

Abstract
Background
Smoking is a well-established risk factor for colorectal cancer (CRC) development. However, the reliability of DNA methylation-based smoking signatures in predicting smoking status and their prognostic value in CRC remain unclear, particularly across different biological sample types.
Results
Five previously validated methylation-based smoking signatures were analyzed in 2237 CRC patients with blood-derived DNA and 2273 patients with tumor tissue-derived DNA. Blood-derived signatures showed strong correlations with self-reported smoking status, effectively differentiating current smokers from never smokers (all p < 0.0001), with excellent discriminative ability (median area under the receiver operating characteristic curve: 0.94). In contrast, tumor tissue-derived signatures exhibited much weaker associations with smoking status. Among non-metastatic CRC patients, blood-derived methylation signatures were significantly associated with increased risks of all-cause and non-CRC-related mortality, but not with CRC-specific mortality. Conversely, two tumor tissue-derived signatures demonstrated stronger associations with CRC-specific mortality compared to blood-derived signatures.
Conclusions
Blood-derived methylation-based smoking signatures are robust indicators for smoking exposure and are associated with increased mortality risk among non-metastatic CRC patients. When applied to tumor tissue, signatures showed stronger associations with CRC-specific mortality.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13148-025-01918-9.
Keywords: Smoking, DNA methylation biomarkers, Colorectal cancer, Prognosis
Background
Colorectal cancer (CRC) remains one of the most common cancers and a leading cause of cancer-related deaths worldwide [1, 2]. Smoking is a well-established risk factor for CRC, influencing both its incidence and prognosis [3–5]. According to several large patient cohort studies and meta-analyses, smoking is strongly associated with risk of mortality among patients with CRC, especially among stage I–III patients [3, 6–8]. Accurate assessment of smoking status is crucial for understanding its impact on clinical outcomes and tailoring personalized monitoring strategies.
Traditionally, smoking status has been assessed through self-reporting [9, 10], which, while widely used, is subject to bias and inaccuracies. Misreporting or underreporting smoking behaviors and smoking amount can lead to misclassification of smoking-related risks [9, 10]. In response to these limitations, several methylation-based smoking signatures derived from blood samples have been proposed to quantify smoking exposure [11–15]. These epigenetic markers offer the potential to capture cumulative exposures to smoking, providing a more comprehensive picture of an individual’s smoking history [12]. Moreover, they can reflect the biological impact of smoking more accurately, accounting for individual variations in response to smoking exposure [12].
Still, the utility of methylation-based smoking signatures in predicting self-reported smoking status and their potential impact on survival outcomes in patients with CRC is yet to be fully explored. Moreover, it is unclear whether these methylation-based scores, originally identified in blood samples, can maintain their relevance when applied to tissue samples. This study aims to investigate the predictive value of methylation-based smoking signatures in both blood and tumor tissue samples among patients with colorectal cancer. By comparing these epigenetic markers with self-reported smoking data, we seek to evaluate their reliability as indicators of smoking exposure and their associations with patient survival.
Methods
Study cohort
We conducted and reported this study according to The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement [16]. The German Darmkrebs: Chancen der Verhütung durch Screening (DACHS, English name "Colorectal cancer: chances for prevention through screening”) study is a large population-based case–control and patient cohort study on CRC conducted in the Rhine-Neckar region in the southwest of Germany from 2003 to 2021. Details of this study have been previously described [17, 18]. Briefly, German-speaking patients aged over 30 with a first histologically confirmed primary CRC diagnosis, physically and mentally capable of a one-hour interview, were recruited from 22 hospitals in the study region.
Data collection
At baseline, trained interviewers collected data on sociodemographic characteristics, lifestyle, medical history, and disease symptoms through a standardized interview. Tumor characteristics and disease stage (TNM 6th edition) were obtained from medical records [19]. Smoking history before CRC diagnosis was recorded, classifying patients as current, former (quitted at least two years before diagnosis), and never smokers, and quantifying lifetime cumulative exposure by pack-years [20].
Peripheral blood samples were collected post-interview and stored at − 80 °C. Methylation assessment of whole-blood DNA was performed in a subsample of 2242 patients diagnosed with CRC between 2003 and 2010 using the Infinium MethylationEPIC BeadChip Kit (Illumina, San Diego, CA, USA), covering over 850,000 CpG sites. Molecular analyses of tumor tissue were conducted using DNA extracted from formalin-fixed, paraffin-embedded samples. Genome-wide methylation analysis was performed on tissue DNA in a subsample of 2316 patients diagnosed with CRC between 2003 and 2013 using the Illumina Human Methylation 450 BeadChip, covering over 485,000 CpG sites.
