

Abstract
Nucleotide excision repair removes damages from the DNA by incising the damaged strand on the 3′ and 5′ sides of the lesion. In Escherichia coli, the two incisions are made by the UvrC protein, which consists of two functional halves. The N-terminal half contains the catalytic site for 3′ incision and the C-terminal half contains the residues involved in 5′ incision. The genome of E. coli contains an SOS-inducible gene (ydjQ) encoding a protein that is homologous to the N-terminal half of UvrC. In this paper we show that this protein, which we refer to as Cho (UvrC homologue), can incise the DNA at the 3′ side of a lesion during nucleotide excision repair. The incision site of Cho is located 4 nt further away from the damage compared with the 3′ incision site of UvrC. Cho and UvrC bind to different domains of UvrB, which is probably the reason of the shift in incision position. Some damaged substrates that are poorly incised by UvrC are very efficiently incised by Cho. We propose that E. coli uses Cho for repair of such damages in vivo. Initially, most of the lesions in the cell will be repaired by the action of UvrC alone. Remaining damages, that for structural reasons obstruct the 3′ incision by UvrC, will be repaired by the combined action of Cho (for 3′ incision) and UvrC (for 5′ incision).
The UvrC protein is the endonuclease responsible for incisions at the 3′ and 5′ sides of a damaged site during nucleotide excision repair in Escherichia coli (1, 2). The protein binds to the UvrB–DNA preincision complex, which is formed by stable binding of the UvrB protein to a damaged site, initially as part of the UvrA2B complex, but from which the UvrA subunits have subsequently dissociated (for reviews see refs. 3 and 4). The two incisions occur in a defined order, first at the 3′ side and then at the 5′ side of the damage. The UvrC protein consists of two functional parts. The N-terminal half contains the catalytic domain for 3′ incision (2), which is homologous to the catalytic domain of the GIY–YIG family of intron-encoded homing endonucleases (Fig. 1; ref. 5). In addition, the N-terminal half of UvrC contains a region that interacts with a homologous domain of the UvrB protein in the UvrBC–DNA incision complex (6, 7). Mutational analyses have shown that interaction by means of these homologous domains is needed for the 3′ incision but not for the 5′ incision (8, 9). A truncated UvrC protein lacking the N-terminal half is capable of 5′ incision on a substrate that is prenicked at the 3′ incision position (2), showing that the C-terminal half of UvrC contains all of the elements for the second incision event.
Figure 1.

Alignment of the N-terminal part of the E. coli UvrC protein with Cho proteins from E. coli (Eco), Salmonella typhimurium (Sty), Salmonella enteritidis (Sen), and Klebsiella pneumoniae (Kpn), and with a 24-kDa protein from Mycoplasma hyopneumoniae (Mhy), a 43-kDa protein from Clostridium acetobutylicum (Cac), and a 69-kDa protein from Mycobacterium tubercylosis (Mtu). Proteins that are almost identical to this 69-kDa protein are also found in other mycobacteria, i.e., Mycobacterium bovis, Mycobacterium avum, Mycobacterium smegmatis, and Mycobacterium leprae (not shown). Residues that are identical or similar to residues in UvrC are highlighted. The UvrB-binding domain of UvrC is underlined.
The complete genome sequence of E. coli (10) revealed the presence of a gene, ydjQ, encoding a protein of 295 aa (molecular mass, 33.7 kDa) that shares significant homology with the N-terminal half of UvrC (Fig. 1). With Northern analysis it was shown that ydjQ is part of the SOS regulon, controlled by the LexA repressor (11). Interestingly the uvrA and uvrB genes are also under control of the SOS response, whereas uvrC is not. In this paper we show that the product of the ydjQ gene, which we refer to as Cho (UvrC homologue) is capable of incising DNA at the 3′ side of a damage in the presence of the UvrA and UvrB proteins. For some types of damage, Cho incision is even more efficient than the 3′ incision by UvrC. We propose that the presence of Cho in E. coli broadens the substrate range of nucleotide excision repair.
Materials and Methods
Bacterial Strains and Plasmids.
