

Abstract
Purpose
Wiedemann–Steiner syndrome (WDSTS) is an autosomal dominant disorder with broad and variable phenotypes including short stature. This study aims to determine the long-term effect of recombinant human growth hormone (rhGH) treatment on WDSTS and summarize the phenotypes and genotypes of WDSTS.
Methods
We analyzed the clinical and genetic features of five patients with WDSTS, and comprehensively reviewed reported WDSTS diagnostic features.
Results
Four patients had short stature, one exhibited early puberty, and all exhibited distinctive facial features, intellectual disabilities, and hypertrichosis. Two patients had subnormal GH peaks. Three patients treated with rhGH for 1.5–4.9 years showed height gains (1.8, 1.1, and 1.9 standard deviations score [SDS]); patient 5 received rhGH and leuprolide for 1 year, with 0.2 SDS in height gain and controlled bone age. Five KMT2A gene variants were identified, four of which were novel. Our review (54 articles including 260 WDSTS cases) revealed that growth retardation, intellectual delay, distinctive facial features, and hirsutism are frequent findings of the condition. Among the 229 KMT2A gene variants described, frameshift variants were the most common (37.7%).
Conclusion
Our findings broaden the KMT2A gene variant, clinical, and molecular spectra used to diagnose and treat WDSTS, and highlight the crucial role of genetic testing in WDSTS diagnosis and the effectiveness of rhGH therapy.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12887-025-05751-0.
Keywords: KMT2A, Wiedemann–Steiner syndrome, Recombinant human growth hormone
Introduction
Wiedemann–Steiner syndrome (WDSTS; OMIM#605130) is a rare autosomal dominant genetic disorder that shows multiple congenital anomalies in different systems. WDSTS was first described by Wiedemann and first defined as a syndrome by Steiner and Marques [1]. Subsequent whole-exome sequencing (WES) revealed that de novo mutations in KMT2A were responsible for WDSTS in five of six studied individuals [2]. WDSTS is characterized by growth retardation, global developmental delay, unique facial features (hypertelorism, long eyelashes, broad and arching eyebrows, bulbous nose, wide nasal bridge, downward-slanting eyes, and vertically narrow palpebral fissures), hirsutism, and intellectual disability. Moreover, WDSTS is often accompanied by cardiac abnormalities, genitourinary anomalies, feeding difficulties, hypotonia, behavioral disorders, and skeletal anomalies [3].
In this study, we analyzed the phenotypic and genetic characteristics of five patients with WDSTS treated at the Children’s Hospital affiliated to Zhengzhou University, as well as the therapeutic effect of recombinant human growth hormone (rhGH). Although several case reports have described the effects of rhGH treatment on WDSTS, most focused on a short follow-up time of only a few months. To the best of our knowledge, this is the first multi-case study to comprehensively determine the effects of long-term (1-4.9 years) rhGH treatment, with one patient being followed up to 18 years of age (when the patient reached adult height). In addition, we provide a comprehensive summary of the clinical phenotypes and genotypes of WDSTS through a global literature review. This combined analysis improves our understanding of the diagnostic features of WDSTS.
Materials and methods
Participants
All patients were treated at the Children’s Hospital affiliated to Zhengzhou University between 2017 and 2023. All had genetically confirmed WDSTS. Data from all patients’ medical charts were reviewed and collected. This included demographic data and key parameters concerning WDSTS, including birth history and development, clinical features, physical examination, evolution of laboratory tests, and imaging examination. They underwent GH provocation testing using two medications in one combined tests in one day, intravenous insulin was administered (0.05–0.1 U/kg) following an overnight fast, levodopa was administered orally(10 mg/kg) and blood samples were obtained at 0, 15, 30, 60, 90 and 120 min to determine GH after the nadir glucose level; GH deficiency is defined as height more than 2 standard deviation below the mean of the corresponding age and gender, with delayed bone age, GHmax below 10 ng/mL in two growth hormone provocation tests and a subnormal IGF-I level.
Molecular genetic testing
For patient 1, nanopore sequencing was conducted at Grandomics Medical Laboratory (patient 1 previously had negative WES results). For patients 2–5, WES was conducted at the Institute of Pediatric Genetic and Metabolic Diseases in our hospital; the variants detected through WES were confirmed through Sanger sequencing of samples from the patients’ parents when the latter were available. The reference sequence used for KMT2A was NM_001197104.2. The Single-Nucleotide Polymorphism database (dbSNP, https://www.ncbi.nlm.nih.gov/snp), Genome Aggregation Database (gnomAD, http://www.gnomadsg.org), 1000Genomes (https://browser.1000genomes.org), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and Human Gene Mutation Database (HGMD, https://www.hgmd.cf.ac.uk/ac/index.php) were used for reference. SIFT, PolyPhen-2, MutationTaster, GERP, and REVEL bioinformatics software for protein function prediction were used to screen for variants that had not been previously reported and that may have effects on protein structure.
rhGH treatment
rhGH was administered at bedtime via subcutaneous injection in the umbilical region or arm at an initial dose of 0.043–0.053 mg/kg/day. Patients were followed up once every 3 months, and the rhGH dose was adjusted according to the height velocity and IGF-1 levels.
Analysis of rhGH treatment effects
During endocrine follow-up, physicians evaluated and recorded the patients’ heights and weights. In addition, scoliosis, bone age, and prime metabolic parameters, such as blood glucose and thyroid function, IGF-1, were regularly monitored. If complications occurred, rhGH treatment would be discontinued. We considered an annual height velocity (HV) < 1 cm/year or bone age > 15 years (or boys > 16 years) as the final adult height (FAH) or approximate FAH, at which point rhGH administration was stopped.
Literature review
Four electronic databases, namely PubMed, Web of Science, China National Knowledge Infrastructure, and WanFang, were searched to summarize the phenotypes and genotypes of WDSTS. The keywords used in the search were “Wiedemann–Steiner syndrome”, “WSS”, “WSDTS”, and “KMT2A”. Clinical data and KMT2A variants were obtained from case reports and cohort studies. The inclusion criteria were genetic confirmation of WDSTS and some WDSTS-related diagnostic phenotypes. Patients without available clinical data and with other genetic diseases were excluded from the review.
