

Abstract
CCR3 is a chemokine receptor initially thought specific to eosinophils but subsequently identified on TH2 cell subsets, basophils, mast cells, neural tissue, and some epithelia. Because of the prominent role of these cells in allergic disease, including asthma, we generated mice deficient in CCR3 to determine its contribution in a model of allergic airway disease. Here we show that CCR3 is important for the basal trafficking of eosinophils to the intestinal mucosa but not the lung. In contrast, CCR3 disruption significantly curtails eosinophil recruitment to the lung after allergen challenge, with the majority of the eosinophils being arrested in the subendothelial space. Further, a role for CCR3 in mast cell homing has been identified; after sensitization and allergen challenge, we find increased numbers of intraepithelial mast cells in the trachea of knockout mice. Physiologically, we find that the net result of these complex cell fates after sensitization and allergen challenge is a paradoxical increase in airway responsiveness to cholinergic stimulation. These data underscore a more complex role for CCR3 in allergic disease than was anticipated.
Asthma is essentially a chronic inflammatory disease of the airways characterized by intermittent reversible airway obstruction and by bronchial smooth muscle cell hyperreactivity to various stimuli. In the last few years, it has become apparent that asthma is a multifactorial, multicellular, and complex disease. Consequently, it has become important to identify the cellular mechanisms involved in airway inflammation to identify the aetiologic factors in asthma and find better therapeutic targets.
Our interest in studying the role of CCR3 in murine models of airway hyperresponsiveness (AHR) was stimulated by a number of observations concerning human asthma. Human asthma is associated with the presence of lung and peripheral eosinophilia and peribronchial infiltration of CD4+ T cells producing type 2 cytokines, in particular IL-4, IL-5, and IL-13 (1–4), the latter of which may underlie some of the polygenic component of allergic asthma associated with elevated IgE. Second, allergic asthma is associated with two distinct phases of bronchoconstriction: an immediate phase on antigen exposure that depends on mast cell-derived mediators and a late phase bronchoconstriction occurring hours after antigen exposure that is associated with accumulation of eosinophils and TH2 lymphocytes in the airways (5, 6).
CCR3 is a β chemokine receptor abundant on eosinophils, mast cells, basophils, and a subset of human TH2-like T lymphocytes (7–11). Noteworthy, all of these cell types are essential for the development of an allergic response. CCR3 recognizes a number of chemokines, including eotaxin 1, 2, 3, RANTES, MIP-1α, as well as MCP-2, 3, and 4. Among these, eotaxin and RANTES have been shown to induce eosinophil migration both in vitro and in vivo in humans and various animal models (7, 12–16). In addition, expression of most of these ligands has been associated with asthma, and recent studies in human asthmatics make CCR3 an attractive target for therapeutic intervention (17–20).
Materials and Methods
Animal Studies.
These were performed according to institutional and National Institutes of Health guidelines for animal use and care. All experiments described were performed by using homozygote CCR3−/− and wild-type littermates, which were third, and seventh generation BALB/c backcrossed mice.
Immunization and Challenge Protocol.
Mice were immunized with 10 μg of ovalbumin (OA) and 1.125 mg of aluminium hydroxide (Imject Alum, Pierce) in 0.2 ml of sterile saline i.p. on days 0, 7, and 14. Sham-immunized mice received aluminium hydroxide alone. On days 21–24, mice were exposed to aerosolized OA (5%) or saline for 40 min.
Analysis of Cells in Blood and Bronchoalveolar Lavage (BAL).
Cell numbers in blood and BAL were analyzed 6 and 24 h after the last aerosol challenge, as described (21). Eosinophil numbers in tissues were determined by measuring eosinophil peroxidase as described (13).
Determination of Airways Hyperresponsiveness in Conscious Animals.
AHR was measured in unrestrained conscious animals by using barometric plethysmography (21, 22) (Buxco Electronics, CT). AHR is expressed as the fold increase in Penh (enhanced pause).
Determination of Airways Hyperresponsiveness in Anaesthetized Animals.
The pulmonary parameters GL (pulmonary conductance) and Cdyn (pulmonary compliance) were measured as described (21, 23).
Histology.
Tissue samples were obtained and immediately fixed in 4% paraformaldehyde as described (24). The tissues were embedded in JB4 glycolmethacrylate, sectioned at 2-μm thickness, and placed on glass slides. The Congo red histochemistry and chloroacetate esterase cytochemistry procedures were used to identify eosinophils and mast cells, respectively (24).
Results
Generation of CCR3-Deficient Mice.
