

Abstract
In Saccharomyces cerevisiae, the heat shock transcription factor (HSF) is thought to be a homotypic trimer that is bound to the promoters of heat shock protein (HSP) genes at both normal and heat shock temperatures. Exposure to heat shock greatly and rapidly induces HSF transcriptional activity without further increasing DNA-binding affinity. It is believed that HSF is under negative regulation at normal growth temperatures, but the detailed mechanism by which HSF is activated is still not clear. We report the analysis of mutations in a conserved arginine (residue 274) at the C-terminal end of the DNA-binding domain (DBD). Two mutations significantly increase both basal activity of HSF at normal temperatures and induced activity on heat shock. We demonstrate by coimmunoprecipitation experiments that the mutations reduce the association between the DNA-binding domain/oligomerization domain and the transcription activation domains. Our studies suggest that the DNA-binding domain of HSF can interact with activation domains directly, and this interaction is important for the repression of HSF activity under normal growth conditions. Destabilizing this interaction by heat or by mutations results in HSF transcriptional activation. We propose that Arg-274 is critical for intramolecular repression of HSF activity in normally growing cells.
Eukaryotic cells respond to heat shock and other physiological stresses by dramatically increasing the expression of a specific set of genes encoding the heat shock proteins (HSPs; for reviews, see refs. 1 and 2). The promoters of these genes possess a cis-acting heat shock element (HSE) that consists of multiple inverted, tandem repeats of the consensus sequence AGAAn, where n can be any nucleotide (3, 4). The heat shock transcription factor (HSF), which binds specifically to HSEs and regulates the heat shock response, has been identified in yeast (5–7), Drosophila (8), human (9), and many other organisms (1).
In the budding yeast Saccharomyces cerevisiae, ScHSF is required for both growth at normal temperatures and viability on stress. In fact, ScHSF is bound to DNA at all temperatures, and heat shock converts the DNA-bound transcriptionally weak ScHSF into a highly active factor (10, 11). Although on heat shock the ScHSF shows some increased binding to secondary, low-affinity HSEs, the overall effect on DNA binding is insignificant and thus not likely to be an important regulatory step in ScHSF activation (12, 13). Other mechanisms, such as phosphorylation and (or) dephosphorylation, that modify the activity of the transcription activation domains, are more likely to be involved in the activation process (6, 11, 14, 15). Point mutation and deletion studies show that most mutations in ScHSF cause an increase in the constitutive transcriptional activity (10, 16–18), indicating that ScHSF is probably under negative regulatory control in the absence of stress.
The functional domains of HSFs have been well characterized. All HSFs from different species contain two conserved domains: a winged helix–turn–helix DNA binding domain (DBD) (19) and a trimerization domain consisting of an α-helical coiled-coil motif (20, 21). Apart from these two domains, homology comparisons reveal no other significant conservation in the activation domains of HSFs. By deletion mapping and analysis of chimeric proteins containing HSF sequences fused to heterologous DNA-binding domains, three ScHSF activation domains were identified. The activation domain II (AII, residue 410–648) is essential for growth during heat shock but not for normal conditions. The roles of activation domain I (AI, residue 1–172) and activation domain III (AIII, residue 589–833) were not clear when they were first identified by Nieto-Sotelo et al. (10), because AIII is dispensable for viability at both normal and stress-temperatures. Later studies showed that the AI domain probably functions in non-shocked cells to allow viability and is responsible for transient activity, whereas the AIII domain is probably responsible for sustained activities at increased temperature (11). All three activation domains are constitutively active when fused to yAP-1 DNA-binding domain (10). Mechanisms of repression at normal growth temperatures remain unknown; however, some evidence suggests that activation domains are restrained by the DNA-binding domain and trimerization domain (10). Deletions of certain residues in the DNA-binding domain increase the overall activity of HSF, indicating that DNA-binding domain may play an important regulatory role in addition to HSE recognition. However, it is not clear that the DNA-binding domain represses ScHSF activity through direct intramolecular interactions with activation domains, or perhaps indirectly with the association of another member of heat shock response circuitry, such as HSP70 or HSP90.
In this paper, we report experiments dealing with a key arginine residue (R274) and its role in HSF regulation. Arg-274 is a highly conserved residue located at the C-terminal end of the HSF DNA-binding domain (1). We show that two mutations significantly increase both constitutive activity of HSF at normal temperatures and induced activity on heat shock. In addition, coimmunoprecipitation experiments reveal that the DBD will bind to all three activation domains and that mutations in Arg-274 reduce this association. Our studies suggest that DNA-binding domain of HSF can interact with activation domains directly, and this interaction is important for the repression of HSF activity under normal conditions.
Materials and Methods
Yeast Strains.
The haploid yeast strain, pYES-URA, in which chromosomal loci of HSF were disrupted, was used. This hsf− strain contains an episomal copy of HSF whose transcription is controlled by the GAL1,10 promoter (5).