Results
Patients’ clinical data
The five patients included three boys and two girls aged between 3.5 and 12.1 years. The prenatal period of the five patients was normal; patients 1, 2, and 5 had normal birth and family histories; patients 3 and 4 had a history of asphyxia at birth; and the birth weights of the five patients were 3.0(P10–25), 3.0(P10–25), 2.6(P3–10), 2.4(P3–10), and 3.2(P10–25) kg, respectively. Patient 1 was admitted to the hospital at 2 years of age because of global developmental delay and received physiotherapy. Patients 1 and 4 had behavioral disorders; both had anxiety about social activities, but their parents refused a psychological assessment of the patients. Patient 3 underwent surgery after being diagnosed with tetralogy of Fallot at 6 months of age and recovered during the follow-up. Patient 4 underwent ophthalmological surgery for tear duct obstruction and strabismus. Patient 5 had a feeding disorder before the age of 2 years. The parents of all patients stated that they had poor academic records, especially in math, and their comprehension was mildly or moderately delayed, which prompted the diagnosis of an intelligence disorder. Their heights before treatment were 87 (− 3.2 standard deviation score[SDS]), 100.7 (− 3.1 SDS), 113 (− 5.4 SDS), 113.6 (− 5.2 SDS), and 142.9 cm (− 0.3 SDS), with midparental height(MPH) of 157.5 (−0.6 SDS), 178.5 (1.0 SDS), 165 (−1.3 SDS), 165 (−1.3 SDS), and 162 (0.3 SDS) cm, respectively. The dominant manifestation of patients 1–4 was short stature, which prompted their referral to our hospital. Subsequently, we observed the following special facial features in all five children: hypertelorism, long eyelashes, broad and arching eyebrows, bulbous nose, wide nasal bridge, down-slanting palpebral fissures, and vertically narrow palpebral fissures. Routine blood and urine examinations revealed normal liver and kidney function, myocardial enzyme levels, and thyroid function. According to a subsequent GH provocation test conducted on patients 1–4, GH peak levels in patients 3 and 4 were 4.0 and 7.68 ng/mL, respectively, and both had low insulin-like growth factor 1 (IGF-1) levels of 71.1(−1.3 SDS), 86.1(−1.3 SDS), 23.1 (−8.7 SDS), 192.3 ng/mL (−2.2 SDS), and 372.7(−0.3 SDS), respectively [4]. However, patients 1 and 2 exhibited normal GH levels, respectively. Patient 5 presented high levels of unstimulated luteinizing hormone (6.1 mIU/mL) and estrogen (62.3 pg/mL). Magnetic resonance imaging of the brain showed pituitary heights were 3.6 mm, 4.5 mm, 3.0 mm, 3.8 mm and 7.6 mm respectively for all patients. Patients 1 and 2 had normal bone age, patient 3 had delayed bone age, patient 5 had advanced bone age, and patient 4 refused to undergo bone age evaluation and treatment due to insufficient fund and long distance. The baseline data were summarized in Table 1. The typical features of patient 1 and 2 were shown in Fig. 1. Other clinical features and important parameters are summarized in Supplementary Table 1.
Table 1.
The baseline data and the height SDS in five patients
| Patient ID | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Gender | F | M | M | M | F |
| Birth weight (kg) | 3(P10–25) | 3(P10–25) | 2.6(P3–10) | 2.4(P3–10) | 3.2(P10–25) |
| CA/BA (years) at initial treatment | 3.5/3.5 | 5.5/5 | 12.1/10 | 11.9/11 | 10.8/12 |
| H(cm)/W (kg) at initial treatment | 87 (− 3.2 SDS)/10 | 100.7 (− 3.1 SDS)/16.2 | 113.0 (− 5.4 SDS)/17.6 | 113.6 (− 5.1 SDS)/30.5 | 142.9 (− 0.3 SDS)/27.5 |
| BMI (kg/m2) | 13.2 | 16.0 | 13.8 | 23.6 | 13.5 |
| Tanner stage | I | I | I | I | II |
| Peak GH (ng/mL) | 11.4 | 19.5 | 4.0 | 7.7 | ND |
| IGF-1 (ng/mL) | 71.1(−1.3SDS) | 86.1(−1.3SDS) | 23.1(−8.7SDS) | 192.3(−2.2SDS) | 372.7(−0.3SDS) |
| LH(U/L) | ND | ND | ND | 3.96 | 6.110 |
| FSH(U/L) | ND | ND | ND | 3.12 | 8.43 |
| E2(pg/mL) | ND | ND | ND | < 5.00 | 62.30 |
| T(ng/mL) | ND | ND | ND | < 0.0250 | 0.286 |
| Pituitary height (mm) | 3.6 | 4.5 | 3.0 | 3.8 | 7.6 |
| Pre-dose of rhGH (mg/kg/day) | 0.050 | 0.053 | 0.043 | ND | 0.053(combined with GnRHa) |
| Treatment duration (years) | 4.5 | 1.5 | 4.9 | ND | 1 |
| CA/BA (years) at last examination | 9/8.5 | 7/6.5 | 18.6/16 | ND | 11.8/12 |
| Height increase (SDS) | 1.8 | 1.1 | 1.9 | ND | 0.2 |
| height velocity at first year(cm) | 9.7 | 9.9 | 8.5 | ND | 8 |
| Annual height velocity (cm/year) | 7.8 | 8.9 | 7.8 | ND | 8 |
| H(cm)/age (years) at last examination | 125.9(−1.4 SDS)/9 | 114(− 2.0 SDS)/7 | 157.9(− 3.5 SDS)/18.6 | ND | 150(− 0.1 SDS)/11.8 |
| MPH(cm) | 157.5(−0.6 SDS) | 178.5(1.0 SDS) | 165(−1.3 SDS) | 165(−1.3 SDS) | 162(0.3 SDS) |
CA Chronological age, BA Bone age, E2 Estradiol, FSH Follicle-stimulating hormone, MPH Midparental height, GH Growth hormone, H Height, IGF-1 Insulin-like growth factor 1, LH Luteinizing hormone, rhGH Recombinant human growth hormone, SDS Standard deviation score, T Testosterone, W Weight, ND No date
Fig. 1.