The strains and plasmids used are listed in Table 1. The cho gene was disrupted by using the system described by Datsenko and Wanner (12). In short, by using PCR with primers Cho1 (TCGCTTCCCGCCTGTTACACTGGATAGATAACCAGCATTCGGAGTCAACAGTGTAGGCTGGAGCTGCTTC) and Cho2 (TCATAGTTACCGGAAAGCAGCGGCTTACAAAGAATTTTATAACCGTCGTGCAT- ATGAATATCCTCCTTAG) and pKD3 as a template, a fragment was generated containing the cam gene flanked by directly repeated FLP recognition target (FRT) sites and a 50-nt homology extension of the DNA regions flanking the chromosomal cho gene (depicted in italics in the primer sequences). The homology regions were chosen such that no sequences of genes flanking cho will be disrupted. After λ Red-mediated recombination in BW25113 containing pKD46, a strain was created in which the cho gene is replaced by the FRT-flanked cam gene. The cho disruption was transferred to KMBL1001 (CS5540) or RW542 (CS5617) by using P1 transduction. Subsequently, the cam cassette was eliminated from the chromosome by using pCP20 expressing the FLP recombinase, resulting in a cho deletion with an FRT scar sequence (CS5541 and CS5618). Next the ΔuvrC∷cam deletion from CS5430 was transferred to the Δcho strains by phage P1 transduction resulting in CS5550 and CS5619. All deletion constructs were verified by PCR.
Table 1.
Bacterial strains and plasmids
| Strain/plasmid | Relevant genotype or characteristics | Source or ref. |
|---|---|---|
| E. coli strains | ||
| KMBL1001 | F− derivative of W1485 (no known mutations) | 13 |
| BW25113 | lacIq rrnBT14ΔlacZWJ16hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | 12 |
| CS5430 | KMBL1001, ΔuvrC∷cam | 14 |
| CS5540 | KMBL1001, Δcho∷cam | This paper |
| CS5541 | KMBL1001, Δcho(FRT) | This paper |
| CS5550 | KMBL1001, Δcho(FRT) ΔuvrC∷cam | This paper |
| RW542 | lexA51(Def) | 11 |
| CS5616 | RW542, ΔuvrC∷cam | This paper |
| CS5617 | RW542, Δcho∷cam | This paper |
| CS5618 | RW542, Δcho(FRT) | This paper |
| CS5619 | RW542, Δcho(FRT), ΔuvrC∷cam | |
| ER2566 | fhuA2[lon] ompT lacZ∷T7 gene 1 endA1 | New England Biolabs |
| Plasmids | ||
| pKD46 | λ Red recombinase, araC-ParaB, Ap | 12 |
| pKD3 | Cm(FRT) | 12 |
| pCP20 | FLP, Ap, Cm | 12 |
| pWU1 | ori pSC101, Km, Tc | 14 |
| pCA179 | pWU1 with uvrC(R42A), Tc | 14 |
| pTYB1 | PT7, intein tag, Ap | New England Biolabs |
| pCA170 | cho cloned into pTYB1 | This paper |
| pWU5 | 1,330-bp cam fragment inserted in pHSS6 | Our laboratory |
| pNP81 | 2,300-bp uvrB fragment inserted in pHSS6 | Our laboratory |
UV Sensitivity Assay.
Plasmids pCA179 (UvrC, R42A) and pWU1 (vector) were introduced into the KMBL1001 derivatives CS5430 and CS5550 and in the RW542 derivatives CS5616 and CS5619. Transformants were grown to an OD600 of 0.3. Next, the cultures were diluted 10-fold and spots of 2 μl were deposited on LB agar plates. The plates were irradiated with the indicated doses of UV light and subsequently incubated overnight at 30°C.
Cloning, Expression, and Purification of the cho Gene Product.