Plasmid Constructions and Protein Expression.
The shuttle vector used in this work, pYES2-FHC, is a 2μ-based high copy plasmid, with FLAG epitope and six-His tag at C-terminal, and with either TRP1 or URA3 selection (22). Expression vector pGEX-2t was used to make glutathione S-transferase (GST)-fusion recombinant proteins. The fusion proteins were expressed in Escherichia coli strain BL21(DE3), and purified with GST affinity resin (Stratagene). The GST-fusion proteins were eluted with 100 mM reduced glutathione in 50 mM Tris (pH 8.0), 0.5 M NaCl, and 1% Triton X-100. Expression vector pCAL-n was used to make calmodulin-binding peptide (CBP)-fusion proteins (Stratagene), and the recombinant proteins were purified by calmodulin affinity resin (Stratagene).
Mutagenesis.
Desired mutant oligos were annealed to single-stranded dU DNA. The mixture was heated to 70°C, and slowly cooled to room temperature. Second strand was synthesized by adding T4 DNA polymerase and T4 DNA ligase. Sequencing of the plasmids confirmed that only the desired mutations were introduced. All of the mutant HSFs were cloned into pYES2-FHC/TRP plasmid with BamHI and XhoI sites, and were selected by SG-TRP + URA + 5-FOA plate.
Transformations.
Yeast cells were transformed by using ALKALI-CATION Yeast Transformation Kit from Bio 101. The protocols are based on lithium acetate procedure.
Yeast Cell Extract Preparation and Western Blotting.
Yeast whole cell extracts were prepared according to Schultz et al. (23). Western blotting analysis was performed with about 100 μg of whole cell extract and anti-FLAG M2 antibody (Eastman Kodak). The immune complexes were visualized by AP (alkaline phosphatase)-based chromogenic method.
Total RNA Isolation and Primer Extension Analysis.
Total RNA from control (25°C) and heat-shocked cells (40°C) was isolated by using RNAqueous Total RNA Isolation Kit from Ambion (Austin, TX). Five nanograms of 5′ end-labeled oligonucleotides (described below) were hybridized to 5≈8 μg of total RNA in TE (10 mM Tris·Cl/l mM EDTA, pH 7.4) containing 200 mM KCl. Total final volume was adjusted to 12 μl. The oligonucleotides were annealed by heating the reaction mixture at 65°C for 30 min, followed by gradual cooling to room temperature. Then avian myeloblastosis virus (AMV) reverse transcriptase was added to the annealing product, and the reaction mixture was incubated at 42°C for 1 h. The samples were electrophoresed on denaturing 8 M urea-6% polyacrylamide gels. Dried gels were exposed to Molecular Dynamics PhosphorImager plates, which were then scanned on the Molecular Dynamics ImageQuant.
The oligonucleotides used for primer extension analysis are as follows: SSA4, 5′-AGCATCGTTCGTCACTTCTGGATCA-3′; and actin, 5′-CCGGCTTTACACATACCAGA-3′.
In Vitro Cross-Linking Assay.
To investigate the oligomerization of wild-type and mutant DBDs, 1 μg of GST-tagged DBDs was cross-linked by addition of 2 mM 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) to D buffer (25 mM Hepes, pH 7.4/100 mM KCl/1 mM EDTA/0.2% Triton X-100) and incubation for 30 min at 25°C. The reactions were quenched by the addition of lysine to 20 mM, and analyzed by 8% SDS/PAGE. Western blotting was then performed by using anti-GST antibody (Amersham Pharmacia), and molecular weight markers (Bio-Rad) were used to determine the approximate sizes of the complexes.
Mobility-Shift Assay.
Mobility-shift assay was performed by using the HSE-containing fragment as the follows: wild-type HSE, GCGCGCCTCGAATGTTCGCGAAAAGA; mutant HSE, GCGCGCCTCGAATGGGCGCGAAAAGA. GAA repeats are shown in bold, and mutated nucleotides in mutant HSE are underlined. Binding assay buffer contains 250 μg/ml BSA, 100 μg/ml poly(dI⋅dC), and 4% Ficoll. γ-32P-labeled HSEs or unlabeled HSE were incubated with 1 μg of wild-type or mutant DBDs at room temperature for 20 min, and the reaction complexes were analyzed by 5% acrylamide/0.5× TBE gel.
Coimmunoprecipitation Assay.
To investigate the interactions between DBDs and activation domains, CBP-tagged activation domains were labeled with [γ-32P]ATP by protein kinase A (PKA). GST-tagged wild-type and mutant DBDs were immobilized by anti-GST antibody-coupled protein G Sepharose (Amersham Pharmacia), then incubated with [γ-32P]AI, -AII, or -AIII (≈50,000 cpm/μl) for 30 min at 25°C. After 3 washes with D buffer containing 0.5 M NaCl and 0.5% Triton X-100, the precipitates were resuspended in SDS loading buffer and analyzed by 12% SDS/PAGE and autoradiography.
