

Abstract
The light-induced electron transfer reaction of flavin cofactor photoactivation in Xenopus laevis (6–4) photolyase has been studied by continuous-wave and time-resolved electron paramagnetic resonance spectroscopy. When the photoactivation is initiated from the fully oxidized form of the flavin, a neutral flavin radical is observed as a long-lived paramagnetic intermediate of two consecutive single-electron reductions under participation of redox-active amino acid residues. By time-resolved electron paramagnetic resonance, a spin-polarized transient radical-pair signal was detected that shows remarkable differences to the signals observed in the related cyclobutane pyrimidine dimer photolyase enzyme. In (6–4) photolyase, a neutral tyrosine radical has been identified as the final electron donor, on the basis of the characteristic line width, hyperfine splitting pattern, and resonance magnetic field position of the tyrosine resonances of the transient radical pair.
Ultraviolet light (λ≤300 nm) damages cellular DNA by formation of cyclobutane pyrimidine dimers (CPDs) and (6–4) photoproducts from adjacent pyrimidine bases on the same DNA strand (1). Such dimers may be restored to their monomeric form through the action of two photoactive (300<λ<500 nm) damage-specific DNA repair enzymes, named CPD photolyase (also called DNA photolyase) (2–6) and (6–4) photolyase (7, 8). Both enzymes are found in various organisms and exhibit a 20–30% amino acid sequence identity (3, 9, 10). Two photoactive pigments are used in the DNA repair pathway. One is invariably a redox-active FAD (11, 12), and the other so-called second chromophore, which acts as a light harvester, is a methenyltetrahydrofolate in most species (11, 13, 14) but 5-deazariboflavin in certain rare species that synthesize this compound (15, 16). It has been proposed that the initial step in the DNA repair mechanism is a photoinduced single electron transfer (ET) from the FAD cofactor, which in the active enzyme is in its fully reduced form (12, 17, 18), FADH−, to the DNA lesion. This mechanism is supported by a previous electron paramagnetic resonance (EPR) study in which a photoinduced spin-polarized radical pair (RP) signal assigned to the flavin–CPD dimer complex was observed (19).
If the enzyme is found in an inactive state with FAD either semireduced as neutral radical, FADH⋅, or fully oxidized, FADox (20), photolyases can undergo reversible ET reactions with the participation of amino acid residues to lower the redox state of the flavin cofactor to FADH−. This photoactivation process has recently attracted much experimental (20–26) and theoretical (27, 28) interest. In Escherichia coli CPD photolyase, cofactor photoactivation proceeds on a nanosecond time scale via a chain of tryptophan residues (W-382, -359, and -306) (29). W-306 is believed to be the final electron donor that is rereduced on a millisecond time scale either by back ET from the flavin or by exogenous reductants (30). In Anacystis nidulans CPD photolyase, however, a further ET step from a tyrosine to the tryptophanyl radical W-306• (E. coli numbering) follows for ≈40% of W• within ≈50 μs, whereas ≈60% of W• decay by charge recombination with the flavin within 1 ms (24, 25). Although the chain of tryptophan residues responsible for photoactivation in E. coli CPD photolyase is highly conserved among different types of photolyases and also the photolyase-homologous cryptochromes (31), it is still unknown whether photoactivation in (6–4) photolyase proceeds along the same pathway.
In the present contribution, we report on the first continuous-wave (cw) and time-resolved (tr) EPR studies of the laser-flash-induced photoactivation of Xenopus laevis (6–4) photolyase with the FAD cofactor initially in the fully oxidized form, FADox. The results reveal remarkable differences with respect to photoactivation in CPD photolyase. The spectral (Δg≤10−4) and temporal (≤100 ns) resolutions of our tr-EPR experiment allow us to directly identify the redox partners of the RP state generated in the course of the light-initiated ET of FAD photoactivation.
Materials and Methods
Sample Preparation.