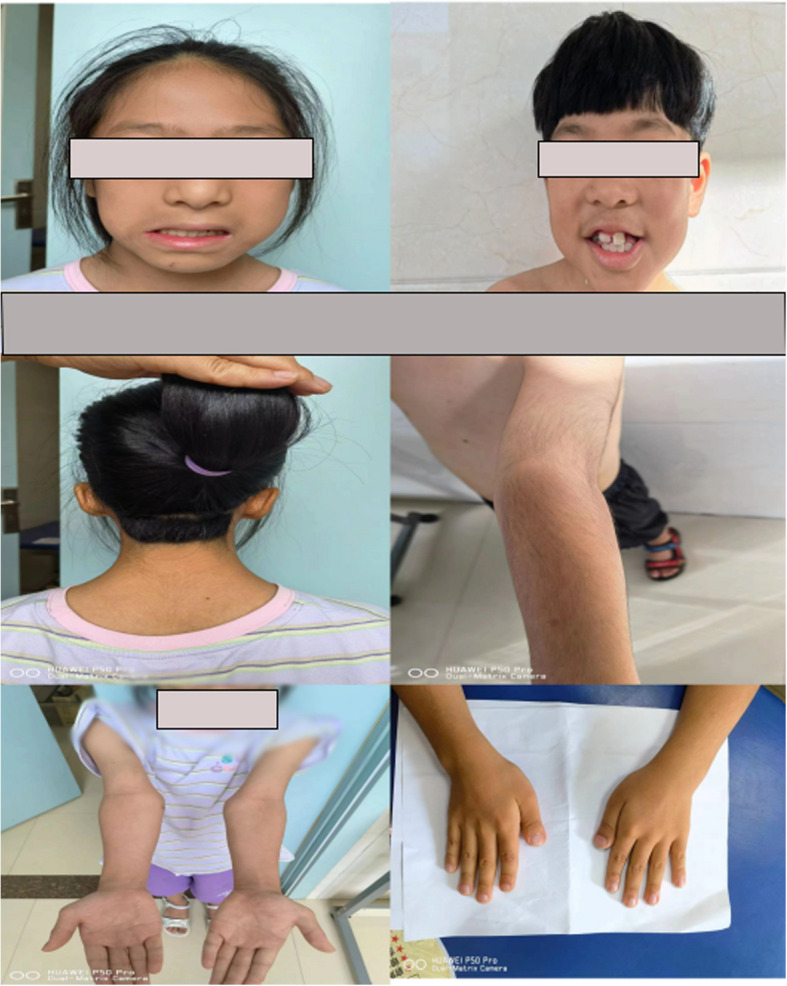
The typical features of patient 1 and 2
rhGH treatment was started at an initial dose of 0.043–0.057 mg/kg/day and lasted 1–4.9 years for patients 1, 2, 3, and 5; patient 5 was also treated with leuprolide. Following treatment, the height SDS increased for four patients (Table 1); the patients’ specific heights are shown in Fig. 2 during treatment. Following rhGH therapy for 4.9 years, until the age of 17 years, patient 3 exhibited a height improvement from − 5.4 to − 3.5 SD, reaching 157.9 cm at 18.6 years of age, which coincided with the MPH. Patients 1–3 achieved the annual HV of 7.8 − 8.9 cm/year. Patient 5 achieved the ideal HV of 8 cm/year and controlled bone age. None of the patients had complications. Patient 4 refused treatment, and the follow-up was interrupted.
Fig. 2.

Specific heights at follow-up for patients during rhGH treatment
All patients were treated with rhGH at initial time in figure, patient 1 stop treatment because she can not visited hospital during COVID-19 and restart treatment after COVID-19, patient 3 stop rhGH when his bone age > 15 years. Numbers in parentheses indicate age(Y) and height(cm). P1 represents patient 1, P2 represents patient 2, P3 represents patient 3, P5 represents patient 5
Novel variants in molecular genetic testing results
Molecular genetic testing showed that all five patients had heterozygous mutations in the KMT2A gene: Del:chr11:118,353,970–118,356,519 (including 9–10 exons), c.11081 del(p.K3694Sfs*3), c.5803-3 T > G, c.6379 C > T(p.R2127*), and c.8588 dup(p.M2863Ifs*6). None of the parents who were tested exhibited these mutations. Moreover, none of these variants has been previously reported in either the searched databases or related literature, except for c.6379 C > T(p.R2127*) [5]. Five variants were predicted using pathogenicity assessment software. According to the ACMG guidelines, the following five variants were preliminarily identified as likely pathogenic or pathogenic variants: Del:chr11:118,353,970–118,356,519 (including 9–10 exons)(PVS1 + PM2), c.11081 del(p.K3694Sfs*3) (PVS1 + PM2), c.5803-3 T > G (PM2 + PM6 + PP3 + PP4), c.6379 C > T(p.R2127*) (PVS1 + PS2 + PM2_Supporting), and c.8588 dup (p.M2863Ifs*6) (PVS1 + PM2).
WDSTS literature review
As of August 2023, 54 articles related to WDSTS and involving 260 patients had been published, including 10 articles published in Chinese [2, 3, 6–57]. The studies included 139 males and 121 females, with an age range spanning from birth to 45 years. The following is a summary of the clinical phenotypes described in WDSTS literature (Supplementary Table 2):
Growth retardation: 74.4% (175/235)
Distinctive facial features (e.g., Cupid’s bow lip and hypertelorism): 24.2–74.3% (32/132 to 171/230)
Neurodevelopmental abnormalities (including magnetic resonance imaging abnormalities and intellectual disability): 16.1–96.3% (19/99 to 232/241)
Hirsutism (e.g., hypertrichosis cubits and thick eyebrows): 57.8–73.7% (126/218 to 166/225)
Otolaryngological abnormalities: 26.0% (44/169)
Ophthalmological abnormalities: 35.1% (76/216)
GH hormone deficiency: 41.0% (25/61)
Skeletal anomalies (e.g., hip dislocation and short, fleshy hands): 12.7–46.8% (17/127 to 36/64)
Feeding difficulties: 60.5% (138/228)
Cardiac disorders: 35.4% (73/206)
Urological deformities: 41.7% (63/151)
Cryptorchidism: 10.2% (11/108)
Recurrent infections: 70.6% (12/17)
The distribution of these phenotypes is shown in Fig. 2A. The analysis of 260 gene variants revealed the following 229 different variants (Supplementary Table 3).