Standardized information on CRC therapy, comorbidities, and recurrence was obtained from physicians during follow-up visits at 3, 5, and 10 years after diagnosis. Vital status, date, and cause of death were collected from local population registries and public health authorities. Only patients with complete smoking and DNA methylation data were included in this study (N = 2237 for blood samples, N = 2273 for tumor tissue samples).
Calculation of methylation-based smoking signatures
We selected five previously validated blood methylation-based smoking signatures (Fig. 1) [11–15]. Four of these used methylation scores calculated as a linear combination of smoking-associated CpGs [12–15], while one predicted smoking status (current, former, and never) based on 121 CpGs and sex [11]. These signatures contained between 4 and 233 CpGs, with one (cg05575921) common to all and four (cg05951221, cg06126421, cg06644428, cg21566642) shared by three. Most CpGs were unique to each signature (Fig. 1).
Fig. 1.
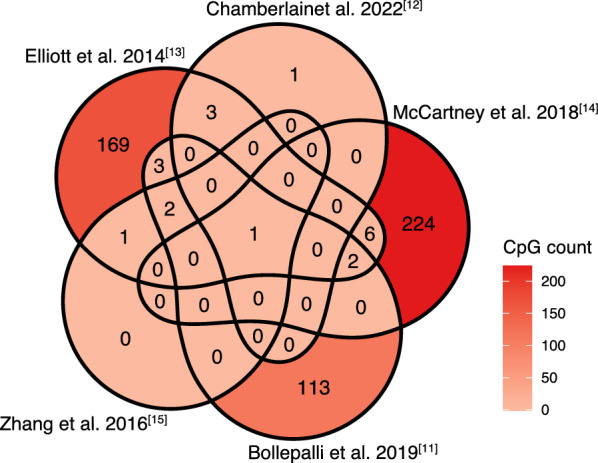
Overlapping CpGs of proposed blood methylation-based smoking signatures
The raw DNA methylation data file from the iScan array scanner was processed using the ‘minfi’ R package. Missing CpG data were imputed with nearest averaging multiple imputation. For each signature, we used the same normalization methods as those used in the original studies that developed the respective score (Supplement eTable 1) [11]. We did not apply additional CpG filtering beyond the original signature definitions. Instead, we extracted the CpG sites corresponding to each signature, based on their availability in our dataset. No further feature selection or filtering was performed.
We evaluated the coverage of CpG sites for each signature on both methylation platforms (450k and 850k arrays) and summarized the proportion of available CpGs per signature (Supplement eTable 1). All CpG sites required for the five signatures were available on the 450k array used for tissue sample. For the 850k array used for blood sample, CpG coverage was complete for two signatures (100%) [12, 14], while two others showed high but incomplete coverage, with 93% (174/187 CpGs) [13] and 91% (111/121 CpGs) [11] available, respectively. For the 4-CpG signature by Zhang et al. [15], only two CpGs (50%) were present on the 850k array.
The ‘EpiSmokEr’ R package [11] was used to predict smoking status and two smoking scores [13, 15]. The other two scores [12, 14] were calculated using linear equations provided by the original publications. The distributions of the five smoking signatures, as well as the individual CpG sites included in these signatures, were compared between blood and tumor tissue samples.
Statistical analysis
All analyses were stratified by the source of DNA methylation data. Primary analyses focused on blood (N = 2237) and tumor tissue (N = 2273) samples, while secondary analyses used data derived from 264 adjacent normal tissue samples. Patient characteristics were summarized using descriptive statistics.
Smoking score distributions across categories of self-reported smoking status were compared using violin plots and Tukey's honestly significant difference test when normality assumptions were met, or the Wilcoxon rank-sum test otherwise. The Bonferroni method was used to account for multiple comparisons. Associations between the smoking scores and pack-years were visualized with scatter plots and measured by the Spearman correlation metrics. We additionally compared the distribution of self-reported smoking status and methylation-based smoking signatures by TNM stage.
Multivariable binary logistic regression was used to quantify the relationship between each epigenetic smoking signature and self-reported smoking status, adjusting for age, sex, body mass index (BMI) 5–14 years earlier, alcohol consumption, physical activity, use of nonsteroidal anti-inflammatory drugs, hormone replacement therapy, and prior large bowel endoscopy. McFadden’s pseudo-R2 was used to assess the explanatory power of each signature. The area under the receiver operating characteristic curve (AUROC) with 95% confidence intervals (2000 stratified bootstrap replicates) was used to measure the discriminative power. To account for the potential confounding effect of treatment-related epigenetic changes, we performed additional sensitivity analyses restricting to patients who had not received chemotherapy or radiotherapy.