X. laevis (6–4) photolyase was overproduced in E. coli, purified as described previously (12, 13), and stored in liquid nitrogen. The concentration of the enzyme was determined on the basis of the FAD cofactor's absorbance at 450 nm (ɛ450 = 1.12 × 104 M−1 cm−1) (12). For these experiments, typically 0.5 mM (6–4) photolyase in a buffer containing 0.3 M NaCl/0.1 M Tris⋅HCl, pH 8.0/50% (vol/vol) glycerol was used. The redox state of the flavin cofactor was checked systematically by measuring the ground-state absorption spectrum from 300 to 800 nm by using a Shimadzu UV-1601PC spectrophotometer. For some experiments, either DTT as an exogenous reductant or potassium ferricyanide as an oxidant was added to the sample at the concentration indicated.
EPR Spectroscopy.
cw-EPR experiments were performed by using a laboratory-built spectrometer consisting of an electromagnet, a Bruker (Rheinstetten, Germany) ER041 MR microwave bridge, in conjunction with a Bruker ER4118X-MD-5W1 dielectric resonator, which was immersed in a laboratory-built helium-gas flow cryostat. The temperature of the sample was controlled to ± 1 K by a Lake Shore Cryotronics (Westerville, OH) 321 autotuning temperature controller. Magnetic-field modulated EPR signals were recorded by using a Stanford Research (Sunnyvale, CA) SR810 DSP lock-in amplifier. The microwave frequency was measured by an EIP (Milpitas, CA) 548 frequency counter and the magnetic field controlled by a Bruker ER035 MR NMR gaussmeter.
tr-EPR experiments were performed by using the same setup but without magnetic field modulation, rather in direct-detection mode, and hence the signals have a nonderivative line shape with A, enhanced absorptive and E, emissive spin polarization. For this purpose, the time-dependent EPR signal was processed in a fast low-noise preamplifier and recorded by using a Tektronix TDS-520A digitizing oscilloscope. A complete data set consists of a series of transient signals taken at equidistant magnetic field points covering the total spectral width. Transient spectra can be extracted from this data set at any fixed time after the laser pulse as slices parallel to the magnetic field axis.
Optical excitation of the sample was provided by a Nd:YAG (neodymium yttrium/aluminum garnet) laser pumped dye laser from Thomson-CSF (Buyancourt Cedex, France) BMI AL.152C, with a wavelength of 440 nm (Coumarin-10), a pulse width of 6 ns, pulse energy 4 mJ, and a laser repetition rate of 1 Hz.
The samples were introduced into the resonator in suprasil quartz tubes (o.d. 2 mm, i.d. 1 mm) in an inert gas atmosphere in the dark. Such small-diameter sample tubes were necessary to avoid microwave absorption of the liquid solution protein samples.
Results and Discussion
Isolation and purification of (6–4) photolyase typically renders the FAD cofactor fully oxidized, which then exhibits characteristic optical absorption bands at 364 and 448 nm with the 448-nm peak having vibronic structure at 428 and 474 nm (ref. 13; see Fig. 1). No absorption is observed in the wavelength region 550 <λ<800 nm. Therefore, the presence of semireduced FAD (flavin semiquinones typically exhibit absorption bands at 585 and 625 nm; refs. 32, 33) before sample irradiation can be excluded. This finding is corroborated by EPR spectroscopy, where no signal of any paramagnetic species is detected under the same conditions (results not shown).
Figure 1.

Absorption spectrum of X. laevis (6–4) photolyase enzyme recorded at 283 K, before sample illumination.
cw-EPR Spectroscopy.
cw-EPR experiments have been performed on a liquid solution (6–4) photolyase sample where DTT has been added as an exogenous reductant to rereduce amino acid radicals, X•+ or XH•+ that are generated by light-initiated ET according to the following scheme:
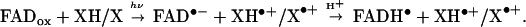 |
1 |
Indirect experimental evidence points to a triplet-initiated ET (23), even though 3FADox has not yet been reported in photolyases. In Eq. 1, XH or X is a redox-active amino acid residue, for example W in FAD photoactivation of E. coli CPD photolyase. After light-induced ET, both FAD•− and XH•+ may change their protonation states to form FADH• (34) and/or X•. This depends on the local pH conditions and the pKa values of the respective cofactors. In CPD photolyase, it is known that the semiquinone form of FAD is present as the neutral flavin radical protonated at N5 (35).