- Frameshift variants: 37.7% (98/260)
- Deletions: 22.3% (58/260)
- Duplications: 13.9% (36/260)
- Insertions and deletions: 1.5% (4/260)
Nonsense mutations: 29.2% (76/260)
Missense mutations: 21.5% (56/260)
Splicing variants: 9.6% (25/260)
Copy-number variants: 1.2% (3/260)
In-frame variants: 0.8% (2/260)
Pathogenic variants frequently occurred in exons 27, 3, and 5, with frequencies of 23.1% (60/260), 19.6% (51/260), and 13.8% (36/260), respectively. The distribution of these variants is shown in Fig. 2B and C. A summary of the 54 papers is provided in Supplementary Table 4.
The X-axis represents the number of participants and the Y-axis represents the clinical features. The colored boxes indicate affected individuals, the dark gray boxes indicate unaffected individuals, and the light gray boxes denote data not available in Fig. 3(A); The localization of the variants in common exons can be seen in Fig. 3(B), whereas Fig. 3(C) shows different variants detected in the gene.
Fig. 3 .

Clinical Phenotypes, Variant Distribution, and Spectrum in 260 patients
The X-axis represents the number of participants and the Y-axis represents the clinical features. The colored boxes indicate affected individuals, the dark gray boxes indicateunaffected individuals, and the light gray boxes denote data not available in (A); The localization of the variants in common exons can be seen in (B), whereas (C) shows different variants detected in the gene
Discussion
WDSTS morbidity ranges from 1 in 100,000 to 1 in 250,000 people [43]. According to a summary of the clinical phenotypes of 260 children, the most common symptoms were growth retardation, intellectual disability, delayed motor or speech development, and distinctive facial features (including hypertelorism, wide nasal bridge, down-slanting palpebral fissures, and hirsutism). Rare phenotypes include sparse hair [35], ectropion of the lateral third of the lower eyelids [34], wide thumbs [39], absent palmar creases [42], pancytopenia, and increased number of eosinophils [25, 55]. The majority of clinical presentations in the five patients in our study were consistent with these common phenotypes. Rare phenotypes in the five patients included neck webbing in patients 1 and 2, and multiple facial nevi in patient 3. In addition, clinical phenotypes varied with age. In neonates and infants, growth retardation, hypotonia, delayed motor or speech development, feeding difficulties, recurrent infections, and accompanying malformations, such as cardiac disorders and sacral dimples, were predominant, while short stature, intellectual disability, distinctive facial features, hirsutism, and mental behavioral disorders were predominant in older children. Genetic testing is necessary for early diagnosis because of the complex clinical phenotypes. Several severe phenotypes, such as abnormal fetal heart tracing accompanying acidosis, persistently poor spontaneous respiratory effort [6], third-degree atrioventricular block, and immune defects, have been reported [14]. Thus, early diagnosis and symptomatic treatment are critical for avoiding poor prognosis.
The KMT2A gene is located on chromosome 11q23, which contains 36 exons and encodes 3,969 amino acids, forming a protein that contains 18 functional domains, the main function of which is to modify the methylation of lysine on histone proteins or DNA, thus regulating the transcription of genes [58]. The KMT2A gene is expressed in many tissues and plays an important role in embryonic, hematopoietic, and neural development [59]. KMT2A truncated variants, such as frameshift variants, nonsense mutations, splice mutations, and loss of large segments, accounted for 77.7% (202/260) of the 260 variants, causing loss of function of lysine methyltransferase 2A, which results in haploinsufficiency [2]. In addition, 53.6% (30/56) of missense mutations occurred in the CXXC domain (zinc finger domain), 10.7% (6/56) in the plant homology domain, 7.1% (4/56) in the nuclear localization signal domain, and 3.6% (2/56) in the trans activator domain. Pathogenic missense mutations are mainly enriched in the CXXC domain, which is related to the main function of the CXXC domain of binding to the CpG island in the promoter [60]. The functionality of proteins involved in binding to CpG islands is impacted by both truncated variants and pathogenic missense mutations in the CXXC domain; however, the effect on the expression of downstream genes remains unknown [60]. Furthermore, over 50% of these variants were concentrated in exons 27, 3, and 5, with exon 27 having the highest incidence, probably because it is the longest exon of the gene and contains 4,249 bases. The five patients in this study exhibited a 2.5 kb deletion, and two frameshift, one splice, and one nonsense mutations, which are typical truncated variants that severely affect the function of KMT2A. A segment deletion observed in patient 1 is very rare in WDSTS, and only three cases have been previously reported, one of which had a deletion in exons 27–28, while the other two had a deletion in exons 2–10. The latter presented dislocation of the hip, and one case presented a dental abnormality; however, one of the cases with a deletion in exons 2–10 was not described in detail. Patient 1 exhibited dental abnormalities and cubitus valgus; therefore, we speculate that segment deletions may have a greater effect on the skeletal system [6, 40, 47]. Patient 2 had a variant site in exon 30 located in the phenylalanine-tyrosine-rich C-terminal domain, which is important for the structural maintenance of KMT2A [61]. This was not reported for exon 30, which expands the KMT2A variation spectrum.
WDSTS has overlapping phenotypes with other syndromes characterized by developmental delay and intellectual disability, such as Coffin–Siris, Noonan, Cornelia de Lange, Rubinstein–Taybi, and Kabuki syndromes. However, these syndromes can be initially distinguished based on the following typical characteristics:
Coffin–Siris syndrome: Hypoplasia of the distal phalanges or nails of the fifth and additional fingers, wide mouth, thick upper lip, and ectropion of the lower lip [62].
Noonan syndrome: High frequency of congenital heart defects, triangular face, small chin, and fullness of the upper eyelid [63].
Cornelia de Lange syndrome: Small hands or limb reduction defects, arched eyebrows, long philtrum, and wide incisors [64].