Results
Effect of a DNA-Binding Domain Mutation on Heat Shock Factor Activity.
By mutagenesis of ScHSF, we isolated a temperature-sensitive mutant that is viable only at room temperature and 30°C, but not at 37°C (Table 1). Sequence analysis showed that a mutation in the DBD of HSF had occurred. The original sequence Arg-Gln-Lys (amino acid 274–276) was altered to Ser-Gln-Gln-His-Ala. This mutant was designated dbd-mut.
Table 1.
Phenotypes of yeast cells expressing mutant ScHSFs
| Wild type | dbd-mut | R274 K | R274 G | R274 E | |
|---|---|---|---|---|---|
| 25° | +++ | ++ | +++ | +++ | — |
| 30° | +++ | + | +++ | +++ | — |
| 37° | +++ | — | +++ | ++ | — |
To compare the transcriptional activities of wild-type and dbd-mut HSFs, the levels of specific HSP gene transcription were analyzed. Transcriptional activities of wild-type and dbd-mut HSFs were determined by directly measuring the level of mRNA transcription from SSA4, the major HSP70-inducible gene, by primer extension. Actin mRNA was monitored simultaneously as an internal control. Surprisingly, although the transcription of SSA4 in wild-type cells was not detectable under non-shock conditions, in the dbd-mut cells, SSA4 was actively transcribed at control temperature (25°C; Fig. 1A, lane 4). In addition, the activity of this mutant ScHSF can be further induced by heat shock: the transcription level of SSA4 in dbd-mut cells is about three times higher than that of wild-type cells at heat shock temperature (40°C; determined by PhosphorImager analysis, Fig. 1, compare lanes 2 and 5). During recovery from heat shock, wild-type cells recover significantly quicker than the dbd-mut cells (data not shown). Whereas in wild-type cells SSA4 is not detectable 60 min after heat shock, in dbd-mut cells, SSA4 transcription returns to pre-heat shock levels.
Figure 1.

Transcriptional activities of mutant HSFs. (A) Transcriptional activities of wild-type and mutant ScHSFs are measured by levels of HSP70 mRNA by using primer extension. Primer extension products are indicated (HSP70, 309–313 nucleotides; actin, 198–200 nucleotides). Lanes 1–3, wild-type HSF; lanes 4–6, dbd-mut HSF; lanes 7–9, R274K HSF; lanes 10–12, R274G HSF. NS, non-shock, cells grown at 25°C. HS, heat shock, cells subjected to heat shock at 40°C for 30 min. R, recovery, cells put back to 25°C for 1 h after heat shock to allow recovery. (B) Western blotting analysis of wild-type and mutant HSF proteins. Yeast cell extracts were subjected to SDS/PAGE and transferred to nitrocellulose filter. The protein blot was probed with anti-FLAG antibody (Sigma) fused to the C terminus of HSFs.
The drastically increased HSP70 expression might account for the lethality of this mutant at heat shock temperatures. It has been reported before that elevated level of Hsp70 can impede cell growth and division (24). With activation of the HS response, normal protein synthesis is suspended, and protective HSPs are produced that rescue the heat-denatured proteins. But cell survival is achieved at the expenses of normal growth and development, presumably because HS response takes precedence over other developmental events and alters the normal programs of gene activity (24). The hyperactive dbd-mut HSF could potentially disrupt the expression of essential housekeeping genes and cause cell death.
Arginine 274 Is Important for Negative Regulation of Heat Shock Factor's Transcription Activity.
Among the amino acids altered in dbd-mut ScHSF, arginine 274 is a highly conserved residue present in HSFs from many different species (1). The crystal structure of Drosophila melanogaster HSF's DNA-binding domain shows that Arg-274 is located in a flexible linker region at the very end of the C terminus. Arg-274 is about 20 residues downstream of the β4 strand, and probably not directly involved in DNA binding. To analyze the specific functional significance of Arg-274, three site-directed mutants were created: R274K, R274G, and R274E, in which the Arg was changed to positively, neutrally, and negatively charged residues, respectively. To study the properties of these mutant HSFs, we used a yeast strain carrying a disrupted chromosomal HSF locus. HSF is an essential gene in yeast, so the strain with a disrupted chromosomal HSF was kept viable with a HSF gene carried on a URA3-containing vector. This strain was transformed with mutant ScHSFs cloned in a TRP1-based plasmid. Selection on 5-fluoroorotic acid allowed the loss of the URA3-containing wild-type ScHSF plasmid, leaving cells with only mutant ScHSF on the TRP1 vector.