After several tens of laser flashes, a stable radical signal is detected (Fig. 2). The spectral position at g = 2.0034 ± 0.0003, the peak-to-peak line width, and the characteristic line shape of the signal are very similar to the spectrum of the flavin cofactor in CPD photolyase (19, 23, 35, 36). In Fig. 2, the spectrum from Gindt et al. (23) is also displayed for comparison. Neutral (protonated at N5) flavin radicals can be distinguished from anionic (unprotonated at N5) flavin radicals by means of their characteristic EPR peak-to-peak line widths of ≈2.0 and ≈1.3 mT, respectively. This difference is because of the presence or absence of the large hyperfine contribution of the proton at N5 (35). Therefore, the observed peak-to-peak line width of (2.0 ± 0.1) mT in (6–4) photolyase is typical of a neutral N5-protonated flavin radical, FADH•. The overall line width and characteristic shape of the signal are because of the partially resolved hyperfine couplings of H5, N5, and N10 of the isoalloxazin moiety of the FAD cofactor.
Figure 2.

cw-EPR signal generated by pulsed laser excitation (1-Hz repetition rate) of X. laevis (6–4) photolyase after 100 nonsaturating laser flashes in the presence of DTT (4 mM) as an exogenous reductant (trace A). EPR data were recorded at T = 278 K in the first-derivative mode. Instrument settings: microwave frequency, 9.6984 GHz; magnetic field modulation amplitude, 0.09 mT; modulation frequency, 100 kHz; time constant, 300 ms; and microwave power, 2 mW. To corroborate the assignment of this signal to a neutral flavin radical, FADH•, the cw-EPR spectrum of the neutral flavin semiquinone form of CPD photolyase is also displayed (dashed line; for details, see ref. 23). For a better comparison of the line width of the FADH• cw-EPR detected signal with the tr-EPR spectrum depicted in Fig. 3, trace A has been integrated to yield trace B. The molecular structure shows the redox-active 7,8-dimethyl isoalloxazine moiety of the FADH• cofactor; R, ribityl side chain and adenine dinucleotide moiety.
On the basis of the observation of FADH•, we conclude that enzymatic photoactivation starting from FADox proceeds in two consecutive light-initiated ET steps with FADH• as a metastable intermediate. Protonation of FAD•− to form FADH• must occur fast on the time scale of cw-EPR (≤10 μs). Continued exposure of the enzyme to light eventually converts the FADH• radical into the biologically active form, FADH−, which is diamagnetic and thus exhibits no EPR signal.
tr-EPR Spectroscopy.
To study the short-lived RP states generated after pulsed laser excitation of (6–4) photolyase (see Eq. 1), tr-EPR experiments at high time resolution have been performed (see Fig. 3). Because observation of EPR in the time domain is concomitant with a decrease in detection sensitivity, transient signals need to be repeatedly accumulated to increase their signal-to-noise ratio. Therefore, to restore identical startup conditions after each laser pulse, potassium ferricyanide (2 mM) has been added as an exogenous oxidant to a DTT-free (6–4) photolyase sample. This procedure (23) has proven to reoxidize FADH• generated according to Eq. 1 after photoactivation and therefore to restore the original conditions (i.e., FADox and XH or X) required for repetitive signal accumulation.§ The acquisition of the tr-EPR signal at a 1-Hz laser-pulse repetition rate required ≈30 h. No discernible cw-EPR signal was detected before sample illumination.
Figure 3.