Cox proportional hazards models were used to evaluate the association between smoking exposure (self-reported and methylation-predicted) and all-cause mortality. The proportional hazards assumption was checked using the scaled Schoenfeld residuals. The median follow-up time was computed using the reverse Kaplan–Meier method. Delayed-entry Cox models were used to address time differences between CRC diagnosis and sample collection [21]. Cause-specific Cox models were applied for CRC-specific and other mortality, with death from other causes considered as a competing risk. Models were adjusted for age, sex, body mass index (BMI) at CRC diagnosis, physical activity, alcohol consumption, TNM stage, tumor location, and treatment with chemotherapy or radiotherapy. The primary survival analysis included stage I-III CRC patients (blood, N = 1911; tumor tissue, N = 1959) [6, 7], with stage IV patients analyzed separately. Interaction terms between each methylation signature, age, and sex were tested for effect modification, followed by stratified analyses if required. Sensitivity analysis was performed among overlapping patients with both blood and tissue samples available.
Regression results for methylation-derived scores were standardized, with one-unit increases corresponding to one standard deviation. Participants with missing values for covariables, which were all very rare (below 2%), were excluded from regression analyses. Statistical significance was set as p value < 0.05 in two-sided testing. All analyses were performed using R version 4.2.0.
Patient and public invlvement
Patients or the public were not involved in the designing, or conduct, or reporting or dissemination plans of our research.
Results
Characteristics of the study cohort
The characteristics of patients with blood methylation data were comparable to those with tumor tissue methylation data (Table 1).
Table 1.
Characteristics of the analyzed study populations
| Characteristics | Blood sample | Tumor tissue sample | P value |
|---|---|---|---|
| (N = 2237)1 | (N = 2273)1 | ||
| Median age (IQR) | 69 (62, 77) | 70 (62, 77) | 0.573 |
| Sex | 0.807 | ||
| Female | 922 (41.2) | 946 (41.6) | |
| Male | 1315 (58.8) | 1327 (58.4) | |
| Smoking status | 0.937 | ||
| Never | 1037 (46.4) | 1048 (46.1) | |
| Former | 858 (38.4) | 883 (38.8) | |
| Current | 342 (15.3) | 342 (15.0) | |
| Pack-years of smoking2 | 0.917 | ||
| 1–10 | 391 (33.6) | 411 (33.6) | |
| 10–19 | 282 (23.5) | 291 (23.8) | |
| 20–29 | 210 (17.5) | 197 (16.1) | |
| > 30 | 308 (25.7) | 317 (25.9) | |
| Missing | 9 | 9 | |
| BMI (kg/m2) 5–14 years earlier | 0.930 | ||
| Underweight (< 18.5) | 12 (0.5) | 11 (0.5) | |
| Normal (18.5–25) | 691 (30.9) | 717 (31.5) | |
| Overweight (25–30) | 1059 (48.1) | 1069 (47.7) | |
| Obese (≥ 30) | 440 (20.0) | 446 (19.9) | |
| Missing | 35 | 30 | |
| BMI (kg/m2) at diagnosis | 0.864 | ||
| Underweight (< 18.5) | 50 (2.2) | 49 (2.2) | |
| Normal (18.5–25) | 822 (36.7) | 846 (37.2) | |
| Overweight (25–30) | 931 (41.8) | 953 (42.1) | |
| Obese (≥ 30) | 424 (19.0) | 417 (18.4) | |
| Missing | 10 | 8 | |
| Alcohol drinking (days/week) | 0.952 | ||
| Never drinker (0) | 674 (30.2) | 684 (30.2) | |
| Light drinker (1–2) | 521 (23.4) | 538 (23.7) | |
| Heavy drinker (≥ 3) | 1036 (46.4) | 1045 (46.1) | |
| Missing | 6 | 6 | |
| Physical activity (lifetime average MET-hours/week) | 0.345 | ||
| Median (IQR) | 191.1 (129.9, 277.3) | 189.8 (129.4, 269.1) | |
| Missing | 39 | 31 | |
| Use of nonsteroidal anti-inflammatory drugs | 548 (24.5) | 591 (26.0) | 0.259 |
| Use of hormone replacement therapy3 | 270 (29.3) | 275 (29.1) | 1.000 |
| Missing | 4 | 2 | |
| Prior large bowel endoscopy | 500 (22.4) | 526 (23.2) | 0.550 |
| Missing | 1 | 1 | |
| TNM stage | |||
| I | 403 (18.1) | 413 (18.2) | 0.990 |