The cho gene was amplified by using primer Cho3 (GGTGGTCATATGGTACGGCGTTTAACTTCTCCGCG), which introduces a NdeI site overlapping the translation start, and primer Cho4 (GGTGGTTGCTC TTCCGCAACTGGCTCGCTGGTCATTCG), which removes the stop codon and introduces a SapI restriction site downstream of the cho gene. The PCR product was digested with NdeI and SapI and cloned in the pTYB1 vector of the T7 IMPACT system (New England BioLabs) (pCA170). ER2566 containing pCA170 was grown at 37°C until OD600 of 0.5, after which isopropyl β-d-thiogalactoside (IPTG, 0.5 mM) was added and the culture was grown overnight at 14°C. The cell pellet of a 1-liter culture was resuspended in 25 ml of lysis buffer (20 mM Hepes, pH 8.0/500 mM NaCl/1 mM EDTA/0.1% IGEPAL; Sigma) followed by sonication. Cell debris was removed by ultracentrifugation, and the supernatant was loaded on a 3-ml chitin column equilibrated in lysis buffer. The column was washed with 5 vol of lysis buffer and then equilibrated in cleavage buffer (20 mM Hepes, pH 8.0/50 mM NaCl/0.1 mM EDTA/30 mM DTT). After 16 h at 16°C, the cleaved Cho protein was eluted with 1 vol of cleavage buffer without DTT. The isolated Cho protein was 95% pure as visualized on a Coomassie brilliant blue-stained protein gel.
Purification of the UvrABC Proteins.
The purifications of the wild-type UvrA, UvrB (15), and UvrC (16) proteins have been described. The proteolytic UvrB* fragment was purified as described (8). The plasmid overproducing the 5′ incision mutant UvrC(D399A), was kindly provided by A. Sancar (1), and the 3′ incision mutant UvrC(R42A) has been described (2). Both proteins were purified by the same procedure as wild-type UvrC.
DNA Substrates.
The 50-bp substrate G1 contains a cholesterol lesion (Chol-S; Fig. 2) at position 27 of the top strand. This substrate was constructed as described (17). Substrate M1 is the same DNA fragment, but with a menthol group (Fig. 2) attached to the N3 of thymine at position 27. Synthesis of the phosphoramidite-protected nucleoside building block with the menthol lesion will be described elsewhere. This building block was incorporated into the DNA fragment by automated oligonucleotide synthesis. The 50-bp cisplatin substrate (Pt1) contains the three top-strand oligonucleotides, Pt5 (AGTTCTATGCGCACCG), 18A-Pt (AATTCCCACTGGAACCCA), and Pt6 (AGCTTGCCGGGCCTCT), and the bottom strand Pt2 (GAGAGGCCCGGCAAGCTTGGGTTCCAGTGGGAATTCGGTGCGCATAGAACT). The position of the Pt-crosslink in 18A-Pt is underlined. The cisplatin treatment and purification of oligo 18A-Pt have been described (18). The 50-bp DNA substrate was constructed as described (18) with the following modifications. After hybridization and ligation of the four oligonucleotides, the DNA was purified with an AutoSeq G-50 column (Amersham Pharmacia) in H2O. The sample was concentrated and loaded on a 15% polyacrylamide gel containing 7 M urea. After electrophoresis and autoradiography, the substrate was eluted from the gel in 0.1 M sodium acetate followed by ethanol precipitation and finally the DNA was dissolved in 20 μl of H2O. The 2-acetylaminofluorene (AAF)-containing substrate (A1) was constructed by annealing of the bottom strand (GAGAGGCCCGGCAAGCTTTGGTTGTTCGAAGTTAAGCGGTGCGCATAGAACT) with the top strand oligonucleotides Pt5, Pt6, and oligo(CTTAACTTCGAACAACCAA) containing an AAF adduct on the G residue (19). After ligation, the substrate was purified as described above for Pt1.
Figure 2.

Structures of the Chol-S and menthol lesions used in this study. The Chol-S lesion consists of a cholesterol moiety attached to the ribose of a nucleoside. The menthol lesion consists of a menthol group attached to the N3 position of a thymine.
Incision Assays.
Plasmid pWU5 (3,480 bp, 50 ng/μl) was UV irradiated with 300 J/m2 and mixed with an equal amount of the nonirradiated supercoiled plasmid pNP81 (4,450 bp). The mixture was incubated with 10 nM UvrA, 100 nM UvrB, and 25 nM UvrC or Cho as indicated in 10 μl of Uvr-endo buffer (50 mM Tris⋅HCl, pH 7.5/10 mM MgCl2/100 mM KCl/1 mM ATP/0.1 μg/μl BSA) at 37°C. The reaction was terminated after 15 min by adding Ficoll and 3% SDS, and the samples were analyzed on a 1% agarose gel containing TBE (90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3).