tr-EPR signal generated by pulsed laser excitation (1-Hz repetition rate) of X. laevis (6–4) photolyase in the presence of potassium ferricyanide (2 mM) as an exogenous oxidant. EPR data were recorded at T = 278 K at 1.4 μs after laser-flash excitation in the direct-detection mode [integrated EPR amplitudes (dots) with A , enhanced absorption and E, emission]. Instrument settings: 9.6980 GHz; microwave power, 2 mW; optical sample excitation same as in Fig. 2. To obtain precise hyperfine line positions, the experimental spectrum was deconvoluted to a sum of 12 Gaussians (drawn lines). The arrows indicate the underlying hyperfine pattern of four nearly equivalent protons (for details, see text). Inset shows the corresponding light-initiated EPR signal observed in E. coli CPD photolyase (data taken from ref. 23).
In the tr-EPR experiment, transient paramagnetic species are detected in their spin-polarized states (A, enhanced absorption or E, emission) shortly after the laser pulse. Typically, the tr-EPR spectrum of a triplet-initiated spin-correlated RP consists of an antiphase line pair for each of the two interacting paramagnetic species (37, 38). These line pairs are symmetrically centered about the resonance field positions of the individual radicals (here FADH• and X•+ or XH•+). The separation of the lines is determined by the anisotropic dipolar and the isotropic exchange interaction between the RP partners. When all interaction anisotropies are completely averaged out, the corresponding EPR spectrum can be easily calculated if the isotropic g values, as well as the hyperfine patterns of the individual radicals and the exchange interaction between them, are known. This is the case, for example, in freely diffusing RPs in liquid solution or in RPs embedded in a protein matrix that undergoes rapid overall diffusive motion. In rigid systems with well-defined RP geometry, such as the photosynthetic reaction center, the calculation of the resulting asymmetric and broad EPR powder pattern is still feasible, provided that all interaction anisotropies and the relative orientations of their principal axes are known (39, 40). The appearance of relatively narrow spectral features in the RP signal of Fig. 3 leads us to the conclusion that some motional dynamics are present in cofactor photoactivation of (6–4) photolyase. The slight but noticeable asymmetry of the spectral pattern, however, indicates that motional averaging is incomplete. This might be because of restricted overall and protein side chain motional dynamics of the 60-kDa enzyme in the viscous solvent. In principle, the simulation of such a system's spectrum is still feasible; however, knowledge of additional parameters such as the principal axes of molecular motion and motional correlation times is required. These are unknown at present; therefore, we refrain from the presentation of a highly ambiguous simulation and rather restrict ourselves to a qualitative analysis of the tr-EPR signal.
The RP tr-EPR spectrum recorded 1.4 μs after the laser pulse is composed of broad spectral wings at 343.0 and 348.6 mT and a narrow E/A-polarized signal. The broad features are centered at g = 2.0034 ± 0.0003, typical of a neutral flavin radical (35), and hence are assigned to the flavin part of the RP (see Fig. 2). Its characteristic (derivative-like) line shape arises from extensive destructive averaging of the broad and overlapping absorptively and emissively polarized signal contributions within the line pair belonging to FADH•. Except for lifetime broadening of the spectrum, which is observed at early times after the laser pulse (t<100 ns), the shape of the tr-EPR spectrum does not significantly change for observation times ≥100 ns, also indicating that FAD•− is protonated within 100 ns of its formation.
The narrow E/A-polarized feature of the spectrum is centered at g = 2.0048 ± 0.0003. Such a high g value is characteristic for a neutral (deprotonated) tyrosyl radical, Y•, for which isotropic g values of 2.0045 ≤ giso ≤ 2.0050 are expected (41–43). A tryptophan radical, by comparison, would show resonances centered at giso ≈2.0025 (43).
Further evidence for a neutral tyrosyl radical being involved in (6–4) photolyase photoactivation is provided by the characteristic line shape of the central E/A feature: even though the line width of a tyrosyl radical interacting with a second paramagnetic species could, in principle, appear narrower than that of an isolated one (because of the overlap of absorptively and emissively polarized spectral contributions at sufficiently weak spin–spin interaction), the line width of the E/A feature in Fig. 3 is too small for a tryptophan radical. The latter typically exhibits a characteristic hyperfine splitting in the range of 0.8–2.8 mT arising from one of the two β protons attached to the indole ring of W• (44). Such a large splitting is not observed here but is present in the RP signal of E. coli CPD photolyase (see Fig. 3 Inset) (23).