| II | 767 (34.5) | 784 (34.6) | |
| III | 741 (33.3) | 745 (32.9) | |
| IV | 314 (14.1) | 325 (14.3) | |
| Missing | 12 | 6 | |
| Tumor location | |||
| Distal colon | 610 (27.3) | 629 (27.7) | 0.523 |
| Proximal colon | 808 (36.1) | 848 (37.3) | |
| Rectum | 818 (36.6) | 795 (35.0) | |
| Missing | 1 | 1 | |
| Treatment with chemotherapy/radiotherapy | 1084 (48.5) | 1062 (46.8) | 0.273 |
| Missing | 3 | 6 | |
IQR Interquartile range, BMI Body mass index, MET Metabolic equivalent of task, TNM Tumor, lymph node, and metastasis staging system. 1The two sample groups contain 2008 overlapping patients. 2Only among former or current smokers. 3Only among female participants
Both cohorts overlapped largely (2008 same patients), with very comparable patient characteristics (all p value > 0.1) sample size (blood: 2237; tumor tissue: 2273), median ages (blood: 69 years; tumor tissue: 70 years), percentages of male (blood: 58.8%, tumor: 58.4%), and percentages of stage IV CRC patients (blood sample: 14.0%, tumor tissue: 14.3%). Approximately 15% of patients were current smokers, and 46% had never smoked. The median follow-up time for stage I-III patients was 10.6 years (IQR 10.2–13.5) for those with a blood sample and 10.5 years (IQR 10.1–12.3) for those with a tumor tissue sample, with 10-year mortality rates of 43.6% and 50.6%, respectively.
Distribution of methylation-based smoking signatures
All five smoking scores, and nearly all constituent CpG sites, showed significantly different distributions between the two tissue types (Supplement eTable 2). With the exception of the score developed by Zhang et al. [15], tumor tissue-derived scores were significantly higher than those derived from blood samples. In addition, the smoking status predicted using tumor tissue-derived scores classified a substantially higher proportion of individuals as current smokers (74.1%) compared to the blood-derived predictor (29.8%). Stage IV patients had a higher proportion of self-reported current smokers and higher blood methylation-based smoking score developed by Zhang et al. (Supplement eFigure 1) [15].
Associations between epigenetic smoking signatures and self-reported smoking status
In patients with blood DNA methylation data, all four methylation-based smoking scores increased progressively across never, former, and current smokers (Fig. 2A, p < 0.0001). A positive correlation was found between these scores and pack-years (median Spearman correlation: 0.58, p < 0.0001; Fig. 2B), with the McCartney et al. [14] score showing the strongest correlation (0.67). In tumor DNA methylation data, a similar but weaker pattern was observed across smoking groups (Fig. 3A), with very weak correlations with pack-years (median Spearman correlation: 0.09; Fig. 3B).
Fig. 2.

A Distribution of blood-derived methylation-based smoking scores across self-reported smoking groups; B Scatterplots showing the association between blood-derived methylation-based smoking scores and self-reported pack-years
Fig. 3.

A Distribution of tumor tissue-derived methylation-based smoking scores across self-reported smoking groups; B Scatterplots showing the association between tumor tissue-derived methylation-based smoking scores and self-reported pack-years
All five blood-derived DNA methylation-based signatures were strongly associated with smoking status in the adjusted model (all p < 0.0001; Table 2), especially for current versus never smokers (median McFadden’s pseudo-R2: 0.56, range 0.37–0.70). Discrimination between current and never smokers was almost perfect (AUROC median: 0.94; range 0.92–0.96), followed by current versus former smoking (0.85, 0.75–0.88). The strongest associations were found for the two signatures developed by McCartney et al. [14] and Bollepalli et al. [11], respectively.
Table 2.