The linear DNA substrates were 5′-terminally labeled in the top strand by using T4 polynucleotide kinase. The DNA substrates (2–4 fmol) were incubated with 2.5 nM UvrA, 100 nM UvrB, and with 50 nM UvrC, UvrC(D399A), UvrC(R42A), or Cho as indicated in 20 μl of Uvr-endo buffer at 37°C. The reaction was terminated after the indicated times by adding 2 μl of 0.5 M EDTA and 1 μl of 10% SDS. After 5 min, 2.4 μl of glycogen (2 μg/μl) was added, followed by ethanol precipitation. The incision products were visualized on 15% or 20% polyacrylamide gels containing 7 M urea. The DNase I ladder used in Fig. 5 was prepared with substrate G1. The DNA substrate was incubated for 6 min at 20°C with a 300-fold dilution of DNase I (10 units/μl, Amersham Pharmacia) in 50 mM Tris⋅HCl, pH 7.5/10 mM MgCl2/50 mM NaCl/2.5 mM CaCl2/0.1 μg/μl BSA. The reaction was terminated as described above.
Figure 5.
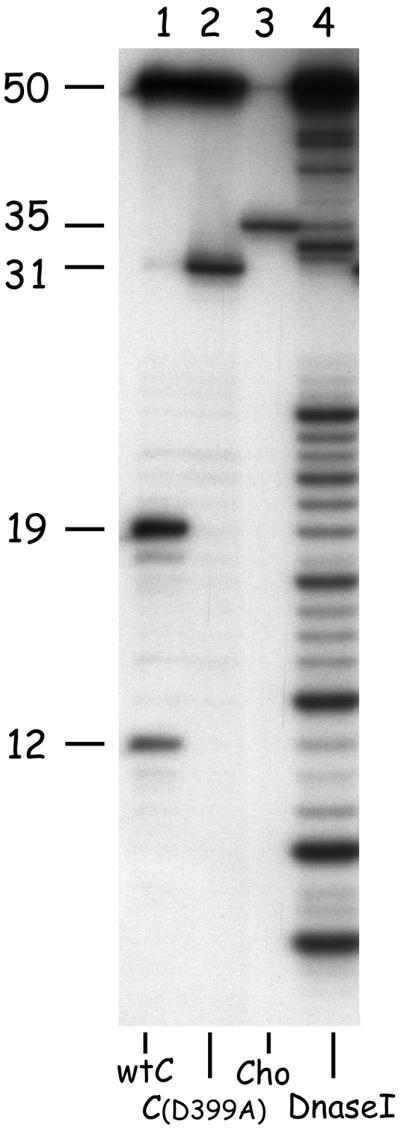
Incision of the 50-bp substrate containing a cholesterol lesion (G1). The substrate was incubated with UvrA, UvrB, and wtUvrC (lane 1) or UvrC(D399A) (lane 2), or Cho (lane 3). A DNase I ladder of substrate G1 was used as a reference (lane 4). The sizes of the different incision products are indicated. Note that for proper determination of the size of the Cho incision product, much less radioactive substrate was put on the gel, and as a result the nonincised 50-mer in lane 3 is hardly visible.
Results
The cho Gene Contributes to UV Survival in Vivo.