A repeating 0.45-mT hyperfine splitting pattern is observed in the narrow feature of the (6–4) photolyase EPR signal (marked with arrows in Fig. 3), which points to a paramagnetic species with two or more magnetically equivalent nuclei. Again, this is consistent with a neutral tyrosine radical but not with a tryptophan radical, where the hyperfine lines of many inequivalent nuclei overlap to give an unresolved signal (with the one exception of the large β proton coupling still resolved; see above). In tyrosyl neutral radicals, because of molecular symmetry, the protons H2 and H6 as well as H3 and H5 are pairwise magnetically equivalent. The two β protons may also be equivalent by geometry or because of motional averaging, resulting in an EPR spectrum that exhibits a well-resolved hyperfine line pattern (42, 43). We therefore conclude that the hyperfine splitting observed in Fig. 3 arises from the H3/H5 protons and β protons, for which almost identical isotropic hyperfine coupling constants of −0.57 and 0.5 mT, respectively, have been recently calculated with density functional theory (45), resulting in the observed quintet-structured pattern for the absorptively and emissively polarized lines belonging to Y•.
To summarize, we have demonstrated that FAD cofactor photoactivation in (6–4) photolyase occurs via two sequential single-electron reduction steps with a tyrosine residue acting as final electron donor. The Y• signal is observed within 100 ns of pulsed laser excitation. Intermediate tryptophanyl radicals in (6–4) photolyase are not observed by our tr-EPR experiments. This indicates either W• lifetimes of <100 ns or the presence of an ET pathway different from the one proposed for FAD photoactivation in CPD photolyases (29).
In contrast to the present results, only FAD reduction by tryptophans has been reported in other photolyases, with the exception of the A. nidulans CPD photolyase. In this enzyme, however, the photo-generated W• is only partially reduced by a tyrosine and on a much longer time scale (t1/2 = 50 μs), whereas the majority of W• is rereduced by back-ET from the flavin (24). It is not clear to date whether this nonuniform behavior in A. nidulans CPD photolyase is caused by sample heterogeneity or results from competing reactions where the reduction of W• by Y is energetically slightly less favorable than the reverse of Eq. 1. Certainly, FAD cofactor photoactivation in photolyases is a versatile process that can use different amino acid residues as electron donors and/or ET pathways to maintain the enzyme's biological activity. Developing a molecular understanding of the function of DNA repair enzymes is very important in itself but even more fundamental when considering the function of the recently discovered cryptochromes (46), of which the photolyases are thought to be the evolutionary precursors (3, 5).
Future studies will be focused on the identification of the redox-active tyrosine residue responsible for FAD photoactivation in (6–4) photolyase and its single-point mutants.
Acknowledgments
We thank Dr. Gerald Richter, Prof. Adelbert Bacher (both of the Technical University of Munich), Prof. Robert Bittl, and Dr. Michael Fuhs (both of the Free University Berlin) for valuable discussions. This work was supported by the VolkswagenStiftung (Grant I/77100), which is gratefully acknowledged.
Abbreviations
- CPD
cyclobutane pyrimidine dimer
- EPR
electron paramagnetic resonance
- RP
radical pair
- cw
continuous wave
- tr
time resolved
- ET
electron transfer
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
EPR spectra identical to the one depicted in Fig. 3 have also been obtained from a (6–4) photolyase sample without added exogenous oxidant (data not shown). However, the EPR signals could be observed only for a limited number of laser excitations and therefore resulted in a limited signal-to-noise ratio.
