediated Rep-dependent site-specific integration in AAVS1 transgenic mice for expression of hFIX. Gene Ther. 2009;16:589–595. doi: 10.1038/gt.2009.9. [DOI] [PubMed] [Google Scholar]
- 39.Goncalves M.A. Adeno-associated virus: from defective virus to effective vector. Virol. J. 2005;2:43. doi: 10.1186/1743-422X-2-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nash K., Chen W., Salganik M., Muzyczka N. Identification of cellular proteins that interact with the adeno-associated virus rep protein. J. Virol. 2009;83:454–469. doi: 10.1128/JVI.01939-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batchu R.B., Shammas M.A., Wang J.Y., Munshi N.C. Dual level inhibition of E2F-1 activity by adeno-associated virus Rep78. J. Biol. Chem. 2001;276:24315–24322. doi: 10.1074/jbc.M008154200. [DOI] [PubMed] [Google Scholar]
- 42.Batchu R.B., Shammas M.A., Wang J.Y., Munshi N.C. Interaction of adeno-associated virus Rep78 with p53: implications in growth inhibition. Cancer Res. 1999;59:3592–3595. [PubMed] [Google Scholar]
- 43.Hermonat P.L., Santin A.D., Batchu R.B. The adeno-associated virus Rep78 major regulatory/transformation suppressor protein binds cellular Sp1 in vitro and evidence of a biological effect. Cancer Res. 1996;56:5299–5304. [PubMed] [Google Scholar]
- 44.Corish P., Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12:1035–1040. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- 45.Ballenghien M., Faivre N., Galtier N. Patterns of cross-contamination in a multispecies population genomic project: detection, quantification, impact, and solutions. BMC Biol. 2017;15:25. doi: 10.1186/s12915-017-0366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jo S.Y., Kim E., Kim S. Impact of mouse contamination in genomic profiling of patient-derived models and best practice for robust analysis. Genome Biol. 2019;20:231. doi: 10.1186/s13059-019-1849-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allay J.A., Sleep S., Long S., Tillman D.M., Clark R., Carney G., Fagone P., McIntosh J.H., Nienhuis A.W., Davidoff A.M., et al. Good manufacturing practice production of self-complementary serotype 8 adeno-associated viral vector for a hemophilia B clinical trial. Hum. Gene Ther. 2011;22:595–604. doi: 10.1089/hum.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hauck B., Murphy S.L., Smith P.H., Qu G., Liu X., Zelenaia O., Mingozzi F., Sommer J.M., High K.A., Wright J.F. Undetectable transcription of cap in a clinical AAV vector: implications for preformed capsid in immune responses. Mol. Ther. 2009;17:144–152. doi: 10.1038/mt.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penaud-Budloo M., Le Guiner C., Nowrouzi A., Toromanoff A., Chérel Y., Chenuaud P., Schmidt M., von Kalle C., Rolling F., Moullier P., Snyder R.O. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 2008;82:7875–7885. doi: 10.1128/JVI.00649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hauck B., Zhao W., High K., Xiao W. Intracellular viral processing, not single-stranded DNA accumulation, is crucial for recombinant adeno-associated virus transduction. J. Virol. 2004;78:13678–13686. doi: 10.1128/JVI.78.24.13678-13686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schultz B.R., Chamberlain J.S. Recombinant adeno-associated virus transduction and integration. Mol. Ther. 2008;16:1189–1199. doi: 10.1038/mt.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakai H., Storm T.A., Kay M.A. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver in vivo. J. Virol. 2000;74:9451–9463. doi: 10.1128/jvi.74.20.9451-9463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sachs A. The role of poly(A) in the translation and stability of mRNA. Curr. Opin. Cell Biol. 1990;2:1092–1098. doi: 10.1016/0955-0674(90)90161-7. [DOI] [PubMed] [Google Scholar]
- 54.Bernstein P., Peltz S.W., Ross J. The poly(A)-poly(A)-binding protein complex is a major determinant of mRNA stability in vitro. Mol. Cell Biol. 1989;9:659–670. doi: 10.1128/mcb.9.2.659-670.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seto E., Shi Y., Shenk T. YY1 is an initiator sequence-binding protein that directs and activates transcription in vitro. Nature. 1991;354:241–245. doi: 10.1038/354241a0. [DOI] [PubMed] [Google Scholar]