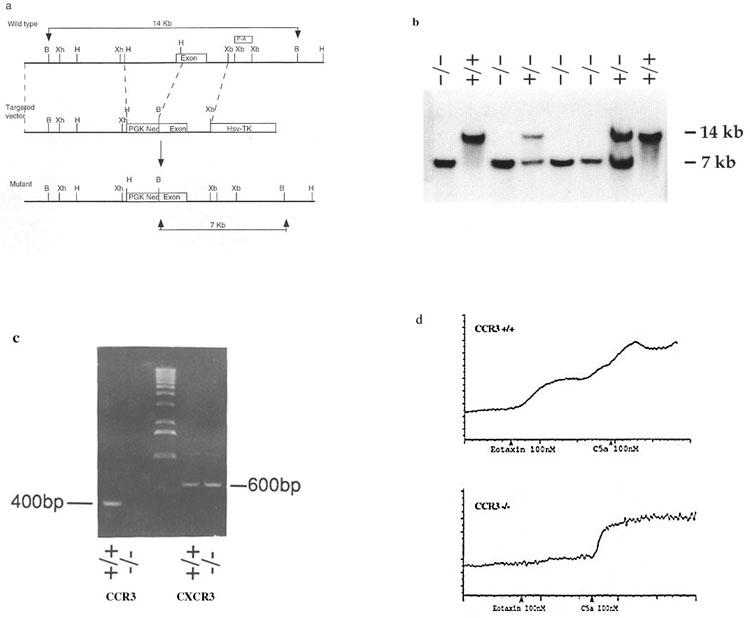
We deleted the CCR3 locus in the germ line of mice through homologous recombination by using standard techniques. The targeting vector and map is shown in Fig. 7a (which is published as supporting information on the PNAS web site, www.pnas.org). Mice with a targeted disruption of the CCR3 allele were identified through Southern analysis of BamHI digested tail DNA (Fig. 7b). To confirm that CCR3 was not present in the targeted animals, reverse transcription–PCR analysis was performed on mouse bone marrow mRNA. Fig. 7c shows absence of CCR3 mRNA in CCR3-deficient mice, but presence of CXCR3 in both wild-type and CCR3 knockout animals. In addition, ribonuclease protection assay confirmed the absence of CCR3 mRNA in thymus, spleen, and lung tissue of knockout animals (data not shown). CCR3−/− mice were fertile, born at the expected Mendelian ratios, and grossly normal when maintained under specific pathogen-free conditions. Further, hematologic parameters, including leukocyte differential counts, platelet, and hematocrit were indistinguishable from wild-type littermates as well.
To confirm loss of functional CCR3 receptor expression, CCR3-deficient and wild-type littermates were backcrossed into IL-5 transgenic mice (25) and IL-5/CCR3−/− and IL-5/CCR3+/+ offspring bled for cells (26) for calcium mobilization analysis. Eotaxin induced increased intracellular calcium responses in peripheral white blood cells from IL-5/CCR3+/+ but not IL-5/CCR3−/− mice, confirming loss of functional receptor activity (Fig. 7d). C5a induced comparable responses in both wild-type and CCR3 knockout animals.
A Central Role for CCR3 in Regulating the Basal Trafficking of Eosinophils.
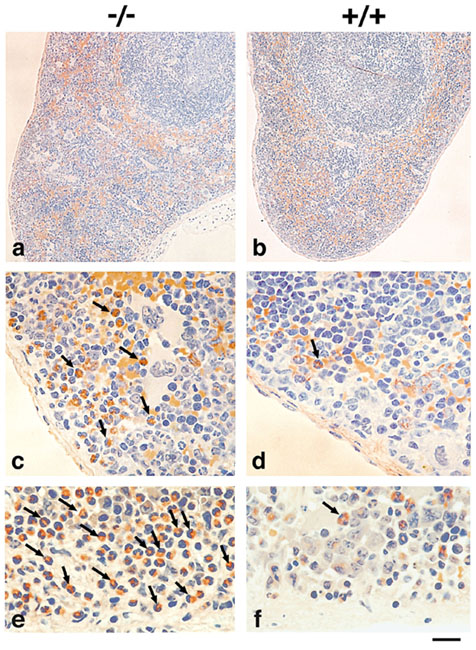
Eotaxin, one of the many CCR3 ligands, has been shown to be important for regulating the baseline levels of eosinophils in the thymus and jejunum (27). The role of CCR3 was therefore examined in eosinophil homing. When eosinophils were quantified in particular tissue compartments, CCR3-deficient mice were found to have a significant 7-fold reduction in the basal eosinophil content of the small intestine, concurrent with a 6-fold increase in the spleen. Lung tissue and thymus content of eosinophils were unchanged (Table 1). Thus, CCR3 seems to be instrumental in the basal trafficking of eosinophils to the mucosal tissues of the gastrointestinal tract but not the lung. Histological analysis of spleen confirmed that there was a marked eosinophilia present in the spleens of CCR3-deficient mice (Fig. 8 a and c, which is published as supporting information) when compared to their littermate controls (Fig. 8 b and d). Closer examination revealed that these eosinophils were located in and around the red pulp areas of the tissue, particularly under the capsule and out in the peripheral sinusoids of the spleen (Fig. 8c). Notably, that the eosinophils were evenly dispersed and not arranged in clusters implied that the increased number of cells in this organ was not because of local proliferation or haematopoiesis.