References
- 1.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 2.Sancar A. Biochemistry. 1994;33:2–9. doi: 10.1021/bi00167a001. [DOI] [PubMed] [Google Scholar]
- 3.Todo T. Mutat Res. 1999;434:89–97. doi: 10.1016/s0921-8777(99)00013-0. [DOI] [PubMed] [Google Scholar]
- 4.Sancar G B. Mutat Res. 2000;451:25–37. doi: 10.1016/s0027-5107(00)00038-5. [DOI] [PubMed] [Google Scholar]
- 5.Deisenhofer J. Mutat Res. 2000;460:143–149. doi: 10.1016/s0921-8777(00)00023-9. [DOI] [PubMed] [Google Scholar]
- 6.Carell T, Burgdorf L T, Kundu L M, Cichon M. Curr Opin Chem Biol. 2001;5:491–498. doi: 10.1016/s1367-5931(00)00239-8. [DOI] [PubMed] [Google Scholar]
- 7.Todo T, Takemori H, Ryo H, Ihara M, Matsunaga T, Nikaido O, Sato K, Nomura T. Nature (London) 1993;361:371–374. doi: 10.1038/361371a0. [DOI] [PubMed] [Google Scholar]
- 8.Zhao X, Mu D. Histol Histopathol. 1998;13:1179–1182. doi: 10.14670/HH-13.1179. [DOI] [PubMed] [Google Scholar]
- 9.Todo T, Ryo H, Yamamoto K, Toh H, Inui T, Ayaki H, Nomura T, Ikenaga M. Science. 1996;272:109–112. doi: 10.1126/science.272.5258.109. [DOI] [PubMed] [Google Scholar]
- 10.Nakajima S, Sugiyama M, Iwai S, Hitomi K, Otoshi E, Kim S-T, Jiang C-Z, Todo T, Britt A B, Yamamoto K. Nucleic Acids Res. 1998;26:638–644. doi: 10.1093/nar/26.2.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancar A, Sancar G B. J Mol Biol. 1984;172:223–227. doi: 10.1016/s0022-2836(84)80040-6. [DOI] [PubMed] [Google Scholar]
- 12.Todo T, Kim S-T, Hitomi K, Otoshi E, Inui T, Morioka H, Kobayashi H, Ohtsuka E, Toh H, Ikenaga M. Nucleic Acids Res. 1997;25:764–768. doi: 10.1093/nar/25.4.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hitomi K, Kim S-T, Iwai S, Harima N, Otoshi E, Ikenaga M, Todo T. J Biol Chem. 1997;272:32591–32598. doi: 10.1074/jbc.272.51.32591. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Liu J, Hsu D S, Zhao S, Taylor J-S, Sancar A. J Biol Chem. 1997;272:32580–32590. doi: 10.1074/jbc.272.51.32580. [DOI] [PubMed] [Google Scholar]
- 15.Eker A P M, Hessels J K C, van de Velde J. Biochemistry. 1988;27:1758–1765. [Google Scholar]
- 16.Kiener A, Husain I, Sancar A, Walsh C. J Biol Chem. 1989;264:13880–13887. [PubMed] [Google Scholar]
- 17.Sancar G B, Jorns M S, Payne G, Fluke D J, Rupert C S, Sancar A. J Biol Chem. 1987;262:492–498. [PubMed] [Google Scholar]
- 18.Payne G, Heelis P F, Rohrs B R, Sancar A. Biochemistry. 1987;26:7121–7127. doi: 10.1021/bi00396a038. [DOI] [PubMed] [Google Scholar]
- 19.Rustandi R R, Jorns M S. Biochemistry. 1995;34:2284–2288. doi: 10.1021/bi00007a024. [DOI] [PubMed] [Google Scholar]
- 20.Heelis P F, Sancar A. Biochemistry. 1986;25:8163–8166. doi: 10.1021/bi00373a006. [DOI] [PubMed] [Google Scholar]
- 21.Heelis P F, Okamura T, Sancar A. Biochemistry. 1990;29:5694–5698. doi: 10.1021/bi00476a008. [DOI] [PubMed] [Google Scholar]
- 22.Kim S-T, Sancar A, Essenmacher C, Babcock G T. Proc Natl Acad Sci USA. 1993;90:8023–8027. doi: 10.1073/pnas.90.17.8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gindt Y M, Vollenbroek E, Westphal K, Sackett H, Sancar A, Babcock G T. Biochemistry. 1999;38:3857–3866. doi: 10.1021/bi981191+. [DOI] [PubMed] [Google Scholar]