Rubinstein–Taybi syndrome: Wide thumbs and multiple distal phalanges, arched eyebrows, downward-slanting eyelids, and a grimacing smile [65].
Kabuki syndrome: Long eyelid fissures, lower palpebral eversion, arched eyebrows, and cutaneous hemangiomas [66].
Nevertheless, the diagnosis of these syndromes is ultimately based on genetic testing. In this study, patients 2–5 were diagnosed through WES. Patient 1 initially showed no meaningful WES results at 4 years of age; however, subsequent nanopore sequencing revealed heterozygous deletion of exons 9–10 of the KMT2A gene. This suggests that copy-number variants should be considered when WES results are inconclusive, and effective testing methods should be selected for precise detection.
Currently, the treatment of WDSTS is mainly symptomatic, including rhGH intervention for short stature; physical and occupational therapies for delayed motor and speech development; surgical intervention for malformations in the cardiac, musculoskeletal, head and neck, urogenital, and other systems; and early psychological intervention or psychoactive drugs for mental behavioral disorders. WDSTS is associated with GH deficiency [3, 22, 29, 44, 47], which hinders KMT2A histone lysine methyltransferase activity, thereby affecting gene transcription and cell proliferation [67], and may ultimately lead to pituitary hypoplasia. Otherwise, zebrafish experiments have revealed that KMT2A variants can lead to the premature differentiation of neuronal progenitor cells, potentially impacting pituitary development [68]. Additionally, case reports have found children with KMT2A variants exhibiting pituitary imaging abnormalities, such as ectopic posterior pituitary and pituitary stalk interruption syndrome [69]. In this study, pituitary screening was conducted on all five pediatric cases, revealing that the pituitary heights of patient 3 and 4 were 3.0 mm and 3.8 mm, respectively, both of which are lower. than normal pituitary height for boys and girls over 6 years old, which is (4.56 ± 0.89) mm and (4.26 ± 1.25) mm [70]. Therefore, children with WDSTS and short stature, especially those with GH deficiency, can be treated with rhGH as it enables achieving a normal adult height.
In our study, four patients were admitted to our hospital because of short stature, with one experiencing early and rapid puberty. After rhGH therapy, patient 3 showed notable height improvement, and patients 1 and 2 achieved improved annual growth rates. Treatment with rhGH combined with GnRHa effectively controlled bone age in patient 5. Previous studies have reported improved height in more than 10 children after rhGH treatment; the growth rate increased to 8–10 cm/year. [3, 20, 29, 39, 44, 47]. Additionally, other studies have documented a female reaching 150.6 cm and a male reaching 164.8 cm (the target height for males was 171.7 cm) after 15 months and 12 years of rhGH or combined rhGH and GnRHa therapy, respectively [19, 39]. In China, the height of a female patient reached 150 cm after 3.6 years of combined rhGH and GnRHa treatment [49]. For children with precocious puberty or advanced bone age, treatment with rhGH combined with GnRHa may be beneficial; however, a larger patient population is needed to confirm this curative effect. Recently, large cohort studies have explored neurocognitive and psychobehavioral disorders in children with WDSTS to address these comorbidities [71–73]. One patient exhibited progress in their severely delayed speech and social disorders following physical and occupational therapy, suggesting that early intervention in children with neurological and psychiatric disorders may improve their quality of life and learning abilities.
In conclusion, WDSTS diagnosis should be considered when growth retardation is accompanied by intellectual disorders, distinct facial features, or hypertrichosis. According to the condition of the patient, the following key examinations can be conducted: provocative GH testing; blood, skeletal, and otolaryngological system examinations; immune function testing; ophthalmic testing; cardiac and abdominal ultrasonographies; brain magnetic resonance imaging; and psychobehavioral assessment. This study highlights the need for genetic testing to achieve a definitive diagnosis of WDSTS. Moreover, genetic testing can help patients receive effective treatments, such as rhGH therapy, surgical correction, rehabilitative training, and psychological interventions, to enhance their quality of life.
Limitation
This study is retrospective and has a limited sample size. There is a possibility of false positives in the growth hormone stimulation test used to diagnose GHD (Growth Hormone Deficiency), and the test has low specificity and poor reproducibility. Moreover, the cutoff value for diagnosing growth hormone deficiency in our center is 10 µg/L, while some international centers adopt a cutoff value of 7 µg/L. The peak GH level of patient 2 between these two values, which may introduce some controversy regarding the diagnosis of GHD.
Supplementary Information
Acknowledgements
This work was supported by the Medical Science and Technology Research Project of Henan Province (Grant number LHGJ20200613) and the Zhengzhou Science and Technology Beneficiary Program Project (Grant number 2022 KJHM0005).
Abbreviations
- CA
Chronological age
- BA
Bone age
- E2
Estradiol
- FAH
Final adult height
- FSH
Follicle-stimulating hormone
- HV
Height velocity
- GH
Growth hormone
- H
Height
- IGF-1
Insulin-like growth factor 1
- LH
Luteinizing hormone
- MPH
Midparental height
- rhGH
Recombinant human growth hormone
- SDS
Standard deviation score
- T
Testosterone
- WDSTS
Wiedemann–Steiner syndrome
- WES
Whole-exome sequencing
Authors’ contributions
Study conception and design were performed by Yaodong Zhang and Yongxing Chen. Patient treatment and clinical data collection were performed by Yongxing Chen, Haiyan Wei, Yingxian Zhang and Jiajia Chen. Literature and clinical data collection were performed by Jiaqian Hu and Xi Wang. Literature collection and clinical data reorganization and analysis were performed by Mengqin Wang and Zixia Zhang. Data and gene variation analyses were performed by Shuxian Yuan and Yixuan Zhao. The first draft of the manuscript was written by Mengqin Wang and was revised by Zixia Zhang, Yaodong Zhang and Yongxing Chen. All authors read and approved the final manuscript.
Funding
This work was supported by the Medical Science and Technology Research Project of Henan Province (Grant number LHGJ20200613) and the Zhengzhou Science and Technology Beneficiary Program Project (Grant number 2022 KJHM0005).
Data availability
Data is provided within the manuscript and supplementary information files.