Table 1.
Basal eosinophil numbers in various tissue compartments
| Eosinophils × 105 per g tissue
|
||||
|---|---|---|---|---|
| Thymus (n = 4) | Lung (n = 7) | Spleen (n = 14) | Small intestine (n = 7) | |
| Wild type | 5.4 ± 1.6 | 4.2 ± 0.1 | 8.3 ± 2.8 | 9.0 ± 1.8 |
| CCR3−/− | 4.5 ± 0.8 | 5.5 ± 1.1 | 48 ± 9.8** | 1.3 ± 0.3** |
Tissue eosinophil content in naïve mice was measured by eosinophil peroxidase assay. Results are expressed as means ± SEM. Significant differences between wild-type and CCR3−/− mice are indicated as
, P < 0.001, as determined by unpaired Student's t test.
CCR3 Contributes to Eosinophil Trafficking in Sensitized-Challenged Lungs but Confers No Protection Against Airway Hyperresponsiveness to MCh.
To assess the physiological response of CCR3-deficient mice in a murine model, littermates were sensitized with low-dose OA in alum or sham-treated (alum alone) and then exposed over 4 days to aerosolized OA or saline, respectively. This is a relatively chronic model in which animals develop a significant pulmonary inflammatory response, consisting mainly of TH2 lymphocytes and eosinophils, and exhibit airway hyperresponsiveness to MCh challenge. In these experiments, mice were killed at 6 and 24 h after the last aerosol challenge, and eosinophil numbers in the lung, blood, and spleen were determined. At both times after allergen challenge, there was a significant increase in eosinophils in the BAL and lung tissue of wild-type sensitized challenged mice compared to that of sham-treated animals (Fig. 1 a and b). In CCR3-deficient mice, we observed a significant reduction (50–70%) in eosinophil trafficking to both the airway lumen (Fig. 1a) and lung tissue (Fig. 1b) at 6 and 24 h after allergen challenge. Interestingly, blood levels of eosinophils were comparable in both groups of animals at these same time points (Fig. 1c). Again, we observed a concurrent increase in eosinophil numbers in the spleen of CCR3-deficient mice, particularly after OA challenge, presumably reflecting that eosinophils not recruited to sensitized and challenged lung home alternatively to the spleen (Fig. 8 e and f; Fig. 1d). The effects on cell trafficking seemed to be specific to the eosinophil, because we observed no differences in the mononuclear cell or neutrophil numbers in lung lavage at these same time points (data not shown). Histological examination of the lung tissue confirmed that eosinophil migration into the airways was severely dampened in OA-challenged CCR3−/− mice (Fig. 2a) compared to that in wild-type controls (Fig. 2b). On closer inspection, we found that the blood vessels in CCR3−/− lungs were full of eosinophils, which surprisingly were located frequently under the endothelial cells on the luminal side of the elastic laminae (Fig. 2c), which was not seen in wild-type challenged mice (Fig. 2d). Therefore, CCR3−/− eosinophils are presumably able to roll, adhere, and pass through the endothelial cell layer but are unable to pass through elastic tissue out into the lung parenchyma.
Figure 1.

Eosinophil accumulation after saline or OA challenge in sham or sensitized mice. Eosinophil accumulation in BAL (a) and lung tissue (b) at 6 and 24 h after saline or OA challenge in CCR3-deficient (solid bars) or wild-type littermate (open bars) mice. Significant differences between wild-type control and CCR3−/− mice are indicated as *, P < 0.04 and **, P < 0.001, as determined by unpaired Student's t test. Results are presented as mean ± SEM (n = 11–19 mice/group). Eosinophil accumulation in blood (c) and spleen (d) at 6 and 24 h after saline or OA challenge in CCR3-deficient (solid bars) or wild-type littermate (open bars) mice. Results are presented as mean ± SEM (n = 6–7 mice/group); **, P < 0.001.
Figure 2.