To test the role of the cho gene in nucleotide excision repair, we constructed a Δcho deletion in the wild-type strain KMBL1001 as described in Materials and Methods. The resulting mutant (CS5540) did not show a detectable difference in survival after UV treatment compared with the wild-type strain (not shown). However, because the cho gene is LexA regulated (11), whereas the uvrC gene is not (20), the expression of the cho gene will be retarded compared with the expression of the uvrC gene. Consequently, most of the UV damages will be removed by the action of UvrC and a possible contribution of Cho cannot be detected. We therefore tested the effect of the cho deletion in a ΔuvrC background (Fig. 3). In the absence of UvrC, indeed, a small difference in UV survival depending on the presence or absence of the cho gene can be seen (Fig. 3A, rows 1 and 2). Apparently cho does contribute to UV survival, but it cannot fully replace the function of the uvrC gene. The protein encoded by the cho gene is homologous to the N-terminal half of the UvrC protein, but being half the size of UvrC it lacks the domain corresponding to the C-terminal half. This finding predicts that the Cho protein might be capable of inducing the 3′ incision, but not the 5′ incision. We therefore tested the effect of cho on UV survival in the presence of the UvrC(R42A) mutant, which is capable of making the 5′ incision but not the 3′ incision (2). The results show that, indeed, the combined action of Cho and UvrC(R42A) leads to an enhanced survival after UV irradiation (Fig. 3A, rows 3 and 4). The effect of cho on UV survival is even more pronounced when the same experiment is repeated in a background lacking a functional LexA protein, and which is therefore expressing the cho gene constitutively (Fig. 3B). Although in this genetic background the UV survival of all strains is already higher as a result of the induction of other SOS-regulated repair systems (e.g., homologous recombination repair), the combined action of Cho and UvrC(R42A) clearly results in an additional repair of UV-induced lesions. These results strongly suggest that Cho is capable of making the 3′ incision on UV-damaged DNA in vivo.
Figure 3.

The effect of the cho gene on UV survival. Small droplets of the different strains were deposited on agar plates, after which they were irradiated with the indicated dose of UV light. (A) UV survival of strains in which the uvrC and/or cho have been deleted and which carry either a plasmid vector (pWU5) or a plasmid expressing the UvrC(R42A) protein (pCA179). (B) The same experiment, but all strains now contain an additional lexA51(Def) mutation.
The Cho Protein Is a UvrAB-Dependent Nuclease.
The cho gene was cloned and the Cho protein was purified as described in Materials and Methods. The incision activity of Cho was tested in vitro by using UV-irradiated supercoiled DNA. Incubation of the DNA mixture with UvrA, UvrB, and UvrC leads to nicking of the UV-irradiated plasmid as a result of the damage-dependent DNA incision by UvrC, whereas the nonirradiated plasmid remains in the supercoiled conformation (Fig. 4, lane 3) A similar result is obtained when the plasmids are incubated with UvrA, UvrB, and Cho (lane 2) showing that Cho specifically incises damaged DNA. No incision by Cho is observed when UvrA or UvrB is omitted from the incubation mixture (lanes 4 and 5), showing that the incision activity of Cho is UvrA and UvrB dependent.
Figure 4.

Incision of UV-irradiated supercoiled DNA by UvrC or Cho. A mixture of supercoiled DNA of UV-irradiated pWU5 (A, 2,150 bp) and nonirradiated pNP81 (B, 4,362 bp) was incubated with the indicated proteins. The positions of the supercoiled (sc) and relaxed (rel) forms of the plasmids are shown.
Cho Induces 3′ but Not 5′ Incision.
We further analyzed the incision activity of Cho by using a defined damaged substrate, which contains a single cholesterol lesion in the center of a 50-bp fragment (G1) and which is labeled at the 5′ side of the damaged strand. In the presence of UvrA, UvrB, and UvrC, two incision products are observed (Fig. 5, lane 1). The 19-nt fragment is the result of the 5′ incision reaction, and the 12-nt fragment results from the extra 5′ incision. This extra 5′ incision is in fact caused by a damage-independent nuclease activity of UvrBC on a nicked DNA substrate (21). A fragment corresponding to the 3′ incision cannot be detected because 3′ incision is immediately followed by the 5′ incision reaction. The 3′ incision product is observed when UvrC(D399A) is used, which has a mutation in the 5′ catalytic site (1). The 31-nt product obtained corresponds to incision at the fifth phosphodiester bond 3′ of the damage (lane 2). Incubation of substrate G1 with UvrA, UvrB, and Cho leads to a product of 35 nt. Apparently the incision position of Cho is shifted 4 nt with respect to the one made by UvrC, and is located at the ninth phosphodiester bond 3′ of the damage. No fragments corresponding to incision at the 5′ side of the damage can be seen, confirming that Cho is capable of 3′ incision only.
Efficiency of Cho Incision on Different DNA Substrates.