Declarations
Ethics approval and consent to participate
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of the Children’s Hospital Affiliated to Zhengzhou University (No. 2024-K-089).
Informed consent was obtained from the parents or legal guardians of any participant included in the study.
Consent for publication
The authors affirm that the participants’ parents provided informed consent for publication of their data.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yaodong Zhang, Email: syek@163.com.
Yongxing Chen, Email: cyx75@126.com.
References
- 1.Steiner CE, Marques AP. Growth deficiency, mental retardation and unusual facies. Clin Dysmorphol. 2000;9:155–6. [DOI] [PubMed] [Google Scholar]
- 2.Jones WD, Dafou D, Mcentagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, et al. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet. 2012;91:358–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheppard SE, Campbell IM, Harr MH, Gold N, Li D, Bjornsson HT, et al. Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome. Am J Med Genet A. 2021;185:1649–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao B, Peng Y, Song W, Peng X, Hu L, Liu Z, Liu Z, Gong C, Ni X. Pediatric continuous reference intervals of serum insulin-like growth factor 1 levels in a healthy Chinese children population - based on PRINCE study. Endocr Pract. 2022;28(7):696–702. [DOI] [PubMed] [Google Scholar]
- 5.Calvel P, Kusz-Zamelczyk K, Makrythanasis P, Janecki D, Borel C, Conne B, et al. A case of Wiedemann-Steiner syndrome associated with a 46, XY disorder of sexual development and gonadal dysgenesis. Sex Dev. 2015;9:289–95. [DOI] [PubMed] [Google Scholar]
- 6.Mendelsohn BA, Pronold M, Long R, Smaoui N, Slavotinek AM. Advanced bone age in a girl with Wiedemann-Steiner syndrome and an exonic deletion in KMT2A (MLL). Am J Med Genet A. 2014;164A:2079–83. [DOI] [PubMed] [Google Scholar]
- 7.Strom SP, Lozano R, Lee H, Dorrani N, Mann J, O’Lague PF, et al. De novo variants in the KMT2A (MLL) gene causing atypical Wiedemann-Steiner syndrome in two unrelated individuals identified by clinical exome sequencing. BMC Med Genet. 2014;15:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunkerton S, Field M, Cho V, Bertram E, Whittle B, Groves A, Goel H. A de novo Mutation in KMT2A (MLL) in monozygotic twins with Wiedemann-Steiner syndrome. Am J Med Genet A. 2015;167A:2182–7. [DOI] [PubMed] [Google Scholar]
- 9.Steel D, Salpietro V, Phadke R, Pitt M, Gentile G, Massoud A, et al. Whole exome sequencing reveals a MLL de novo mutation associated with mild developmental delay and without ‘hairy elbows’: expanding the phenotype of Wiedemann-Steiner syndrome. J Genet. 2015;94:755–8. [DOI] [PubMed] [Google Scholar]
- 10.Stellacci E, Onesimo R, Bruselles A, Pizzi S, Battaglia D, Leoni C, et al. Congenital immunodeficiency in an individual with Wiedemann-Steiner syndrome due to a novel missense mutation in KMT2A. Am J Med Genet A. 2016;170:2389–93. [DOI] [PubMed] [Google Scholar]
- 11.Sun Y, Hu G, Liu H, Zhang X, Huang Z, Yan H, et al. Further delineation of the phenotype of truncating KMT2A mutations: the extended Wiedemann-Steiner syndrome. Am J Med Genet A. 2017;173:510–4. [DOI] [PubMed] [Google Scholar]
- 12.Miyake N, Tsurusaki Y, Koshimizu E, Okamoto N, Kosho T, Brown NJ, et al. Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin Genet. 2016;89:115–9. [DOI] [PubMed] [Google Scholar]
- 13.Min Ko JM, Cho JS, Yoo Y, Seo J, Choi M, Chae JH, et al. Wiedemann-Steiner syndrome with 2 novel KMT2A mutations. J Child Neurol. 2017;32:237–42. [DOI] [PubMed] [Google Scholar]
- 14.Bogaert DJ, Dullaers M, Kuehn HS, Leroy BP, Niemela JE, De Wilde H, et al. Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes. Sci Rep. 2017;7:3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aggarwal A, Rodriguez-Buritica DF, Northrup H. Wiedemann-Steiner syndrome: novel pathogenic variant and review of literature. Eur J Med Genet. 2017;60:285–8. [DOI] [PubMed] [Google Scholar]
- 16.Enokizono T, Ohto T, Tanaka R, Tanaka M, Suzuki H, Sakai A, et al. Preaxial polydactyly in an individual with Wiedemann-Steiner syndrome caused by a novel nonsense mutation in KMT2A. Am J Med Genet A. 2017;173:2821–5. [DOI] [PubMed] [Google Scholar]
- 17.Lebrun N, Giurgea I, Goldenberg A, Dieux A, Afenjar A, Ghoumid J, et al. Molecular and cellular issues of KMT2A variants involved in Wiedemann-Steiner syndrome. Eur J Hum Genet. 2018;26:107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mietton L, Lebrun N, Giurgea I, Goldenberg A, Saintpierre B, Hamroune J, et al. RNA sequencing and pathway analysis identify important pathways involved in hypertrichosis and intellectual disability in patients with Wiedemann-Steiner syndrome. NeuroMolecular Med. 2018;20:409–17. [DOI] [PubMed] [Google Scholar]
- 19.Feldman HR, Dlouhy SR, Lah MD, Payne KK, Weaver DD. The progression of Wiedemann-Steiner syndrome in adulthood and two novel variants in the KMT2A gene. Am J Med Genet A. 2019;179:300–5. [DOI] [PubMed] [Google Scholar]
- 20.Li N, Wang Y, Yang Y, Wang P, Huang H, Xiong S, et al. Description of the molecular and phenotypic spectrum of Wiedemann-Steiner syndrome in Chinese patients. Orphanet J Rare Dis. 2018;13:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baer S, Afenjar A, Smol T, Piton A, Gérard B, Alembik Y, et al. Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: a study of 33 French cases. Clin Genet. 2018;94:141–52. [DOI] [PubMed] [Google Scholar]
