

All cells depend on nutrients acquired from the extracellular environment to support the biochemical processes required for cell growth. Although unicellular organisms such as bacteria and yeast have developed strategies to survive nutrient limitation, the growth of these organisms is constrained by nutrient availability. In contrast, mammalian cells exist in a nutrient-rich environment where cell growth is not limited by substrate availability, but rather by the levels of extrinsic growth factors (1, 2). Because of the limits imposed by growth factor control, cellular proliferation is restricted even under nutrient conditions that are highly favorable for unlimited cell growth. However, it is becoming apparent that nutrient availability also plays an important role in controlling mammalian cell growth. An example of this form of growth control is found in the work of Angelini et al. (3) reported in this issue that describes how dendritic cells control the proliferation of T cells by regulating the availability of the amino acid cysteine.
Previous studies in lymphocytes identified cysteine as rate limiting for cell growth and proliferation after antigen receptor engagement (4). Cysteine and cystine, the free thiol- and disulfide-bonded forms of the amino acid, respectively, exist in an equilibrium defined by redox conditions. In the oxidizing environment of the extracellular space, the cysteine equivalents exist primarily in the form of cystine (5). In contrast, a reducing environment is maintained intracellularly, and thus the cysteine within the cell exists predominantly in the free thiol form (6). Cystine and cysteine are not taken up by the same amino acid transporter but require the expression of distinct transport systems (7). Despite the limited amount of cysteine in the plasma, lymphocytes do not express the cystine transporter and cannot take up cystine (7, 8). Furthermore, although cysteine is not generally considered an essential amino acid, lymphocytes have been reported to lack cystathionase, an essential enzyme for the production of cysteine from methionine, and thus cysteine is an essential amino acid for lymphocytes (9).
During the specific immune response, T cells require two signals for proliferation. The primary stimulus arises from antigen engagement of the T cell receptor and the second signal, referred to as costimulation, results from interactions between surface molecules on antigen-presenting cells such as macrophages or dendritic cells and surface molecules on T cells (Fig. 1). If the T cell receptor is engaged and costimulatory signals are not provided, minimal proliferation is observed and cells may be tolerized to the antigen rather than activated. It has been shown that interactions between macrophages and T cells result in cysteine production by macrophages and that this cysteine production is required for T cell proliferation (4, 10). Unlike lymphocytes, macrophages possess a transporter for cystine, referred to as system x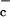 . When activated, for example by (bacterial) lipopolysaccharide (LPS) or tumor necrosis factor (TNF)-α, macrophages serve as a sort of cysteine pump, taking up cystine and releasing cysteine into the microenvironment where it is available to nearby T cells (4). Cystine uptake by macrophages is inhibited by glutamate, and glutamate has been shown to compromise T cell activation (8). Inclusion of 2-mercaptoethanol (2-ME) in the culture medium, which will reduce extracellular cystine to cysteine, or serial additions of cysteine can overcome glutamate inhibition and even obviate the need for antigen-presenting cells when costimulatory signals are provided by other means (10, 11).
. When activated, for example by (bacterial) lipopolysaccharide (LPS) or tumor necrosis factor (TNF)-α, macrophages serve as a sort of cysteine pump, taking up cystine and releasing cysteine into the microenvironment where it is available to nearby T cells (4). Cystine uptake by macrophages is inhibited by glutamate, and glutamate has been shown to compromise T cell activation (8). Inclusion of 2-mercaptoethanol (2-ME) in the culture medium, which will reduce extracellular cystine to cysteine, or serial additions of cysteine can overcome glutamate inhibition and even obviate the need for antigen-presenting cells when costimulatory signals are provided by other means (10, 11).
Figure 1.

Antigen-presenting cells increase extracellular cysteine levels thereby allowing the proliferation of activated T cells. Most of the cysteine (cys-SH) equivalents in the extracellular space exist as the oxidized form, cystine (cys-S-S-cys). T cells are unable to take up cystine and depend on antigen-presenting cells such as dendritic cells and macrophages to supply them with cysteine. Antigen-presenting cells take up cystine from the extracellular space by using the system x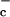 transporter, convert cystine to cysteine intracellularly, and release cysteine into the extracellular space where it is available to T cells. In addition, dendritic cells secrete thioredoxin, which can convert extracellular cystine to cysteine.
transporter, convert cystine to cysteine intracellularly, and release cysteine into the extracellular space where it is available to T cells. In addition, dendritic cells secrete thioredoxin, which can convert extracellular cystine to cysteine.