The 3′ incision of substrate G1 by Cho appears much more efficient than the 3′ incision by UvrC (Fig. 5, lanes 2 and 3, and Fig. 6, G1). We also compared the efficiencies of the two nucleases on a variety of other damaged substrates (Fig. 6). Substrate M1, containing a menthol lesion (Fig. 2), is also recognized by the Uvr proteins. Again, the Cho incision is much more efficient than the UvrC incision on this substrate (Fig. 6, M1). With two other damage-containing fragments, however, the difference between UvrC and Cho is not as apparent. Substrate A1, containing an AAF adduct, is equally incised by UvrC and Cho (Fig. 6, A1), and for the cisplatin-containing substrate incision by UvrC even seems to be somewhat higher than the Cho incision (Fig. 6, Pt1). Apparently depending on the type of substrate, either UvrC or Cho is better suited to carry out the 3′ incision event.
Figure 6.

Incision of substrates containing a Chol-S adduct (G1), a menthol adduct (M1), a cisplatin crosslink (Pt1), or an AAF adduct (A1). The substrates were incubated with UvrA, UvrB, and wtUvrC (first lane of each gel), or UvrC(D399A) (second lane of each gel), or Cho (third lane of each gel).
Cho Does Not Require the UvrC-Binding Domain of UvrB.
It has been shown that for 3′ incision (but not for 5′ incision), the UvrC protein needs to interact with the C-terminal domain of the UvrB protein (8, 9). The preincision complex formed by UvrB*, a proteolytic fragment of UvrB lacking this domain, can therefore not be incised at the 3′ incision position by UvrC (Fig. 7, lane 2). The same preincision complex containing UvrB*, however, is incised by Cho with the same efficiency as the wild-type UvrB–DNA complex (lanes 3 and 4). Apparently the Cho interacts with UvrB by means of another protein domain, which might be the reason that the incision position of Cho is different from that of UvrC. The different modes of UvrB binding of Cho and UvrC might also cause the difference in incision efficiencies between UvrC and Cho on the different damaged substrates.
Figure 7.

The effect of the UvrB* protein on incision of the 50-bp substrate containing a cholesterol lesion (G1) (lanes 2–4) and the combined action of Cho and UvrC(R42A) on the same substrate (lanes 5–8). The position of the incision products resulting from the 3′ incision by Cho (35 nt), the 5′ incision by UvrC (19 nt), and the extra 5′ incision (12 nt) are indicated.
Substrates Incised by Cho Can Be Further Processed by UvrC.
As shown above, some damages are incised much better by Cho than by UvrC. For an in vivo relevance of Cho incision in nucleotide excision repair, however, it is essential that the damaged DNA that is incised by Cho at the 3′ side can be further processed by incision at the 5′ side by the UvrC protein. To test this theory, we incubated substrate G1 with a mixture of Cho and UvrC(R42A). The UvrC(R42A) mutant is incapable of inducing the 3′ incision, and hence the 5′ incision is also not made (Fig. 7, lane 7). The 5′ incision product is observed when UvrC(R42A) is incubated together with an equimolar amount of Cho (lane 8), showing that UvrC does make the 5′ incision after the Cho-induced 3′ incision. A similar result was found when G1 was incubated with UvrB* and a mixture of wtUvrC and Cho (not shown), confirming our previous observations (8) that UvrC can induce 5′ incision on UvrB*–DNA preincision complexes that are incised at the 3′ side. Because on substrate G1 the 3′ incision by Cho is more efficient than the 3′ incision by wtUvrC, the ultimate incision by the combined action of equimolar amounts of Cho and wtUvrC on G1 is higher than the incision by UvrC alone (compare lanes 5 and 6). This finding strongly suggests that in vivo the combined action of Cho and UvrC will also lead to a more efficient repair of some types of damage.
Discussion
In this paper we have shown that the product of the ydjQ gene, which we refer to as the Cho protein, is capable of incising UvrB–DNA preincision complexes at the 3′ side of the lesion. The Cho protein incises the DNA farther away from the lesion than the UvrC protein. Because the catalytic domains of the two proteins are very similar, this shift in incision position is most likely caused by binding of UvrC and Cho to different domains of the UvrB protein, as we could show with the truncated UvrB* protein. Incision by Cho does not require the C-terminal domain of the UvrB protein, and indeed alignment of Cho and UvrC reveals that the domain of UvrC that interacts with this part of UvrB is not present in Cho (Fig. 1).