Associations between methylation-based smoking scores and self-reported smoking status
| Methylation score | Blood sample (N = 2237) | Tumor tissue (N = 2273) | ||||
|---|---|---|---|---|---|---|
| aOR (95% CI)1 | AUROC (95% CI) | R2 | aOR (95% CI)1 | AUROC (95% CI) | R2 | |
| Current vs. Never | ||||||
| Chamberlainet al. [12] | 10.15 (7.58, 13.59) | 0.92 (0.90, 0.94) | 0.50 | 1.09 (0.95, 1.26) | 0.53 (0.50, 0.57) | < 0.01 |
| McCartney et al. [14] | 27.38 (17.82, 42.06) | 0.96 (0.94, 0.97) | 0.70 | 1.45 (1.26, 1.68) | 0.58 (0.55, 0.62) | 0.02 |
| Elliott et al. [13] | 19.85 (13.55, 29.08) | 0.94 (0.93, 0.96) | 0.56 | 1.47 (1.28, 1.69) | 0.62 (0.59, 0.65) | 0.03 |
| Zhang et al. [15] | 14.52 (10.51, 20.07) | 0.94 (0.92, 0.96) | 0.57 | 1.11 (1.05, 1.18) | 0.58 (0.55, 0.62) | 0.02 |
| Bollepalli et al. [11]2 | 382.97 (163.26, 898.34) | 0.92 (0.90, 0.94) | 0.56 | 1.70 (1.19, 2.43) | 0.56 (0.53, 0.59) | 0.01 |
| Former vs. Never | ||||||
| Chamberlainet al. [12] | 2.28 (1.94, 2.67) | 0.66 (0.63, 0.68) | 0.06 | 1.08 (0.98, 1.20) | 0.53 (0.51, 0.56) | < 0.01 |
| McCartney et al. [14] | 8.50 (6.64, 10.88) | 0.80 (0.78, 0.81) | 0.22 | 1.23 (1.11, 1.36) | 0.55 (0.52, 0.57) | 0.01 |
| Elliott et al. [13] | 3.79 (3.14, 4.56) | 0.73 (0.71, 0.76) | 0.13 | 1.27 (1.14, 1.41) | 0.56 (0.54, 0.59) | 0.01 |
| Zhang et al. [15] | 3.1 (2.62, 3.66) | 0.74 (0.71, 0.76) | 0.13 | 1.07 (1.03, 1.12) | 0.54 (0.52, 0.57) | 0.01 |
| Bollepalli et al. [11]2 | 2.12 (1.66, 2.72) | 0.70 (0.68, 0.72) | 0.13 | 1.15 (0.70, 1.89) | 0.55 (0.53, 0.57) | 0.01 |
| Current vs. Former | ||||||
| Chamberlainet al. [12] | 4.75 (3.86, 5.84) | 0.85 (0.82, 0.87) | 0.32 | 0.97 (0.84, 1.12) | 0.50 (0.47, 0.54) | < 0.01 |
| McCartney et al. [14] | 6.14 (4.92, 7.67) | 0.88 (0.85, 0.90) | 0.38 | 1.08 (0.94, 1.24) | 0.54 (0.50, 0.57) | < 0.01 |
| Elliott et al. [13] | 4.86 (3.96, 5.95) | 0.85 (0.82, 0.87) | 0.30 | 1.13 (0.98, 1.30) | 0.56 (0.53, 0.60) | < 0.01 |
| Zhang et al. [15] | 4.99 (4.05, 6.15) | 0.83 (0.81, 0.86) | 0.28 | 1.03 (0.97, 1.09) | 0.54 (0.50, 0.57) | < 0.01 |
| Bollepalli et al. [11]2 | 7.16 (4.89, 10.48) | 0.75 (0.73, 0.77) | 0.18 | 1.00 (0.52, 1.92) | 0.51 (0.49, 0.54) | < 0.01 |
OR Odds ratio, CI Confidence interval, AUROC Area under the receiver operating characteristic curve. McFadden R2 measured the explained variation by blood methylation-based smoking panels. R2 stands for McFadden’s pseudo-R2. 1The multivariable logistic model was adjusted for age, sex, body mass index 5–14 years earlier, alcohol consumption, physical activity, use of nonsteroidal anti-inflammatory drugs, hormone replacement therapy, and prior large bowel endoscopy. 2The association between predicted binary smoking status (e.g., predicted current vs. never smokers) and corresponding self-reported binary outcomes was assessed
In contrast, when applying the smoking signatures in tumor methylation data, associations with smoking status were much weaker (Table 2). The point estimates in adjusted logistic models were all below 2, with small McFadden’s pseudo-R2 (≤ 0.001) and low AUROC values (≤ 0.65). In sensitivity analyses restricted to patients who had not received chemotherapy or radiotherapy, the discriminatory performance of methylation-based smoking signatures for self-reported smoking status remained consistent (Supplement eTable 3).