Trafficking of eosinophils into the airways after allergen challenge in CCR3−/− and wild-type mice. Eosinophil accumulation is selectively reduced in the lungs of OA-challenged CCR3−/− mice (a) compared to those of wild-type littermate controls (b). On closer inspection, we find that the eosinophils in CCR3−/− mice are trapped within the blood vessels and located under the endothelial cells (c Inset). In wild-type mice, the eosinophils have migrated out into the lung tissue intermixed with lymphocytes (d). Original magnifications: a and b, ×100 μm; c and d, ×20 μm.
We used whole-body plethysmography to measure the physiological response of sensitized and sham-treated mice to inhaled MCh. Airway hyperresponsiveness was assessed 24 h after the last allergen exposure by recording respiratory pressure curves and was expressed as enhanced pause (Penh), a calculated value that correlates with measurement of airway resistance, obstruction, and intrapleural pressure in the same mouse (22). As shown in Fig. 3, sham-treated wild-type or CCR3-deficient mice displayed little or no response to inhaled MCh. When sham-treated wild-type mice are compared with sensitized/challenged wild-type littermates, airway hyperresponsiveness to inhaled MCh is markedly enhanced. CCR3-deficient allergen-challenged mice also displayed an enhanced AHR to MCh compared to sham-treated animals; however, the degree of bronchoconstriction was significantly greater than that observed in wild-type OA-challenged mice.
Figure 3.

Assessment of AHR to inhaled MCh in conscious mice. Sham-treated wild-type (○) or CCR3-deficient (●) mice were exposed to aerosolized saline, and OA-sensitized wild-type (□) or CCR3-deficient (■) mice were exposed to aerosolized OA on days 21–24. Airway responses to increasing concentrations of MCh were assessed 22–26 h after the last aerosol exposure by using barometric whole-body plethysmography. The dose–response curves of MCh-induced increases in Penh are shown. Results are expressed as the means ± SEM (n = 8–12 mice/group) of the percent increase in Penh compared with the baseline Penh values after saline exposure. Significant differences between wild-type sham-treated and wild-type sensitized/challenged mice are indicated as *, P < 0.02–P < 0.001, and significant differences between sensitized/challenged wild-type and sensitized/challenged CCR3−/− mice are indicated as **, P < 0.005, as determined by unpaired Student's t test.
To confirm these paradoxical results, we went on to assess airway responsiveness to MCh in anaesthetized tracheostomized mechanically ventilated mice. This well established system allows the simultaneous measurement of pulmonary conductance (GL) and compliance (Cdyn) in response to intravenous infusion of a bronchoconstrictor (23). Concomitantly, using two different parameters, we found similar results: allergen-challenged CCR3-deficient mice exhibited enhanced airway responses to MCh that were significantly greater than that seen in wild-type-challenged animals (Fig. 4). Interestingly, this effect seemed to be more pronounced with respect to pulmonary compliance. Sham-treated wild-type and CCR3-deficient mice displayed similar responses to MCh. Thus, despite a marked decrease in eosinophil trafficking to the lung, CCR3 disruption confers no protection but rather exacerbates MCh-induced AHR.
Figure 4.

Assessment of AHR to intravenous MCh in anaesthetized mice. Sham-treated wild-type (○) or CCR3-deficient (●) mice were exposed to aerosolized saline, and OA-sensitized wild-type (□) or CCR3-deficient (■) mice were exposed to aerosolized OA on days 21–24. Approximately 22–26 h after the last aerosol exposure, mice were anaesthetized, intubated, and mechanically ventilated and airway responses to increasing doses of intravenous MCh assessed. The dose–response curves for (a) pulmonary conductance (GL) and (b) pulmonary compliance (Cdyn) are shown. Results are expressed as the means ± SEM (wild-type, n = 10–16; CCR3−/−, n = 10–18 mice/group) of the percent minimal decrease in pulmonary conductance or compliance obtained after MCh challenge compared with the baseline value just before challenge. Significant differences between sham-treated wild-type and sensitized/challenged wild-type mice are indicated as *, P < 0.01–0.002, and significant differences between sensitized/challenged wild-type and sensitized/challenged CCR3−/− mice are indicated as **, P < 0.04, as determined by unpaired Student's t test.
In other studies, both TH2 lymphocytes and their elaborate cytokines, notably IL-4, IL-5, and IL-13, have been associated with AHR (28–31). The possibility that increased responsiveness to MCh in CCR3-deficient mice is associated with a TH2 profile cytokine expression was investigated. We prepared RNA from the lungs of OA-challenged CCR3-deficient and wild-type littermates. By reverse transcription–PCR, we determined that allergen challenge induced a comparable TH2 response, with respect to IL-4 and IL-13 levels, in both CCR3-deficient and wild-type littermate controls (Fig. 9 a and b, which is published as supporting information). In addition, serum levels of specific OA–IgE were similar (OA wild-type 556 ± 62 versus OA CCR3−/− 678 ± 131 ng/ml, n = 10–11). We can only conclude from these studies that increased airway responsiveness occurs in the absence of a TH2 bias.