The Cho protein is capable of making the 3′ incision only, implying that for completion of the nucleotide excision repair reaction the UvrC protein is still required. What might than be the function of the Cho protein in E. coli? A clue on Cho function comes from the observation that some damaged substrates (e.g., G1 and M1) are poorly incised by the UvrC protein. Because the same substrate is efficiently incised by Cho, the poor incision by UvrC cannot be explained by an inability to form preincision complexes on this type of damage. It must therefore be the 3′ incision reaction of UvrC itself that is inhibited by the type of lesion. We have recently proposed a model where on binding of UvrB to a lesion the damaged nucleotide is flipped out of the DNA helix, away from the protein (22). It is conceivable that such a flipped nucleotide, especially when conjugated to a very bulky group, might obstruct the functioning of the UvrC protein. Cho could then avoid this obstruction by binding to a different domain of UvrB. The opposite situation might also occur, depending on the spatial orientation of the flipped lesion, i.e., inhibition of incision by Cho but not by UvrC. Indeed, we did find that the incision of the cisplatin substrate by UvrC is somewhat more efficient than incision by Cho, and there might be other types of damage where this difference is more pronounced. The cho gene has been shown to be under control of the SOS response (11), whereas the uvrC gene is not (20). We argue that under normal conditions, i.e., without exposure to high concentrations of damaging agents, the basal levels of the UvrA, UvrB, and UvrC proteins in the cell suffice to remove the occasional lesion in the DNA. If, however, a specific lesion remains because UvrC is not able to induce 3′ incision as argued above, a replication block at this damage to which UvrB will probably remain bound will trigger the SOS response, resulting in expression of the Cho protein. The Cho protein in its turn will attempt to incise the preincision complex. When this incision is successful the UvrC protein will induce the second incision and the repair process can be completed. Indeed, the 5′ incision by UvrC does not seem to be influenced by the type of lesion, because for several different damaged substrates it has been shown that prenicking the DNA at the 3′ incision position leads to a very efficient 5′ incision by UvrC (refs. 17, 23, and 24, and unpublished results). Taken together, the combined action of UvrC and Cho broadens the substrate range of nucleotide excision repair in E. coli.
The cho gene is a very recent gene, because homologues can be found only in bacteria that are closely related to E. coli, such as Salmonella typhimurium, Salmonella enteritidis, and Klebsiella pneumoniae (Fig. 1). A search in the database of finished and unfinished genome projects revealed that in three more distant bacterial species, other uvrC/cho homologues are present in addition to the “normal” uvrC gene. Mycoplasma hyopneumoniae contains a potential gene encoding a protein that is smaller than Cho, but which seems to contain a similar catalytic site (Fig. 1). This protein has a region of homology with the UvrB-binding domain of UvrC (Fig. 1), which could imply that the protein is binding to the same domain of UvrB as UvrC. Clostridium acetobutylicum and different species of Mycobacterium also encode proteins with homology with the 3′ catalytic domain of UvrC and Cho, next to a clear homology with the UvrB-binding domain of UvrC (Fig. 1). The C. acetobutylicum protein contains an additional domain of 125 aa, which has no obvious homology with other proteins. Interestingly, the Mycobacterium protein also contains two additional domains, one of which (the N-terminal 239 aa) has a striking homology with the ɛ proofreading subunit of DNA polymerase III. This finding could mean that in this protein a putative damage-dependent 3′ incision activity is coupled to an exonuclease activity and that in these bacterial species yet another branch of the nuclear excision repair pathway has evolved.
Acknowledgments
We thank Lars Jansen for providing the AAF-containing oligonucleotide and John Turner for synthesis of the menthol-containing oligonucleotide. Jaap van Tuyn and Martijn Visscher are acknowledged for strain constructions. This work was supported by the J. A. Cohen Institute for Radiopathology and Radiation Protection (to I.R.S.) and a European Community grant on Quality of Life and Management of Living Resources (QLG1-CT-1999-00008).