R274E mutant is completely lethal; all of the cells died after plating onto 5-FOA plates. R274G mutant grows normally at 30°C but has a slightly slower growth rate at 37°C, whereas R274K mutant exhibits wild-type phenotype (Table 1). Primer extension analysis of HSP70 shows that the transcriptional activity of the R274K mutant HSF is properly regulated by temperature (Fig. 1A, lanes 7–9). On the other hand, although R274G mutant does not show growth defects, the activity of R274G-HSF is higher than that of wild type both at non-shock (25°C) and heat shock (39°C) temperatures (Fig. 1A, lanes 9–12). The abnormal behavior of dbd-mut and R274G HSF demonstrate that Arg-274 is very important for the negative regulation of HSF activity. Without the positive charge at this position, the activity of ScHSF is significantly elevated at control temperatures.
The viability of the mutant strains correlates with the severity of the mutations. The presence of a negatively charged residue probably destabilizes the conformation of HSF significantly, causing cell death. On the other hand, R274G is a relatively mild alteration. PhosphorImager quantitation shows that SSA4 mRNA level in heat-shocked R274G mutant cells is about half as much as in heat-shocked dbd-mut cells. Yeast cells may be able to tolerate moderately high levels of HSP70 expression, resulting in a slightly slower growth rate.
To rule out the possibility that different levels of HSP70 transcription are due to different levels of HSF expression, Western blotting analysis was used to detect the expression of HSF in wild and mutant cells. Equal levels of HSF were expressed in all these cells (Fig. 1B), indicating that it is the Arginine 274 mutation that activates HSF under non-shock temperatures.
The transcriptional activities of mutant HSFs were also inspected by β-galactosidase enzyme activity assays by using a yeast strain transformed with (HSE)4/LacZ reporter gene (data not shown). The results confirmed that dbd-mut and R274G HSFs have higher activity both at non-shock and heat shock temperatures, whereas the R274K HSF mutant was very similar to wild type.
Mutation of Arginine 274 Causes a Reduction in DNA Binding Affinity.
One possible explanation for the hyperactivity of dbd-mut and R274G HSF is increased DNA-binding affinity. Although several earlier studies have shown that the level of DNA binding does not correlate with transcription levels before or after heat shock (2), it was important to measure the DNA binding affinities of mutant HSFs. Wild-type and mutant HSFs from residues 167 to 424 (including the DNA-binding domain and the helical oligomerization domain; Fig. 4A) were fused to a GST-tag, and the recombinant proteins were purified by GST-affinity chromatography (Stratagene). As determined by gel mobility shift assays, all four recombinant DBDs bind to 32P-labeled-HSE sequence (Fig. 2, lanes 1, 6, 11, and 16), but none of the examined recombinant DBDs bound to 32P-labeled-mutant HSE, in which the nGAAnnTTCn consensus was mutated to nGGAnnGGCn (lanes 5, 10, 15, and 20). In addition, binding to [32P]HSE is effectively competed away by unlabeled HSE (Fig. 2, lanes 4, 9, 14, and 19), demonstrating the specificity of wild-type HSE recognition by recombinant DBDs. Interestingly, we observed that the wild-type DBD has the highest affinity for HSE (lanes 1–3); R274G also exhibits high DNA-binding affinity, but is slightly lower than wild type. Both the dbd-mut and R274E show a significant decrease in HSE binding affinity. This result clearly demonstrates that the derepression observed by the mutant HSFs is not due to an increased DNA binding activity. Furthermore, from the structural information available on the Drosophila HSF and Kluyveromyces lactis HSF, R274 does not interact with the DNA directly (1, 25); therefore, the decreased DNA-binding affinity must result from secondary effects of R274 mutations.
Figure 4.

Binding affinities between DNA-binding domain and activation domains correlate with HSF transcriptional activities. (A) Schematic diagram of domain organization of S. cerevisiae HSF. Amino acid endpoints for each region, as well as their proposed functions, are indicated. (AI, AII, and AIII represent three constitutive activation domains. DBD, DNA-binding domain; IRL, isoleusine repeat (oligomerization domain). This diagram is adapted from the work of Nieto-Sotelo et al. (10). (B) Coimmunoprecipitation of activation domains with wild-type and mutant DBDs. Recombinant wild-type and mutant DBDs were immobilized by anti-GST antibody-coupled protein G Sepharose. Recombinant activation domain I (AI), activation domain II (AII), and activation domain III (AIII) were labeled with [γ-32P]ATP and PKA catalytic subunit. 32P-labeled AI (lanes 1–5), AII (lanes 6–10), and AIII (lanes 11–15) were precipitated by immobilized DBDs. Equal amounts of anti-GST antibody-coupled protein G Sepharose without DBD were used as control to assess nonspecific interactions (lanes 1, 6, and 11). (C) Western blotting analysis of immobilized wild-type and mutant DBDs used in coimmunoprecipitation.
Figure 2.