Comparison between self-reported and epigenetic smoking signatures in relation to mortality risk
Among 1911 stage I-III patients with blood methylation data (Fig. 4A), self-reported current smokers had a higher all-cause mortality (hazards ratio [HR] 1.23, 95% confidence interval [CI] 0.98–1.55) and non-CRC-related mortality (1.45, 1.08–1.96) compared with never smokers in the fully adjusted models. However, self-reported current smoking was not a risk factor for CRC-specific mortality. Interestingly, self-reported former smokers had a lower CRC-specific mortality compared with never smokers (0.74, 0.57–0.96).
Fig. 4.

Comparison between self-reported smoking and methylation-based smoking derived from blood and tumor tissue in relation to mortality risk among patients with stage I-III CRC. aHR = adjusted hazards ratio; CI = confidence interval; and CRC = colorectal cancer. The continuous methylation-based scores were standardized. The multivariable Cox regression model was adjusted for age, sex, BMI at diagnosis, physical activity, alcohol consumption, TNM stage, tumor location, and treatment with chemotherapy or radiotherapy
All five methylation-based smoking signatures derived from blood were associated with higher all-cause mortality and non-CRC-related mortality in fully adjusted models. The increased risk of current smokers compared with never smokers, as predicted by the methylation classifier by Bollepalli et al. [11], showed the strongest magnitude of association (all-cause mortality: 1.28, 1.07–1.54; non-CRC-related mortality 1.46, 1.15–1.85). However, significant risk increases were not observed for CRC-specific survival, except for the score developed by Zhang et al. [15] (1.12, 1.01–1.26).
In the 1959 stage I-III CRC patients with tumor methylation data (Fig. 4B), associations of self-reported smoking and mortality were similar to the group with blood samples. However, the methylation signatures were not associated with all-cause or non-CRC-related mortality when applied to tumor tissue. However, two methylation scores, developed by Elliott et al. [13] and Zhang et al. [15], respectively, were significantly associated with CRC-specific mortality (1.19, 1.06–1.33 and 1.19, 1.07–1.33, respectively).
The observed associations were similar when the four continuous methylation scores were dichotomized (Supplement eTable 4). In stage IV CRC patients (Supplement eTable 5), most methylation-based smoking scores were not associated with increased mortality risk, except for the score by Zhang et al. [15], which was strongly associated with non-CRC-related mortality when derived from tumor tissue (2.46, 1.27–4.75).
Significant interactions were observed between four blood-derived methylation scores [12–15] and sex for both all-cause mortality and non-CRC-related mortality (Supplement eTable 6). Additionally, all but the McCartney score [14], showed interactions with age in analyses of non-CRC-related mortality. In tumor tissue samples, only the score by Elliott et al. [13] showed significant interactions with both sex and age for all-cause and non-CRC-related mortality. Stratified analyses revealed higher mortality risk in male and younger patients (Supplement eTable 7). Sensitivity analyses in 2008 overlapping patients with both blood and tissue methylation data showed consistent results with the main analysis (Supplement eTable 8 and eTable 9).
Methylation-based smoking signatures derived from adjacent normal tissue
None of the five methylation-based smoking signatures showed clear associations with self-reported smoking status when applied to 264 adjacent normal tissue samples (Supplement eTable 10). The signatures were also not associated with mortality risk among stage I-III CRC patients in adjacent normal tissue (eTable 11).
Discussion
In this large case–control and patient cohort study, we assessed five validated blood methylation-based smoking signatures in predicting smoking status and mortality among CRC patients using blood and tissue samples. Methylation smoking signatures in blood strongly correlated with self-reported smoking, while associations in tumor tissue were weaker. All signatures derived from blood were associated with higher all-cause and non-CRC-related mortality in non-metastatic CRC patients, with a 4-CpG score [15] also associated with CRC-specific mortality. In tumor tissue samples, this score [15] and another 187-CpG score [13] were associated with higher CRC-specific mortality.
All five blood-derived methylation signatures demonstrated strong associations with self-reported smoking status and showed excellent discriminative ability, particularly in differentiating current smokers from never smokers. The limited overlap between CpGs across the evaluated signatures may stem from differences in study populations, measurement platforms, and statistical methods used during their development. The methylation score developed by McCartney et al. [14], which contained the highest number of CpGs (234, of which 224 are unique), performed the best. Similarly, the signatures from Zhang et al. [15], despite comprising only four CpGs, displayed almost perfect discrimination. This suggests that more parsimonious signatures may offer robust performance while reducing redundancy. These results were consistent with findings from the original studies that developed these scores [11–15], as well as other external cohorts [11, 22, 23]. While tumor tissue-derived signatures had weaker associations, they were still mostly statistically significant, highlighting that blood-derived methylation signatures of smoking can even be verified in tumor tissue.