- 24.Aubert C, Mathis P, Eker A P M, Brettel K. Proc Natl Acad Sci USA. 1999;96:5423–5427. doi: 10.1073/pnas.96.10.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aubert C, Brettel K, Mathis P, Eker A P M, Boussac A. J Am Chem Soc. 1999;121:8659–8660. [Google Scholar]
- 26.Aubert C, Vos M H, Mathis P, Eker A P M, Brettel K. Nature (London) 2000;405:586–590. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 27.Cheung M S, Daizadeh I, Stuchebrukhov A A, Heelis P F. Biophys J. 1999;76:1241–1249. doi: 10.1016/S0006-3495(99)77287-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee E, Medvedev E S, Stuchebrukhov A A. J Phys Chem B. 2000;104:6894–6902. [Google Scholar]
- 29.Park H-W, Kim S-T, Sancar A, Deisenhofer J. Science. 1995;268:1866–1872. doi: 10.1126/science.7604260. [DOI] [PubMed] [Google Scholar]
- 30.Li Y F, Heelis P F, Sancar A. Biochemistry. 1991;30:6322–6329. doi: 10.1021/bi00239a034. [DOI] [PubMed] [Google Scholar]
- 31.Kanai S, Kikuno R, Toh H, Ryo H, Todo T. J Mol Evol. 1997;45:535–548. doi: 10.1007/pl00006258. [DOI] [PubMed] [Google Scholar]
- 32.Beinert H. J Am Chem Soc. 1956;78:5323–5328. [Google Scholar]
- 33.Massey V, Parker G. Biochemistry. 1966;5:3181–3189. doi: 10.1021/bi00874a016. [DOI] [PubMed] [Google Scholar]
- 34.Müller F. Top Curr Chem. 1983;108:71–107. doi: 10.1007/3-540-11846-2_3. [DOI] [PubMed] [Google Scholar]
- 35.Kay C W M, Feicht R, Schulz K, Sadewater P, Sancar A, Bacher A, Möbius K, Richter G, Weber S. Biochemistry. 1999;38:16740–16748. doi: 10.1021/bi991442u. [DOI] [PubMed] [Google Scholar]
- 36.Jorns M S, Sancar G B, Sancar A. Biochemistry. 1984;23:2673–2679. doi: 10.1021/bi00307a021. [DOI] [PubMed] [Google Scholar]
- 37.Closs G L, Forbes M D E, Norris J R. J Phys Chem. 1987;91:3592–3599. [Google Scholar]
- 38.Hore P J, Hunter D A, McKie C D, Hoff A J. Chem Phys Lett. 1987;137:495–500. [Google Scholar]
- 39.Stehlik D, Bock C H, Petersen J. J Phys Chem. 1989;93:1612–1619. [Google Scholar]
- 40.Kothe G, Weber S, Ohmes E, Thurnauer M C, Norris J R. J Phys Chem. 1994;98:2706–2712. [Google Scholar]
- 41.Dixon W T, Murphy D. J Chem Soc Faraday Trans 2. 1976;72:1221–1230. [Google Scholar]
- 42.Un S, Tang X-S, Diner B A. Biochemistry. 1996;35:679–684. doi: 10.1021/bi9523769. [DOI] [PubMed] [Google Scholar]
- 43.Bleifuss G, Kolberg M, Pötsch S, Hofbauer W, Bittl R, Lubitz W, Gräslund A, Lassmann G, Lendzian F. Biochemistry. 2001;40:15362–15368. doi: 10.1021/bi010707d. [DOI] [PubMed] [Google Scholar]
- 44.Himo F, Eriksson L A. J Phys Chem B. 1997;101:9811–9819. [Google Scholar]
- 45.Himo F, Gräslund A, Eriksson L A. Biophys J. 1997;72:1556–1567. doi: 10.1016/S0006-3495(97)78803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cashmore A R, Jarillo J A, Wu Y-J, Liu D. Science. 1999;284:760–765. doi: 10.1126/science.284.5415.760. [DOI] [PubMed] [Google Scholar]