DNA-binding assay. Gel mobility shift assay of wild-type and mutant DBDs to labeled HSE is shown. A mutant HSE with nGAAnnTTCn repeats changed to nGAAnnGGCn was also used to ensure binding specificity (lanes 5, 10, 15, and 20). Increasing amounts of unlabeled HSE were added for competition assay. The numbers shown at the bottom of each lane are the ratios of unlabeled HSE to [32P]HSE. Major DBD–HSE complexes are shown by the lower arrow. Mutant DBDs also form higher-order complexes (upper arrow). Note that microgram quantities of HSFs are used in these experiments because the recombinant proteins bind to the HSE less well than the native HSF.
In addition to the major DNA–protein complex, mutant DBDs and HSE also form higher molecular weight complexes (Fig. 2, lanes 6, 11, and 16, upper arrow). This high molecular weight complex is not seen in wild-type DBD–DNA complex. To examine the molecular interaction causing the higher molecular weight complex, wild-type and mutant DBDs were cross-linked in the absence of DNA and analyzed by SDS/PAGE. Western blotting analysis of cross-linking products of DBDs shows that wild-type DBD exists in an equilibrium of monomers, dimers, and trimers (Fig. 3, lane 1), whereas dbd-mut and R274E exist predominantly as trimers and hexamers (lanes 2 and 4, based on their apparent molecular weight). Monomer forms of these two DBDs are almost absent. The oligomerization state of R274G is in between, with all four forms present (lane 3). This result provides additional evidence that Arg-274 mutation causes the overall conformational changes of DBD and oligomerization domain, and this conformational change leads to a greater tendency to form high molecular weight oligomers. It is likely that mutation of the arginine perturbs the organization of oligomerization domain, increasing the tendency of HSF to form either high molecular weight oligomers or an alternate trimer conformation, perhaps reflecting an “activated” state.
Figure 3.
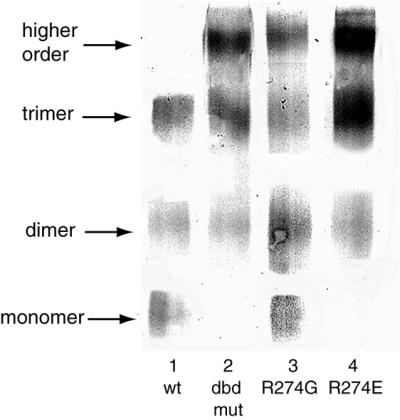
Oligomerization state of wild-type and mutant HSFs. One microgram of recombinant wild-type and mutant DBDs was cross-linked by DSS and analyzed by Western blotting analysis. Wild-type DBD exited as an equilibrium of monomer, dimer, and trimer (lane 1). Mutant DBDs also formed hexamer (lanes 2, 3, and 4).
DNA-Binding Domains of Mutant ScHSFs Have Reduced Affinity to the Activation Domains.
Previous deletion studies of ScHSF have revealed the presence of certain functional domains (Fig. 4A; ref. 10), including a DNA-binding domain, a helical oligomerization domain, and three transcription activation domains, AI, AII, and AIII, whose activities are repressed at normal temperatures. It has been reported that DNA-binding domain may play an important regulatory role of repressing transcriptional activation in addition to its obvious role in mediating DNA binding (16, 22). Many mutations in DNA binding domain increase HSF's activity without enhancing the DNA binding properties. For example, a M232V mutation of ScHSF has a constitutively activating phenotype (16).
The hyperactivity exhibited by dbd-mut and R274G ScHSFs suggests that, under normal conditions, activation domains of HSF are probably masked by the DNA-binding and oligomerization domains. In case of these two mutated ScHSFs, loss of the positively charged Arg causes the derepression of the activation domains. To study the physical interactions between activation domains and DNA-binding domain, we separately cloned the AI (residue 1–172), AII (residue 410–648), AIII (residue 589–833), and DNA-binding and oligomerization domains (DBD, residue 167–424) into different prokaryotic expression vectors, and examined the interactions between them by immunoprecipitation.
Wild-type and mutant DBDs were fused to GST-tag and purified as described before, and then immobilized by anti-GST antibody coupled to protein G Sepharose. AI, AII, and AIII were fused to CBP-tag, and the recombinant AI, AII, and AIII were purified through calmodulin-coupled chromatography. Purified activation domains were labeled in vitro with PKA and [γ-32P]ATP at a PKA target sequence within the CBP. The AI, AII, and AIII were incubated with immobilized wild-type and mutant DBDs, and the precipitated products were analyzed by SDS/PAGE. Fig. 4B shows that HSF DBD binds to all three activation domains. The wild-type DBD showed the highest level of association with the activation domains compared with the mutant DBDs (Fig. 4B, lanes 2, 7, and 12). The binding properties of the mutant DBDs to the activation domains correspond well with their phenotypes and transcription activities. The DBD derived from the lethal mutant R274E demonstrated the lowest level of binding to all three activation domains, whereas R274G is only moderately less able to bind AII and AIII than is wild-type DBD. Fig. 4C shows that the same amounts of DBDs were used in these experiments. These coimmunoprecipitation results confirm the idea that DBD interacts with the activation domains, and these interactions may be important for the repression of HSF activity at non-shock temperatures.