We have, for the first time, demonstrated that blood-derived methylation-based smoking signatures are associated with a higher risk of all-cause mortality among patients with stage I-III CRC. These associations were independent of factors such as age, sex, TNM stage, and other important cofounders. This finding is in line with previous studies on the impact of self-reported smoking on overall survival [3, 5–7]. Our cause-specific analyses showed that the increased risk observed for all-cause mortality was largely due to mortality unrelated to CRC. Indeed, CRC patients who smoke are more likely to develop and die from comorbidities such as cardiovascular and lung diseases [24, 25].
The evidence on the impact of smoking on CRC-specific mortality is mixed. Some studies have found a statistically significantly increased risk of CRC-specific death among current smokers [6, 7], while others have not [26–28]. In our study, only the blood- or tumor-derived 4-CpG smoking signature from Zhang et al. [15] and the tumor-derived 187-CpG smoking score from Elliott et al. [13] were significantly associated with increased CRC-specific mortality. All four CpGs in the Zhang et al. [15] score were also included in the Elliott et al. [13] score. Notably, the cg05575921 CpG site in the AHRR gene, a tumor suppressor gene, was consistently associated with smoking and an increased risk of cancer mortality [29–31]. Previous research has shown that the expression of the AHRR gene in human colorectal cancer tissue correlates with CD40/CD40L signaling and histological grade [32]. Interestingly, among stage IV patient, the 4-CpG smoking score was also strongly associated with increased risk of non-CRC-related mortality. This further demonstrates the prognostic relevance of this score.
Counterintuitively, our study found that former smokers tend to have lower CRC-specific mortality compared with never smokers. Similarly, a US study reported a protective, though non-significant, association between self-reported former smoking and CRC-specific mortality (HR 0.89, 95%CI 0.72–1.10) [6]. Previous research consistently showed that quitting smoking confers mortality benefits [33–35]. Several factors may explain this finding. Former smokers may develop heightened health consciousness, leading to healthier behaviors, earlier detection, and more diligent medical attention, all of which can improve outcomes [36]. Additionally, if smoking cessation is decades ago, some of the harmful effects and epigenetic changes caused by smoking may reverse or stabilize [7, 37, 38].
Blood-derived methylation signatures outperformed tumor tissue-derived signatures in predicting smoking status, overall mortality, and non-CRC-related mortality. This was expected, as all five signatures were originally developed using blood samples [11–15]. One key explanation lies in the well-known tissue-specific methylation variability of DNA methylation [39]. Blood-derived methylation signatures likely reflect the systemic and cumulative biological effects of smoking, such as chronic inflammation and immune response, which may occur early and persist throughout life [40, 41]. In contrast, methylation changes in tumor tissue may be more variable and dynamic and context-dependent. They can be influenced by factors such as tumor microenvironment (e.g., the presence of stromal cells, immune infiltration), treatment, and disease progression [42, 43]. These differences in cellular composition between tumor tissue and peripheral blood may act as a confounding factor, potentially diluting or obscuring the direct effects of smoking-related epigenetic signals captured in blood. This highlights the importance of accounting for tissue heterogeneity when interpreting the transportability and prognostic utility of methylation-based signatures across different biospecimen types.
Although different methylation platforms were used (EPIC 850k for blood samples and 450k for tumor samples), methodological differences are unlikely to explain the superior performance of blood-based signatures. Because both arrays are based on the same core technology and were preprocessed and normalized using the same pipelines. Only differences in CpG coverage could theoretically influence signature performance. However, all relevant CpG sites were available in the tumor tissue samples, whereas some signatures had partial CpG coverage in blood. Despite this, the blood-derived signatures demonstrated stronger and more consistent associations with smoking behaviors and mortality risks, suggesting that incomplete CpG coverage did not compromise the utility of the blood-based methylation scores.
Nonetheless, two tumor tissue-derived signatures from Elliott et al. [13] and Zhang et al. [15] showed stronger associations with CRC-specific mortality compared to blood-derived signatures. This may be explained by specific components of these scores, such as the cg05575921 site in the AHRR gene, that not only reflect smoking exposure but also capture epigenetic changes directly involved in tumor biology, including aspects of the tumor microenvironment, aggressiveness, metastasis potential, and treatment response [32, 44]. However, external validation in independent cohorts with larger sample size and more diverse clinical characteristics is needed to confirm its robustness and clinical utility. If validated, they may offer promising utility as biomarkers for integrating exposure history with tumor behavior in CRC prognostic assessment.