Increased Intraepithelial Mast Cells Correlate with Enhanced Airway Hyperresponsiveness in CCR3-Deficient Mice.
Mast cells play a central role in the early phase of the allergic asthmatic response, but their role in late-phase response and development of allergen-induced AHR remains controversial. The expression of CCR3 on both mast cell progenitors and their mature counterparts in humans prompted us to investigate the effect of CCR3 disruption on mast cell trafficking and activation within the airways of sham and OA-challenged mice. When we examined airway tissue sections, we found that, on average, 3-fold more mast cells were present in the submucosa of the trachea and large bronchi of both sham CCR3−/− sham (not shown) and OA-challenged mice (Fig. 5a) when compared to their respective wild-type controls (Fig. 5b). Strikingly, when tracheal tissue was examined, we found that there were significantly more mast cells within the airway epithelium of OA-challenged CCR3−/− animals (Fig. 5 c and e) compared to that of wild-type challenged controls (Fig. 5d). In some sections, these mast cells appeared to be partially degranulated. Intraepithelial mast cells were not detected in sham-treated mice. Longitudinal sections of trachea were prepared from sham and OA-challenged CCR3-deficient and wild-type mice and the number of mast cells within the submucosal and intraepithelial layers determined. Cell counts confirmed that there were significantly increased numbers (range 4- to 12-fold) of intraepithelial mast cells in OA-challenged mice compared to wild-type littermates (Fig. 6, Table 2).
Figure 5.

Mast cell localization within the airways of CCR3−/− and wild-type mice after allergen challenge. Increased numbers of mast cells were found in the submucosa (a) and epithelia of the large bronchi (c and e) of OA-challenged CCR3-deficient mice compared to those of wild-type controls (b and d, respectively). The chloroacetate esterase procedure was used to identify mast cells in the tissues. Original magnifications: a and b, ×100 μm; c–e, ×20 μm.
Figure 6.

Submucosal and intraepithelial mast cell numbers in mouse trachea after saline or allergen challenge. Longitudinal sections of mouse trachea were stained with chloroacetate esterase, and the mast cell numbers in the submucosa (open bars) and intraepithelium (solid bars) were counted. Results are expressed as means ± SEM (sham, n = 4; OA challenged, n = 7) of the mast cells counted per 10 high-powered fields. The significant difference between intraepithelial mast cells in OA-challenged wild-type and OA-challenged CCR3−/− mice is indicated as *, P < 0.002, as determined by unpaired Student's t test.
Table 2.
Mast cell localization within murine trachea (mean/10hpf)
| Submucosal | Intraepithelial | |
|---|---|---|
| Sham wild type | 0.60 ± 0.10 | 0 |
| Sham CCR3−/− | 0.95 ± 0.12 | 0.17 ± 0.08 |
| OA wild type | 0.60 ± 0.06 | 0.19 ± 0.05 |
| OA CCR3−/− | 0.80 ± 0.10 | 1.00 ± 0.20* |
Results shown are expressed as the means ± SEM (sham-treated mice, n = 4; OA-challenged mice, n = 7). The significant difference between OA-challenged wild-type and OA-challenged CCR3−/− mice is indicated as
, P < 0.002, as determined by unpaired Student's t test.
Discussion
Because CCR3 expression has been associated with TH2 lymphocytes, eosinophils and mast cells, it was reasonable to assume that the gene may be central to the induction of the inflammation associated with asthma. Our studies indicate that CCR3 is important in the basal trafficking of eosinophils to the intestine but not the lung; these observations are consistent with studies done in eotaxin–knockout mice (27). In the studies reported here, we find that activated eosinophil trafficking to sensitized lungs is largely CCR3-dependent, a finding of some controversy as regards the ligand eotaxin knockouts (32, 33); this likely relates to the CCR3–ligand redundancies. Nonetheless, the data shown clearly indicate that eosinophils also have non-CCR3-dependent mechanisms of eosinophil recruitment to allergic lungs.