Considering the fact that three Arg-274 mutations lowered the binding affinities for all three activation domains, this residue may be important for maintaining the overall structure of the DBD and oligomerization domain, but probably does not mediate specific interactions between the DBD and the activation domains. Circular dichroism studies of DBD show that Arg-274 is within a flexible linker region without any ordered secondary structure (26). It has been suggested that this linker region might be important to adjust the 3-fold symmetric HSF trimers to the 180° symmetry between inverted HSE repeats. So it is likely that loss of the positively charged Arg-274 causes some overall conformational changes of DBD and oligomerization domain, resulting in the decreased association with the activation domains.
dbd-mut HSF Is Activated at a Slower Rate than Wild-Type HSF and May Be Analogous to Partially Activated HSF.
The kinetics of ScHSF activation suggest that dbd-mut ScHSF may already be in the active conformation mimicking activated ScHSF. Fig. 5 shows the transcription of SSA4 when wild-type and dbd-mut cells were heat shocked for 0 min, 5 min, 10 min, 15 min, 30 min, and 3 h, respectively. The induction of SSA4 transcription is very rapid in wild-type cells. The amount of SSA4 mRNA is greatly increased after 5 min of heat shock, and reaches peak level only after 15 min of heat shock. Beyond 15 min, SSA4 transcription is maintained at a stable level (Fig. 5A, lanes 1–4). Long periods of heat shock (3 h, lane 5) decrease SSA4 transcription, probably because expression of HSP70 negatively regulates HS response (27). In contrast, 5 min of heat shock did not induce further activation of dbd-mut HSF at all (lane 7). mRNA level of SSA4 remains unchanged. This observation suggests that dbd-mut mimics an activated HSF, so short-term heat shock will not further increase its transcriptional activity. Longer periods of heat shock (15 and 30 min, lanes 8 and 9) probably thermally destabilize the structure of dbd-mut HSF, causing further exposure of activation domains, and in turn the further activation of mutant HSF. Three hours of heat shock cause massive death of mutant cells, which can be observed from the obviously decreased transcription of actin (lane 10). The kinetics of transcriptional activation were further examined by PhosphorImaging, and the results were plotted in Fig. 5B. The ratios of SSA4 transcription to actin are displayed as a function of time. Clearly, the wild type responds rapidly and peaks at 15 min. The dbd-mut does not show the initial increase in transcription; rather, a delayed stimulation is observed at 10 min.
Figure 5.

Kinetics of wild-type and dbd-mut HSF activation. (A) Primer extension analysis of the activation kinetics of wild-type and dbd-mut HSFs. Cells were harvested after 0, 5, 15, 30, or 180 min of heat shock, and HSP70 mRNA levels were measured by primer extension. Actin mRNA was measured at the same time as internal control. dbd-mut HSF (lanes 6–10) is activated slower than wild type (lanes 1–5). (B) Kinetics plot of wild-type and dbd-mut HSFs activation. The units are arbitrary. The ratio of HSP70 mRNA transcription to actin mRNA transcription at 5 min of heat shock in wild-type cells was set to 1, and all of the other quantitative data (obtained from PhosphorImager plate) were normalized accordingly. The x axis is drawn logarithmically. The transcriptional activities of dbd-mut HSF after 30 min of heat shock were not shown, because progressive cell death caused inaccurate measurement.
Discussion
The activation domains of ScHSF are repressed in non-stressed cells. It is known that sequences present in the DBD, oligomerization domain, and a short conserved element, CE2, are required to maintain repression (1). Within the DNA-binding domain of ScHSF, mutation of residue M232 to V results in constitutive activation at non-stress temperatures (16). In this report, we show that mutation of R274, located in a solvent-exposed position of the linker region between DNA-binding domain and oligomerization domain, leads to derepression at normal growth temperature. These conserved residues must play an important role in the maintenance of the inactive state of the transcription factor.