- 22.Stoyle G, Banka S, Langley C, Jones EA, Banerjee I. Growth hormone deficiency as a cause for short stature in Wiedemann-Steiner syndrome. Endocrinol Diabetes Metab Case Rep. 2018;2018:18–0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan AJS, Cytrynbaum C, Hoang N, Ambrozewicz PM, Weksberg R, Drmic I, et al. Expanding the neurodevelopmental phenotypes of individuals with de novo KMT2A variants. NPJ Genom Med. 2019;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramirez-Montaño D, Pachajoa H. Wiedemann-Steiner syndrome with a novel pathogenic variant in KMT2A: a case report. Colomb Med (Cali). 2019;50:40–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Xiang B, Chen H, Chen X, Cai T. A novel deletion mutation in KMT2A identified in a child with ID/DD and blood eosinophilia. BMC Med Genet. 2019;20:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grangeia A, Leão M, Moura CP. Wiedemann-Steiner syndrome in two patients from Portugal. Am J Med Genet A. 2020;182:25–8. [DOI] [PubMed] [Google Scholar]
- 27.Negri G, Magini P, Milani D, Crippa M, Biamino E, Piccione M, et al. Exploring by whole exome sequencing patients with initial diagnosis of Rubinstein-Taybi syndrome: the interconnections of epigenetic machinery disorders. Hum Genet. 2019;138:257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen M, Liu R, Wu C, Li X, Wang Y. A novel de novo mutation (p.Pro1310Glnfs*46) in KMT2A caused Wiedemann-Steiner Syndrome in a Chinese boy with postnatal growth retardation: a case report. Mol Biol Rep. 2019;46:5555–9. [DOI] [PubMed] [Google Scholar]
- 29.Arora V, Puri RD, Bijarnia-Mahay S, Verma IC. Expanding the phenotypic and genotypic spectrum of Wiedemann-Steiner syndrome: first patient from India. Am J Med Genet A. 2020;182:953–6. [DOI] [PubMed] [Google Scholar]
- 30.Jezela-Stanek A, Ciara E, Jurkiewicz D, Kucharczyk M, Jędrzejowska M, Chrzanowska KH, et al. The phenotype-driven computational analysis yields clinical diagnosis for patients with atypical manifestations of known intellectual disability syndromes. Mol Genet Genomic Med. 2020;8:e1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jinxiu L, Shuimei L, Ming X, Jonathan LC, Xiangju L, Wenyuan D. Wiedemann-steiner syndrome with a de novo mutation in KMT2A: a case report. Med (Baltim). 2020;99:e19813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giangiobbe S, Caraffi SG, Ivanovski I, Maini I, Pollazzon M, Rosato S, et al. Expanding the phenotype of Wiedemann-Steiner syndrome: craniovertebral junction anomalies. Am J Med Genet A. 2020;182:2877–86. [DOI] [PubMed] [Google Scholar]
- 33.Hirst L, Evans R. Wiedemann-Steiner syndrome: a case report. Clin Case Rep. 2021;9:1158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Zhang G, Lu Y, Luo X, Wu W. Trio-WES reveals a novel de novo missense mutation of KMT2A in a Chinese patient with Wiedemann-Steiner syndrome: A case report. Mol Genet Genomic Med. 2021;9:e1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo S, Bi B, Zhang W, Zhou R, Chen W, Zhao P, et al. Three de novo variants in KMT2A (MLL) identified by whole exome sequencing in patients with Wiedemann-Steiner syndrome. Mol Genet Genomic Med. 2021;9:e1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CL, Chuang CK, Chiu HC, Tu RY, Lo YT, Chang YH, et al. Wiedemann-Steiner syndrome with a pathogenic variant in KMT2A from Taiwan. Children (Basel). 2021;8(11):952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nardello R, Mangano GD, Fontana A, Gagliardo C, Midiri F, Borgia P, et al. Broad neurodevelopmental features and cortical anomalies associated with a novel de novo KMT2A variant in Wiedemann-Steiner syndrome. Eur J Med Genet. 2021;64:104133. [DOI] [PubMed] [Google Scholar]
- 38.Demir S, Gürkan H, Öz V, Yalçıntepe S, Atlı EI, Atlı E. Wiedemann-Steiner syndrome as a differential diagnosis of Cornelia de Lange syndrome using targeted next-generation sequencing: A case Report. Mol Syndromol. 2021;12:46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Fede E, Massa V, Augello B, Squeo G, Scarano E, Perri AM, et al. Expanding the phenotype associated to KMT2A variants: overlapping clinical signs between Wiedemann-Steiner and Rubinstein-Taybi syndromes. Eur J Hum Genet. 2021;29:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchanan K, Greenup E, Hurst ACE, Sunil B, Ashraf AP. Case report: 11-ketotestosterone may potentiate advanced bone age as seen in some cases of Wiedemann-Steiner syndrome. Front Endocrinol (Lausanne). 2022;13:1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iijima H, Yanagi K, Kaname T, Kubota M. Feeding disorder in a patient with Wiedemann-Steiner syndrome. Pediatr Int. 2022;64:e15203. [DOI] [PubMed] [Google Scholar]
- 42.Carman KB, Kaplan E, Aslan CN, Kocagil S, Cilinigr O, Yarar C. Wiedemann-Steiner syndrome: A rare differential diagnosis of neurodevelopmental delay and dysmorphic features. J Pediatr Genet. 2022;11:162–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H, Zhang G, Yu S, Wu W. Wiedemann-Steiner syndrome: case report and review of literature. Children (Basel). 2022;9:1545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim MR, Yoo EG, Rhie S, Seo GH, Jung MK. Growth hormone deficiency in a boy with Wiedemann-Steiner syndrome: a case report and review. Ann Pediatr Endocrinol Metab. 2023;28(Suppl 1):S25–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benítez Ríos FA, Rodríguez-Fernández LF, Arciniegas NJ, Santiago Cornier A, Carlo S. Pathogenic presentation of a variant of uncertain significance in a Puerto Rican patient witWiedemann-Steiner syndrome. Cureus. 2023;15:e37330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahly AN, Srour M, Buhas D, Scheffer IE, Myers KA. The epileptology of Wiedemann-Steiner syndrome: electroclinical findings in five patients with KMT2A pathogenic variants. Eur J Paediatr Neurol. 2023;44:46–50. [DOI] [PubMed] [Google Scholar]