One of the most interesting histological findings in OA-challenged CCR3−/− airways was the presence of large numbers of presumably “trapped” eosinophils within the blood vessels (see Fig. 2c). This was not evident in OA-challenged wild-type animals (Fig. 2d). That CCR3−/− eosinophils can migrate so far suggests that there are multiple chemokine/chemoattractant factors other than eotaxin/CCR3 involved in the sequestration of eosinophils out of the bone marrow and into the blood stream. Notably, a small percentage of eosinophils do make it all the way out into the lung tissue and airway lumen of OA-challenged CCR3-deficient mice. The most striking observation in our studies was where these “trapped” eosinophils had arrested within the blood vessel: under the endothelial cells and on top of elastic lamina (Fig. 2c). Extravazation of leucocytes out of the blood vessels and to the site of inflammation involves a complex series of cell adhesion molecules stimulated by chemoattractants. Curiously, the chemoattractant(s) necessary to get the eosinophils adherent to pulmonary vascular endothelia and cause migration to the subendothelial space is (are) not acting via CCR3. However, the final extravasation through the basement membrane and eventual migration out into the tissue seems to be hindered. Our results suggest that eotaxin/CCR3 is largely required for this final migratory process. Anatomically, the subendothelial space contains smooth muscle cells and fibroblasts, both cell sources of eotaxin (34, 35). It is possible that these cells provide the chemokine gradient that enables cell migration through the vessel wall. Therefore, in the absence of CCR3, the cells remain trapped within this tissue.
One of the most striking and important observations in our studies was the presence of increased numbers of mast cells in the airways of CCR3−/− mice. Histological examination of airway tissue revealed that there were dramatic increases in the numbers of intraepithelial mast cells in CCR3-deficient mice after allergen challenge. Cell counts of chloroacetate esterase tracheal sections confirmed that there was indeed 4- to 12-fold more intraepithelial mast cells in OA challenged CCR3-deficient mice than in wild-type littermates. Mast cells and their products are important mediators in allergic reactions. Crosslinking of IgE antibodies on mast cells by antigen triggers the release of inflammatory mediators, which play an important role in the early phase of asthmatic reactions; however, the role of mast cells in the late-phase response and the associated airway hyperresponsiveness is still unclear. Studies done to date with mast–cell-deficient mice provide conflicting results and support previous hypotheses that AHR may be induced by two different pathways, one dependent on IgE-receptor activation of mast cells and the other via an IL-5-/eosinophil-dependent process (36–39).
In our studies, a 4- to 12-fold increase in intraepithelial mast cells may well contribute to the enhanced airway responsiveness observed to cholinergic stimulation. We hypothesize that sensitization and allergen challenge of CCR3-deficient mouse airways induce both the differentiation and subsequent activation of intraepithelial mast cells to release a host of mediators, which enhance airway responsiveness to MCh challenge. Immunohistochemical staining of challenged lung sections provides evidence for mast cell activation. Mast cell protease, mMCP-1, was detected in the smaller lower airways, alveolar spaces, and on alveolar macrophages from both CCR3-deficient and wild-type mice (data not shown). Although the origin of mast cell secretory products may be the larger airways, it seems that these products have trickled down into the smaller airways and alveolar spaces, as evidenced by mMCP-1 staining, and could thereby account for differences seen in lung function. Presumably, increased numbers of intraepithelial mast cells would explain the enhanced airway responsiveness seen in CCR3-deficient mice and therefore counteract any protection otherwise mediated via eotaxin/CCR3 deficiency.
Interestingly, preliminary data suggest that in the absence of intraepithelial mast cells, CCR3-deficient mice are completely protected from allergen-induced AHR (40). CCR3 knockout mice that are immunized via the epicutaneous route, subsequently boosted twice over a 7-week period, and subjected to a single aerosol allergen challenge do not develop AHR to inhaled MCh compared to wild-type controls (see ref. 41 for experimental protocol). Histological airway examination of tissue sections from these experiments reveals no increase in intraepithelial mast cells in the sections from OA-challenged CCR3-deficient mice. The observation that i.p. versus epicutaneous sensitizations have dramatic differences in CCR3-dependent phenotype, with respect to the mast cell, has profound implications for understanding the biology of the allergic response. Further, these data imply that previous studies in the murine model that use i.p. sensitization should be revisited with the epicutaneous protocol.
At present, we have no explanation why we find increased mast cells in the airways of CCR3−/− mice in our model of allergic airway disease. Notably, this increase seems to be specific to the airway, because we find no evidence for increased mast cells in the small intestine, skin, and spleen of knockout mice. What causes mast cell progenitors to home to certain tissues and differentiate is not entirely known. That human mast cells retain the expression of CCR3 throughout development (9) suggests that eotaxin/other CCR3 ligands may play an important role in mast cell homing and differentiation. Presumably, mast cell progenitors move towards the mucosal surfaces of the lung, namely the submucosa and airway epithelium, where they preferentially differentiate into mature intraepithelial mucosal mast cells and, in the absence of CCR3, are retained in the tissue. Alternative chemoattractant receptors on progenitor mast cells include CXCR2, CXCR4, and CCR5 (9). In the absence of CCR3, it is conceivable that either of these receptors could change the homing of mast cell precursors. Further studies investigating mast cell progenitors in this model may clarify these issues.