- 56.O'Shea-Greenfield A., Smale S.T. Roles of TATA and initiator elements in determining the start site location and direction of RNA polymerase II transcription. J. Biol. Chem. 1992;267:6450. [PubMed] [Google Scholar]
- 57.Chang L.S., Shi Y., Shenk T. Adeno-associated virus P5 promoter contains an adenovirus E1A-inducible element and a binding site for the major late transcription factor. J. Virol. 1989;63:3479–3488. doi: 10.1128/JVI.63.8.3479-3488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Austen M., Lüscher B., Lüscher-Firzlaff J.M. Characterization of the transcriptional regulator YY1. The bipartite transactivation domain is independent of interaction with the TATA box-binding protein, transcription factor IIB, TAFII55, or cAMP-responsive element-binding protein (CPB)-binding protein. J. Biol. Chem. 1997;272:1709–1717. doi: 10.1074/jbc.272.3.1709. [DOI] [PubMed] [Google Scholar]
- 59.Brimble M.A., Cheng P.H., Winston S.M., Reeves I.L., Souquette A., Spence Y., Zhou J., Wang Y.D., Morton C.L., Valentine M., et al. Preventing packaging of translatable P5-associated DNA contaminants in recombinant AAV vector preps. Mol. Ther. Methods Clin. Dev. 2022;24:280–291. doi: 10.1016/j.omtm.2022.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piovesan A., Caracausi M., Antonaros F., Pelleri M.C., Vitale L. GeneBase 1.1: a tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database. 2016;2016 doi: 10.1093/database/baw153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zaytsev K., Fedorov A., Korotkov E. Classification of Promoter Sequences from Human Genome. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms241612561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacAlpine D.M., Almouzni G. Chromatin and DNA replication. Cold Spring Harbor Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ehrenhofer-Murray A.E. Chromatin dynamics at DNA replication, transcription and repair. Eur. J. Biochem. 2004;271:2335–2349. doi: 10.1111/j.1432-1033.2004.04162.x. [DOI] [PubMed] [Google Scholar]
- 64.Hayashi M.T., Masukata H. Regulation of DNA replication by chromatin structures: accessibility and recruitment. Chromosoma (Berl.) 2011;120:39–46. doi: 10.1007/s00412-010-0287-4. [DOI] [PubMed] [Google Scholar]
- 65.Jain N.K., Ogden P.J., Church G.M. Comprehensive mutagenesis maps the effect of all single-codon mutations in the AAV2 rep gene on AAV production. eLife. 2024;12 doi: 10.7554/eLife.87730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Faust S.M., Bell P., Cutler B.J., Ashley S.N., Zhu Y., Rabinowitz J.E., Wilson J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Investig. 2013;123:2994–3001. doi: 10.1172/JCI68205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greig J.A., Martins K.M., Breton C., Lamontagne R.J., Zhu Y., He Z., White J., Zhu J.X., Chichester J.A., Zheng Q., et al. Integrated vector genomes may contribute to long-term expression in primate liver after AAV administration. Nat. Biotechnol. 2024;42:1232–1242. doi: 10.1038/s41587-023-01974-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen D.P., Wei J.Y., Warren J.C., Huang C. Tuning mobile phase properties to improve empty and full particle separation in adeno-associated virus productions by anion exchange chromatography. Biotechnol. J. 2024;19 doi: 10.1002/biot.202300063. [DOI] [PubMed] [Google Scholar]
- 70.Quan P.L., Sauzade M., Brouzes E. dPCR: A Technology Review. Sensors (Basel) 2018;18 doi: 10.3390/s18041271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cao L., Cui X., Hu J., Li Z., Choi J.R., Yang Q., Lin M., Ying Hui L., Xu F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2017;90:459–474. doi: 10.1016/j.bios.2016.09.082. [DOI] [PubMed] [Google Scholar]
- 72.Suo T., Liu X., Feng J., Guo M., Hu W., Guo D., Ullah H., Yang Y., Zhang Q., Wang X., et al. ddPCR: a more accurate tool for SARS-CoV-2 detection in low viral load specimens. Emerg. Microb. Infect. 2020;9:1259–1268. doi: 10.1080/22221751.2020.1772678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prantner A., Maar D. Genome concentration, characterization, and integrity analysis of recombinant adeno-associated viral vectors using droplet digital PCR. PLoS One. 2023;18 doi: 10.1371/journal.pone.0280242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated during this study are available in the Gene Expression Omnibus (GEO) database, under accession codes: GSE280788 and GSE280789.