Abbreviation
- AAF
2-acetylaminofluorene
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lin J-J, Sancar A. J Biol Chem. 1992;267:17688–17692. [PubMed] [Google Scholar]
- 2.Verhoeven E E A, van Kesteren M, Moolenaar G F, Visse R, Goosen N. J Biol Chem. 2000;275:5120–5123. doi: 10.1074/jbc.275.7.5120. [DOI] [PubMed] [Google Scholar]
- 3.Sancar A. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 4.Goosen N, Moolenaar G F, Visse R, van de Putte P. In: Nucleic Acids and Molecular Biology: DNA Repair. Eckstein F, Lilley D M J, editors. Berlin: Springer; 1998. pp. 103–123. [Google Scholar]
- 5.Aravind L, Walker D R, Koonin E V. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sohi M, Alexandrovich A, Moolenaar G F, Visse R, Goosen N, Vernede X, Fontecilla-Camps J, Champness J, Sanderson M R. FEBS Lett. 2000;465:161–164. doi: 10.1016/s0014-5793(99)01690-7. [DOI] [PubMed] [Google Scholar]
- 7.Alexandrovich A, Sanderson M R, Moolenaar G F, Goosen N, Lane A. FEBS Lett. 1999;451:181–185. doi: 10.1016/s0014-5793(99)00542-6. [DOI] [PubMed] [Google Scholar]
- 8.Moolenaar G F, Franken K L M C, Dijkstra D M, Thomas-Oates J E, Visse R, van de Putte P, Goosen N. J Biol Chem. 1995;270:30508–30515. doi: 10.1074/jbc.270.51.30508. [DOI] [PubMed] [Google Scholar]
- 9.Moolenaar G F, Franken K L M C, van de Putte P, Goosen N. Mutat Res. 1998;385:195–203. doi: 10.1016/s0921-8777(97)00042-6. [DOI] [PubMed] [Google Scholar]
- 10.Blattner F R, Plunkett G, 3rd, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez de Henestrosa A R, Ogi T, Aoyagi S, Chafin D, Hayes J J, Ohmori H, Woodgate R. Mol Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- 12.Datsenko K A, Wanner B L. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachman B J. Bacteriol Rev. 1972;36:525–557. doi: 10.1128/br.36.4.525-557.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moolenaar G F, Moorman C, Goosen N. J Bacteriol. 2000;182:5706–5714. doi: 10.1128/jb.182.20.5706-5714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visse R, de Ruijter M, Moolenaar G F, van de Putte P. J Biol Chem. 1992;267:4820–4827. [PubMed] [Google Scholar]
- 16.Moolenaar G F, Schoot Uiterkamp R, Zwijnenburg D A, Goosen N. Nucleic Acids Res. 1998;26:462–468. doi: 10.1093/nar/26.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moolenaar G F, Monaco V, van der Marel G A, van Boom J H, Visse R, Goosen N. J Biol Chem. 2000;275:8038–8043. doi: 10.1074/jbc.275.11.8038. [DOI] [PubMed] [Google Scholar]
- 18.Visse R, de Ruijter M, Brouwer J, Brandsma J A, van de Putte P. J Biol Chem. 1991;266:7609–7617. [PubMed] [Google Scholar]
- 19.Jansen L E T, Verhagen R A, Brouwer J. J Biol Chem. 1998;273:33111–33114. doi: 10.1074/jbc.273.50.33111. [DOI] [PubMed] [Google Scholar]
- 20.Moolenaar G F, van Sluis C A, Backendorf C, van de Putte P. Nucleic Acids Res. 1987;15:4273–4289. doi: 10.1093/nar/15.10.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moolenaar G F, Bazuine M, van Knippenberg I C, Visse R, Goosen N. J Biol Chem. 1998;273:34896–34903. doi: 10.1074/jbc.273.52.34896. [DOI] [PubMed] [Google Scholar]
- 22.Moolenaar G F, Höglund L, Goosen N. EMBO J. 2001;20:6140–6149. doi: 10.1093/emboj/20.21.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin J-J, Phillips A M, Hearst J E, Sancar A. J Biol Chem. 1992;267:17693–17700. [PubMed] [Google Scholar]
- 24.Zou Y, Walker R, Bassett H, Geacintov N E, Van Houten B. J Biol Chem. 1997;272:4820–4827. doi: 10.1074/jbc.272.8.4820. [DOI] [PubMed] [Google Scholar]