Supplementary Material
Acknowledgments
We thank Joanne Brewer and Christy Nilsson for technical assistance and the staff of Animal Resources Children's Hospital for animal care. This work was supported in part by Grant HL39759 from the National Institutes of Health. A.A.H. was supported by National Institutes of Health Grant HL10463-01.
Abbreviations
- AHR
airway hyperresponsiveness
- OA
ovalbumin
- MCh
methacholine
- BAL
bronchoalveolar lavage
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bousquet J, Chanez P, Lacoste J Y, Barneon G, Ghavanian M N, Enander I, Venge P, Ahlstedt S, Simony-Lafontaine J, Godard P, et al. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 2.Robinson D S, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley A M, Corrigan C J, Durham S R, Kay A B. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 3.Corrigan C J, Haczku A, Gemou-Engesaeth V, Doi S, Kikuchi Y, Takatsu K, Durham S R, Kay A B. Am Rev Resp Dis. 1993;147:540–547. doi: 10.1164/ajrccm/147.3.540. [DOI] [PubMed] [Google Scholar]
- 4.Minty A, Chalon P, Derocq J M, Dumont X, Guillemot J C, Kaghad M, Labit C, Leplatois P, Liauzun P, Miloux B, et al. Nature (London) 1993;362:248–250. doi: 10.1038/362248a0. [DOI] [PubMed] [Google Scholar]
- 5.Pepys J, Hutchcroft B J. Am Rev Resp Dis. 1975;112:829–859. doi: 10.1164/arrd.1975.112.6.829. [DOI] [PubMed] [Google Scholar]
- 6.Bradley B L, Azzawi M, Jacobson M, Assoufi B, Collins J V, Irani A A, Schwartz L B, Durham S R, Jeffery P K, Kay A B. J Allergy Clin Immunol. 1991;88:661–674. doi: 10.1016/0091-6749(91)90160-p. [DOI] [PubMed] [Google Scholar]
- 7.Ponath P D, Qin S, Ringler D J, Clark-Lewis I, Wang J, Kassam N, Smith H, Shi X, Gonzalo J, Newman W, et al. J Clin Invest. 1996;97:604–612. doi: 10.1172/JCI118456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daugherty B L, Siciliano S J, Demartino J, Malkowitz L, Sirontino A, Springer M S. J Exp Med. 1996;183:2349–2354. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ochi H, Hirani W M, Yuan Q, Friend D S, Ausin K F, Boyce J A. J Exp Med. 1999;190:267–280. doi: 10.1084/jem.190.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uguccioni M, Mackay C R, Ochensberger B, Loetscher P, Rhis S, LaRosa G J, Roa P, Ponath P D, Baggioloini M, Dahinden C A. J Clin Invest. 1997;100:1137–1143. doi: 10.1172/JCI119624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sallusto F, MacKay C R, Lanzavecchia A. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 12.Jose P J, Griffiths-Johnson D A, Collins P D, Walsh D T, Moqbel R, Totty N F, Truong O, Hsuan J J, Williams T J. J Exp Med. 1994;179:881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humbles A A, Conroy D M, Marleau S M, Rankin S R, Palframan R T, Proudfoot A E I, Wells T N C, Li D, Jeffery P J, Griffiths-Johnson D A, Williams T J, Jose P J. J Exp Med. 1997;186:601–612. doi: 10.1084/jem.186.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalo J-A, Jia G-Q, Aguirre A, Friend D S, Coyle A J, Jenkins N A, Lin G S, Katz H, Litchman A, Copeland N, et al. Immunity. 1996;4:1–14. doi: 10.1016/s1074-7613(00)80293-9. [DOI] [PubMed] [Google Scholar]
- 15.Rot A, Krieger M, Brunner T, Bischoff S C, Schall T J, Dahinden C A. J Exp Med. 1992;176:1489–1495. doi: 10.1084/jem.176.6.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalo J, Llyod C M, Kremer L, Finger E, Martinez-A C, Siegelman M H, Cybulsky M I, Gutierrez-Ramos J. J Clin Invest. 1996;98:2332–2345. doi: 10.1172/JCI119045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattoli S, Stacey M A, Sun G, Bellini A, Marini M. Biochem Biophys Res Commun. 1997;236:299–301. doi: 10.1006/bbrc.1997.6958. [DOI] [PubMed] [Google Scholar]
- 18.Lamkhioued B, Renzi P M, Abi-Younes S, Garcia-Zepada E A, Allakhverdi Z, Ghaffar O, Rothenberg M D, Luster A D, Hamid Q. J Immunol. 1997;159:4593–4601. [PubMed] [Google Scholar]