The current report by Angelini et al. (3) shows that dendritic cells, the antigen-presenting cells thought to be involved in initial antigen presentation to T cells during an immune response, also take up cystine and release cysteine and that this function is required for T cell activation. The ability to increase extracellular cysteine levels is influenced by the maturation state of the dendritic cell. Dendritic cell maturation caused by LPS or TNF-α stimulation or as a result of CD40 crosslinking increases cysteine production. This process is also influenced by crosstalk between dendritic cells and activated T cells. Maximal cysteine production by dendritic cells requires antigen-specific interactions between dendritic cells and T cells. The importance of antigen-specific contacts between T cells and dendritic cells was confirmed by the observation that dendritic cell secretion of thioredoxin is stimulated only when these interactions occur. As cysteine is rapidly oxidized to cystine in the extracellular space, thioredoxin secretion provides a mechanism for reducing cystine and maintaining cysteine in the extracellular space during an immune response (Fig. 1). In addition, the redox state of other critical immune regulators will be affected by secreted thioredoxin. Based on the results of Angelini et al., thioredoxin may be a biologically relevant functional equivalent to the 2-ME added to culture media by scientific investigators to stimulate lymphocyte growth.
So what are the ramifications of the findings of Angelini et al. (3) for immune responses taking place in vivo? The ability of T cells to kill invaders must be in delicate balance with tolerance toward self-antigens. Signals received from antigen-presenting cells are thought to play an important role in regulating whether T cells are fully activated or tolerized in response to a given antigen. It is possible that the control of cysteine availability by macrophages and dendritic cells plays a role in this determination. Unless antigen-presenting cells make cysteine available, antigen-stimulated T cells will be unable to increase protein synthesis, proliferate, or secrete immunoregulatory cytokines. These findings may help clarify how one type of antiinflammatory agent controls T cell-mediated autoimmune disease. Sulfasalazine is commonly used in the treatment of inflammatory bowel disease, a T cell-mediated autoimmune disease. This drug has recently been found to specifically inhibit the x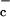 cystine transporter (12). It is generally believed that sulfasalazine is effective in inflammatory bowel disease because it inhibits the inflammatory response that results from the cellular destruction mediated by autoreactive T cells. However, if sulfasalazine inhibits the production of cysteine by macrophages and dendritic cells, it will also inhibit the growth and proliferation of the autoreactive T cells that initiate the inflammatory response. Sulfasalazine is also given as a treatment for rheumatoid arthritis, another T cell-mediated autoimmune disease. Elevated levels of thioredoxin have been found in the synovial fluid of patients with rheumatoid arthritis, suggesting a connection to the regulation of cysteine availability in this disease as well (13).
cystine transporter (12). It is generally believed that sulfasalazine is effective in inflammatory bowel disease because it inhibits the inflammatory response that results from the cellular destruction mediated by autoreactive T cells. However, if sulfasalazine inhibits the production of cysteine by macrophages and dendritic cells, it will also inhibit the growth and proliferation of the autoreactive T cells that initiate the inflammatory response. Sulfasalazine is also given as a treatment for rheumatoid arthritis, another T cell-mediated autoimmune disease. Elevated levels of thioredoxin have been found in the synovial fluid of patients with rheumatoid arthritis, suggesting a connection to the regulation of cysteine availability in this disease as well (13).
Cysteine is not the only amino acid whose levels are regulated by antigen-presenting cells as a means to control T cell activation. Extracellular tryptophan levels are also adjusted by macrophages, and this process has been reported to play an important role in the generation of T cell tolerance (Fig. 2) (14). Macrophages that differentiate under the influence of macrophage colony-stimulating factor (M-CSF) are able to suppress T cell activation. M-CSF-matured macrophages, but not fresh monocytes, were found to produce the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO). By chemically inhibiting IDO or by supplying excess tryptophan, the suppression of T cell proliferation by M-CSF-matured macrophages was prevented. The tryptophan-degrading activity of the macrophages is not constitutive, but rather regulated by interactions with activated T cells. Thus, crosstalk between antigen-presenting cells and T cells is also important for controlling tryptophan catabolism. IFN-γ treatment of macrophages can also result in IDO induction and tryptophan degradation. Macrophages themselves escape growth limitation caused by tryptophan catabolism in part by up-regulating the level of tryptophan tRNA synthetase (15). The ability of macrophages to degrade tryptophan and thus suppress T cell activation has been reported to play a critical role in maternal fetal tolerance. When IDO was inhibited, T cell-mediated maternal rejection of allogeneic concepti was observed in mice (16, 17). This system provides an example of the in vivo relevance of immunosuppression by tryptophan catabolism.