Although not directly involved in DNA binding, Arginine 274 is highly conserved and found in K. lactis HSF, Schizosaccharomyces pombe HSF, Arabidopsis thaliana HSF, Drosophila HSF, mouse HSF1/2, and human HSF1/2 (1). Our results demonstrate that a conservative change of Arg-274 to lysine has little effect on ScHSF transcriptional activity. In contrast, a mutation that reverses that charge of R274 to glutamic acid results in lethality. These observations suggest that the positive charge at this position is important for normal activity of ScHSF. The DBDs from dbd-mut and R274G mutants showed reduced affinities for all three activation domains and tended to form higher order structures. Overall, these observations suggest that Arg-274 is critical for maintaining the inactive structure of ScHSF, and mutations in this residue result in significant conformational changes. The portion of the DBD where Arg-274 is located has no ordered secondary structure as determined by circular dichroism (26). Therefore, despite the fact that Arg-274 is a highly conserved residue, it may be in a flexible portion of ScHSF. The value of a flexible linker between the DBD and oligomerization domain may be to assist DNA binding. If ScHSF binds to DNA as a trimer, three DBDs would be rotated 120° relative to each another. The HSEs however, are palindromic repeats with 180° symmetry. The linker region perhaps maintains the correct positioning of the DBD and oligomerization domain, so that a trimer can fit into HSEs (28). Without a positive charge at residue 274, as seen with the mutants described in this paper, the correct positioning of DBD and oligomerization domain might be disturbed, and the mutants cannot bind to DNA with high affinity.
The structural integrity of the DBD and the oligomerization domain seems to be very important for their negative regulatory functions. Recent work from Hardy et al. (28) showed that deletions of conserved residues within an α-helical bulge in DBD increases the overall activity of ScHSF, but when these conserved residues were mutated, there was little effect observed. This result demonstrates that the presence of a bulge in DBD, rather than specific residues, is critical for the negative regulation. Similarly, structural disturbance resulted from Arg-274 mutation can account for the reduced affinities for activation domains and increased HSF transcriptional activities. Unfortunately, without the structural information of the full-length HSF trimer complexed with DNA, we do not know at this time the detailed function of this arginine.
It has been shown that human HSF1 purified from HeLa cell extract can be activated by heat shock (29), and recombinant mouse HSF1 can acquire DNA-binding on in vitro heat shock (30). All these studies suggest that HSF may be capable of sensing heat shock directly. Perhaps heat shock induces structural changes of HSF, especially within DBD and oligomerization domain, destabilizing the interactions between DBD and activation domains and thus increasing transcriptional activity. The fact that the transcriptional activities of wild-type, dbd-mut, and R274G ScHSFs correspond very well with their respective affinities for activation domains supports this idea. dbd-mut and R274G HSFs are activated even at 25°C. We suggest that this is the result of a less tight association between the DBD and activation domains. In other words, structural changes caused by the Arg-274 mutation might mimic the structural changes induced by heat shock.
We also demonstrated here that increased transcriptional activity of mutant ScHSFs is not due to increased DNA-binding affinity. A substantial amount of data already indicates that the degree of DNA-binding does not correlate with transcriptional activities of ScHSF. For example, SSA4, the major HSP70 in S. cerevisiae, is expressed at extremely low level during steady-state growth at 23°C, but its expression is greatly enhanced on upshift to 39°C (31). However, no further increase in DNA-binding at the SSA4 promoter was observed (32). In our case, although dbd-mut showed reduced DNA-binding affinity compared with wild type, its transcriptional activity, both at non-shock and heat shock temperatures, was actually higher. This observation further confirms that, at least for strong HSEs, like those present in the SSA4 promoter, increased DNA-binding is not the major regulatory process involving in HSF activation. In fact, conformational changes and destabilization of certain intramolecular interactions can even overcome the reduced DNA-binding affinity and activate HSF. For some promoters with weaker, secondary HSEs, such as the HSP 82 promoter, heat shock does lead to an increased occupancy of HSF, and the increased DNA-binding contributes to the overall activation process. However, it is not likely that the moderate increase in DNA-binding fully accounts for the very significant increase in transcriptional activity. Therefore, derepression of the activation domains is probably the most important mechanism involved in the activation process. Our results support the hypothesis that DNA-binding domain of ScHSF has an important role in negative regulation, and that the activation process involves a dynamic dissociation of DBD/oligomerization domain from the activation domains. While this manuscript was being evaluated, related studies employing alanine-scanning mutations in the DBD of ScHSF also demonstrate the interactions between the DBD and the activation domains (33). Structural studies of full-length HSF will provide insights that should demonstrate how mutation of Arg-274 causes conformational changes of the DBD and oligomerization domain.
Acknowledgments
We thank Dr. Xiangdong Fang for suggestions on protein purification. We thank Dr. Duncan Odom for helpful comments on the manuscript. This work was supported by National Institutes of Health Grant GM47381 (to C.S.P.).