- 19.Nakamura H, Weiss S T, Israel E, Luster A D, Drazen J M, Lilly C M. Am J Respir Crit Care Med. 1999;160:1952–1956. doi: 10.1164/ajrccm.160.6.9811089. [DOI] [PubMed] [Google Scholar]
- 20.Teran L M, Noso N, Carroll M, Davis D E, Holgate S T, Schroder J M. J Immunol. 1996;157:1806–1812. [PubMed] [Google Scholar]
- 21.Humbles A A, Lu B, Nilsson C A, Lilly C, Israel E, Fujiwara Y, Gerard N P, Gerard C. Nature (London) 2000;406:998–1001. doi: 10.1038/35023175. [DOI] [PubMed] [Google Scholar]
- 22.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen G L, Irvin C G, Gelfand E W. Am J Respir Crit Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 23.Martin T R, Gerard N P, Galli S J, Drazen J M. J Appl Physiol. 1998;64:2318–2323. doi: 10.1152/jappl.1988.64.6.2318. [DOI] [PubMed] [Google Scholar]
- 24.Friend D S, Gurish M F, Austin K F, Hunt J, Stevens R L. J Immunol. 2000;157:1806–1812. doi: 10.4049/jimmunol.165.1.344. [DOI] [PubMed] [Google Scholar]
- 25.Dent L A, Strath M, Mellor A L, Sanderson C J. J Exp Med. 1990;172:1425–1431. doi: 10.1084/jem.172.5.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teixeira M M, Wells T N C, Lukacs N L, Proudfoot A I, Kunkel S L, Williams T J, Hellewell P G. J Clin Invest. 1997;100:1657–1666. doi: 10.1172/JCI119690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews A N, Friend D S, Zimmermann N, Sarafi M N, Luster A D, Pearlman E, Wert S E, Rothenberg M E. Proc Natl Acad Sci USA. 1998;95:6273–6278. doi: 10.1073/pnas.95.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corry D B, Folkesson H G, Warnock M L, Erle D J, Matthay M A, Wiener-Kronish J P, Locksley R M. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foster P S, Hogan S P, Ramsay A J, Matthaei K I, Young I G. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben T Y, Karp C L, Donaldson D D. Science. 1999;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 31.Grünig G, Warnock M, Wakil A E, Venkayya R, Brombacher F, Rennick D M, Sheppard D, Mohrs M, Donaldson D D, Locksley R M, et al. Science. 1999;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothenberg M E, MacLean J A, Pearlman E, Luster A D, Leder P. J Exp Med. 1997;185:785–790. doi: 10.1084/jem.185.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Loy J, Ryseck R, Carrasco D, Bravo R. Blood. 1998;92:3912–3923. [PubMed] [Google Scholar]
- 34.Ghaffar O, Hamid Q, Renzi P M, Allakhverdi Z, Molet S, Hogg J C, Shore S A, Luster A D, Lamkhioued B. Am J Respir Crit Care Med. 1999;159:1933–1942. doi: 10.1164/ajrccm.159.6.9805039. [DOI] [PubMed] [Google Scholar]
- 35.Teran L M, Mochizuki M, Bartels J, Valencia E L, Nakajima T, Hirai K, Schroder J M. Am J Respir Cell Mol Biol. 1999;20:777–786. doi: 10.1165/ajrcmb.20.4.3508. [DOI] [PubMed] [Google Scholar]
- 36.Takeda K, Hamelmann E, Joetham A, Shultz L D, Larsen G L, Gelfand E W. J Exp Med. 1997;186:449–454. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagai H, Yamaguchi S, Maeda Y, Tanaka H. Clin Exp Allergy. 1996;26:642–647. [PubMed] [Google Scholar]
- 38.Kobayashi T, Miura T, Haba T, Sato M, Serizawa I, Nagai H, Ishizaka K. J Immunol. 2000;164:3855–3861. doi: 10.4049/jimmunol.164.7.3855. [DOI] [PubMed] [Google Scholar]
- 39.Williams C M M, Galli S J. J Exp Med. 2000;192:455–462. doi: 10.1084/jem.192.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma, W., Bryce, P., Humbles, A. A., Laouini, D., Yalcindag, A., Alenius, H., Friend, D. S., Oettgen, H. C., Gerard, C. & Geha, R. S. (2002) J. Clin. Invest., in press. [DOI] [PMC free article] [PubMed]
- 41.Spergel J M, Mizoguchi E, Brewer J P, Martin T R, Bhan A K, Geha R S. J Clin Invest. 1998;101:1614–1622. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}