Figure 2.

Antigen-presenting cells limit T cell proliferation by degrading tryptophan. Macrophages that have been cultivated in the presence of M-CSF can suppress the proliferation of activated T cells. This inhibition results from the tryptophan-degrading activity of the intracellular enzyme IDO. T cells are thus limited for tryptophan, and proliferation is blocked in the G1 stage of the cell cycle.
The use of amino acid restriction to control the proliferation of lymphoid cells has been validated clinically through the use of l-asparaginase for the treatment of acute lymphoblastic leukemia (18). The high cure rate of lymphocytic leukemia in children treated with l-asparaginase in combination with other chemotherapeutics provides a compelling demonstration of how effective and relatively selective reduced amino acid levels can be in regulating cellular proliferation. Despite the success of l-asparaginase, there has been a growing belief that leukemic cells were uniquely susceptible to amino acid limitation and that this mechanism of cellular growth control did not pertain to other cells types. As demonstrated in this issue by Angelini et al. (3), the regulation of local levels of amino acids does have a regulatory role in the immune response. Whether this form of growth control is unique to immune cells or is used by other cellular systems remains to be determined.
Acknowledgments
This work was supported by the Abramson Family Cancer Research Institute and by grants from the National Cancer Institute. A.L.E. is supported by the Helen Hay Whitney Foundation.
Footnotes
See companion article on page 1491.
References
- 1.Raff M C. Nature (London) 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 2.Raff M C. Cell. 1996;86:173–175. doi: 10.1016/s0092-8674(00)80087-2. [DOI] [PubMed] [Google Scholar]
- 3.Angelini G, Gardella S, Ardy M, Ciriolo M R, Di Trapani G, Clarke F, Sitia R, Rubartelli A. Proc Natl Acad Sci USA. 2002;99:1491–1496. doi: 10.1073/pnas.022630299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gmunder H, Eck H P, Benninghoff B, Roth S, Droge W. Cell Immunol. 1990;129:32–46. doi: 10.1016/0008-8749(90)90184-s. [DOI] [PubMed] [Google Scholar]
- 5.Mansoor M A, Svardal A M, Ueland P M. Anal Biochem. 1992;200:218–229. doi: 10.1016/0003-2697(92)90456-h. [DOI] [PubMed] [Google Scholar]
- 6.Arner E S, Holmgren A. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 7.Bannai S. Biochim Biophys Acta. 1984;779:289–306. doi: 10.1016/0304-4157(84)90014-5. [DOI] [PubMed] [Google Scholar]
- 8.Gmunder H, Eck H P, Droge W. Eur J Biochem. 1991;201:113–117. doi: 10.1111/j.1432-1033.1991.tb16263.x. [DOI] [PubMed] [Google Scholar]
- 9.Eagle H, Washington C, Friedman S M. Proc Natl Acad Sci USA. 1966;56:156–163. doi: 10.1073/pnas.56.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwata S, Hori T, Sato N, Ueda-Taniguchi Y, Yamabe T, Nakamura H, Masutani H, Yodoi J. J Immunol. 1994;152:5633–5642. [PubMed] [Google Scholar]
- 11.Ishii T, Hishinuma I, Bannai S, Sugita Y. J Cell Physiol. 1981;107:283–293. doi: 10.1002/jcp.1041070215. [DOI] [PubMed] [Google Scholar]
- 12.Gout P W, Buckley A R, Simms C R, Bruchovsky N. Leukemia. 2001;15:1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 13.Maurice M M, Nakamura H, van der Voort E A, van Vliet A I, Staal F J, Tak P P, Breedveld F C, Verweij C L. J Immunol. 1997;158:1458–1465. [PubMed] [Google Scholar]
- 14.Munn D H, Shafizadeh E, Attwood J T, Bondarev I, Pashine A, Mellor A L. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellor A L, Munn D H. Immunol Today. 1999;20:469–473. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 16.Mellor A L, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn D H. Nat Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 17.Munn D H, Zhou M, Attwood J T, Bondarev I, Conway S J, Marshall B, Brown C, Mellor A L. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 18.Crowther D. Nature (London) 1971;229:168–171. doi: 10.1038/229168a0. [DOI] [PubMed] [Google Scholar]