- 47.Lin Y, Chen X, Xie B, Guan Z, Chen X, Li X, et al. Novel variants and phenotypic heterogeneity in a cohort of 11 Chinese children with Wiedemann-Steiner syndrome. Front Genet. 2023;14:1085210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao ZJ, Jiang Q, Chen XL, Chen Q, Ji XN, Mao YY, et al. Study of de novo point mutations in known genes among patients with unexplained intellectual disability or developmental delay. Zhonghua Yi Xue Za Zhi. 2018;98:3426–32. [DOI] [PubMed] [Google Scholar]
- 49.Shangguan HQ, Hu XY, Shen YP, et al. Wiedemann-Steiner syndrome caused by novel mutation of KMT2A gene: one case report and literature review. Chin J Endocrinol Metab. 2019;01:26–31. [Google Scholar]
- 50.Dai LF, Fang F, Tian XJ, et al. Wiedemann-Steiner syndrome caused by KMT2A gene variation in one child. Chin J Appl Clin Pediatr. 2019;13:1027–9. [Google Scholar]
- 51.Wang JL, Huang K, Wu W, Zhu MQ, Lin H, Wu DW, et al. A case of Wiedemann-Steiner syndrome caused by a novel variation of the KMT2A gene. Zhonghua Er Ke Za Zhi. 2021;59:516–8. [DOI] [PubMed] [Google Scholar]
- 52.Xue H, Feng Y, Zhang C, Ma L, Wu J, Li Q, et al. Wiedemann-Steiner syndrome due to novel nonsense variant of KMT2A gene in a case. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021;38:138–40. [DOI] [PubMed] [Google Scholar]
- 53.Wu R, Tang W, Qiu K, Zhang X, Meng Z. Identification of a novel frameshift variant in the KMT2A gene of a child with Wiedemann-Steiner syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2022;39:630–3. [DOI] [PubMed] [Google Scholar]
- 54.Yan YC, Chen BB. A case of Wiedemann-Steiner syndrome with obstructive sleep apnea syndrome. J Wenzhou Med University. 2022;52:1017–9. [Google Scholar]
- 55.Ai Q, Chen Y, Chen S. Clinical features and genetic analysis of a case of Wiedemann-Steiner syndrome due to variant of KMT2A gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2023;40:222–5. [DOI] [PubMed] [Google Scholar]
- 56.He MY, Zeng FF, Wu Y, Miao Q. A case of Wiedemann-Steiner syndrome characterized by amenorrhoea, hypertrichosis, short stature, intellectual disability. Zhonghua Nei Ke Za Zhi. 2023;62:438–41. [DOI] [PubMed] [Google Scholar]
- 57.Liu SY, Li F, Ma HW. Clinical and genetic analysis of Wiedemann-Steiner syndrome caused by KMT2A gene mutation in three cases. J Clin Pediatr. 2023;41:618–23. [Google Scholar]
- 58.Castiglioni S, Di Fede E, Bernardelli C, Lettieri A, Parodi C, Grazioli P, et al. KMT2A: umbrella gene for multiple diseases. Genes (Basel). 2022;13:514–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reynisdottir T, Anderson KJ, Boukas L, Bjornsson HT. Missense variants causing Wiedemann-Steiner syndrome preferentially occur in the KMT2A-CXXC domain and are accurately classified using AlphaFold2. PLOS Genet. 2022;18:e1010278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Song Y. Structure, function and inhibition of critical protein–protein interactions involving mixed lineage leukemia 1 and its fusion oncoproteins. J Hematol Oncol. 2021;14:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vasko A, Drivas TG, Schrier Vergano SA. Genotype-phenotype correlations in 208 individuals with Coffin-Siris syndrome. Genes (Basel). 2021;12:937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zenker M, Edouard T, Blair JC, Cappa M. Noonan syndrome: improving recognition and diagnosis. Arch Dis Child. 2022;107:1073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selicorni A, Mariani M, Lettieri A, Massa V. Cornelia de Lange syndrome: from a disease to a broader spectrum. Genes (Basel). 2021;12:1075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lacombe D, Bloch-Zupan A, Bredrup C, Cooper EB, Houge SD, García-Miñaúr S, et al. Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement. J Med Genet. 2024;61:503–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Candia F, Fontana P, Paglia P, Falco M, Rosano C, Piscopo C, et al. Clinical heterogeneity of Kabuki syndrome in a cohort of Italian patients and review of the literature. Eur J Pediatr. 2022;181:171–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ikeda D, Chi S, Uchiyama S, Nakamura H, Guo YM, Yamauchi N, et al. Molecular classification and overcoming therapy resistance for acute myeloid leukemia with adverse genetic factors. Int J Mol Sci. 2022;23:5950–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lyra A, Rodart IF, Barros L, Silva TSE, da Rocha AJ, Kochi C, Longui CA. Trio-based whole exome sequencing in patients with ectopic posterior pituitary. Front Pediatr. 2024;2(12):1334610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ritchie FD, Lizarraga SB. The role of histone methyltransferases in neurocognitive disorders associated with brain size abnormalities. Front Neurosci. 2023;10(17):989109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xi ZY, Wang SS. Research Progress on MRI characteristics of the pituitary gland in children and related diseases. Chinese Journal of Neuromedicine. 2019;18(12):1284–8. [Google Scholar]
- 71.Ng R, Bjornsson HT, Fahrner JA, Harris J. Unique profile of academic learning difficulties in Wiedemann-Steiner syndrome. J Intellect Disabil Res. 2023;67:101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ng R, Bjornsson HT, Fahrner JA, Harris J. Anxiety in Wiedemann-Steiner syndrome. Am J Med Genet A. 2023;191:437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ng R, Harris J, Fahrner JA, Bjornsson HT. Individuals with Wiedemann-Steiner syndrome show nonverbal reasoning and visuospatial defects with relative verbal skill sparing. J Int Neuropsychol Soc. 2023;29:512–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is provided within the manuscript and supplementary information files.