Abbreviations
- HSP
heat shock protein
- HSE
heat shock element
- HSF
shock transcription factor
- DBD
DNA-binding domain
- GST
glutathione S-transferase
- CBP
calmodulin-binding peptide
- PKA
protein kinase A
References
- 1.Wu C. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- 2.Mager W H, Dekruijff A J J. Microbiol Rev. 1995;59:506–531. doi: 10.1128/mr.59.3.506-531.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amin J, Ananthan J, Voellmy R. Mol Cell Biol. 1988;8:3761–3769. doi: 10.1128/mcb.8.9.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perisic O, Xiao H, Lis J T. Cell. 1989;59:797–806. doi: 10.1016/0092-8674(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 5.Wiederrecht G, Seto D, Parker C S. Cell. 1988;54:841–853. doi: 10.1016/s0092-8674(88)91197-x. [DOI] [PubMed] [Google Scholar]
- 6.Sorger P K, Pelham H R B. Cell. 1988;54:855–864. doi: 10.1016/s0092-8674(88)91219-6. [DOI] [PubMed] [Google Scholar]
- 7.Jakobsen B K, Pelham H R B. EMBO J. 1991;10:369–375. doi: 10.1002/j.1460-2075.1991.tb07958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clos J, Westwood J T, Becker P B, Wilson S, Lambert K, Wu C. Cell. 1990;63:1085–1097. doi: 10.1016/0092-8674(90)90511-c. [DOI] [PubMed] [Google Scholar]
- 9.Kingston R E, Schuetz T J, Larin Z. Mol Cell Biol. 1987;7:1530–1534. doi: 10.1128/mcb.7.4.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieto-Sotelo J, Wiederrecht G, Okuda A, Parker C S. Cell. 1990;62:807–817. doi: 10.1016/0092-8674(90)90124-w. [DOI] [PubMed] [Google Scholar]
- 11.Sorger P K. Cell. 1990;62:793–805. doi: 10.1016/0092-8674(90)90123-v. [DOI] [PubMed] [Google Scholar]
- 12.Erkine A M, Magrogan S F, Sekinger E A, Gross D S. Mol Cell Biol. 1999;19:1627–1639. doi: 10.1128/mcb.19.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakobsen B K, Pelham H R B. Mol Cell Biol. 1988;8:5040–5042. doi: 10.1128/mcb.8.11.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Y H, Kroeger P E, Morimoto R I. Mol Cell Biol. 1995;15:4309–4318. doi: 10.1128/mcb.15.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoj A, Jakobsen B K. EMBO J. 1994;13:2617–2624. doi: 10.1002/j.1460-2075.1994.tb06552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonner J J, Heyward S, Fackenthal D L. Mol Cell Biol. 1992;12:1021–1030. doi: 10.1128/mcb.12.3.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morimoto R I. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 18.Morano K A, Liu P C C, Thiele D J. Curr Opin Microbiol. 1998;1:197–203. doi: 10.1016/s1369-5274(98)80011-8. [DOI] [PubMed] [Google Scholar]
- 19.Vuister G W, Kim S J, Orosz A, Marquardt J, Wu C, Bax A. Nat Struct Biol. 1994;1:605–614. [PubMed] [Google Scholar]
- 20.Sorger P K, Nelson H C M. Cell. 1989;59:807–813. doi: 10.1016/0092-8674(89)90604-1. [DOI] [PubMed] [Google Scholar]
- 21.Peteranderl R, Nelson H C M. Biochemistry. 1992;31:12272–12276. doi: 10.1021/bi00163a042. [DOI] [PubMed] [Google Scholar]
- 22.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz M C, Choe S Y, Reeder R H. Proc Natl Acad Sci USA. 1991;88:1004–1008. doi: 10.1073/pnas.88.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards M J. Cell Stress Chaperones. 1998;3:213–220. doi: 10.1379/1466-1268(1998)003<0213:athsrh>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Littlefield O, Nelson H C M. Nat Struct Biol. 1999;6:464–470. doi: 10.1038/8269. [DOI] [PubMed] [Google Scholar]
- 26.Flick K E, Gonzalez L, Harrison C J, Nelson H C M. J Biol Chem. 1994;269:12475–12481. [PubMed] [Google Scholar]
- 27.Morimoto R I, Sarge K D, Abravaya K. J Biol Chem. 1992;267:21987–21990. [PubMed] [Google Scholar]
- 28.Hardy J A, Walsh S T R, Nelson H C M. J Mol Biol. 2000;295:393–409. doi: 10.1006/jmbi.1999.3357. [DOI] [PubMed] [Google Scholar]
- 29.Larson J S, Schuetz T J, Kingston R E. Biochemistry. 1995;34:1902–1911. doi: 10.1021/bi00006a011. [DOI] [PubMed] [Google Scholar]
- 30.Goodson M L, Sarge K D. J Biol Chem. 1995;270:2447–2450. doi: 10.1074/jbc.270.6.2447. [DOI] [PubMed] [Google Scholar]
- 31.Lindquist S, Craig E A. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 32.Sorger P K, Lewis M J, Pelham H R B. Nature (London) 1987;329:81–84. doi: 10.1038/329081a0. [DOI] [PubMed] [Google Scholar]
- 33.Bulman A L, Hubl S T, Nelson H C M. J Biol Chem. 2001;276:40254–40262. doi: 10.1074/jbc.M106301200. [DOI] [PubMed] [Google Scholar]