We found that methylation-based smoking signatures derived from adjacent normal tissue were neither associated with self-reported smoking nor with mortality risk in CRC patients. This might be due to the small sample size (only 264 samples) and resulting limited statistical power in this study. Alternatively, adjacent normal tissue may retain the epigenetic patterns similar to healthy tissue and might not show the extensive methylation changes typically associated with smoking, which are more likely to be found in blood (indicative of systemic exposure) or in tumor tissue, where smoking could have contributed to tumorigenesis [44].
We observed significant interactions between blood-derived methylation signatures and sex for both overall mortality and non-CRC-related mortality. It could be partly explained by the higher prevalence of ever smokers among males (68%) compared to females (32%). Additionally, sex-specific hormonal environments, immune responses, and genetic expression profiles might alter the impact of methylation changes on disease progression and mortality [45, 46]. For instance, sex hormones such as estrogen are known to have protective effects against inflammation and oxidative stress, potentially mitigating the adverse effects of smoking-induced aberrant methylation patterns [47]. The stronger associations observed in younger patients might be attributable to age-related epigenetic drift, which may obscure the relationship between specific methylation changes and cancer outcomes in older individuals [48].
To our knowledge, this is the first study to systematically evaluate the prognostic relevance and tissue transportability of established blood-derived smoking-related DNA methylation signatures in CRC. Our approach allowed for a direct comparative evaluation of the same methylation-based smoking scores across blood, tumor, and normal tissues in a large population-based patient cohort with long follow-up time. However, this study has some limitations. First, different methylation platforms were used for blood (Illumina MethylationEPIC 850 k BeadChip) and tissue (Illumina HumanMethylation450 BeadChip) samples. This decision was made based on the availability of the most up-to-date array technology at the time of measurement. While this might introduce concerns regarding the technical variability and comparability, both arrays are based on the same core technology, including bisulfite conversion, probe chemistry, and scanning methods [49]. Furthermore, we applied the same preprocessing and normalization pipelines across both platforms to ensure methodological uniformity.
Second, although we adjusted for several potential confounders, residual confounding by unmeasured variables (e.g., comorbid of lung disease, detailed treatment regimens) could still influence the outcomes. Third, the sample size for adjacent normal tissue was significantly smaller compared to blood and tumor tissue samples, which may limit the statistical power to detect true associations in adjacent normal tissue. Fourth, the patient samples for the blood-based analyses and the tissue-based analyses were not the same, even though they largely overlapped and were almost identical in size. Lastly, the DACHS cohort is limited to German-speaking participants from a specific geographic region in Germany, which may limit the generalizability of our findings.
Further research is needed to explore the utility of these methylation-based smoking signatures across diverse populations and various cancer types to establish their generalizability and applicability in different clinical settings. Investigating the biological mechanisms underpinning the differences in methylation signatures between blood and tumor tissue could provide insights into the distinct roles of systemic versus localized epigenetic changes in CRC progression and mortality. Integrating promising methylation-based smoking signatures (e.g., the 4-CpG score from Zhang et al. [15]) with other biomarkers and clinical variables may enhance predictive models for CRC prognosis and contribute to the development of comprehensive, multi-modal predictive tools.
Conclusions
In conclusion, our results highlight the utility of blood-derived methylation signatures as effective biomarkers for refining the quantification of smoking exposure and assessing its impact on mortality risk in non-metastatic CRC patients. While the transferability of these signatures to tissue samples is limited, several signatures [11, 13, 15] still demonstrate relevance to CRC-specific mortality. These findings underscore the potential of methylation-based smoking markers as noninvasive, objective tools for assessing smoking exposure and its impact on cancer prognosis.
Supplementary Information
Acknowledgements
The authors would like to express their gratitude to Dr. Emery Olivier, Dr. Sébastien Nusslé, and Dr. Jonviea D. Chamberlain for their valuable support in providing detailed calculation for the Epitob signature. Special thanks to Dr. Ziwen Fan and Dr. Joshua Stevenson-Hoare for their statistical guidance. The authors would like to appreciate the study participants, the interviewers who collected the data, and the medical documentalists who processed the data and follow-up information. The authors appreciate the cooperation with hospitals and pathology institutes that recruited patients for this study, provided tumor tissue samples or performed pathology analyses.
Abbreviations
- CRC
Colorectal cancer
- STROBE
The Strengthening the Reporting of Observational Studies in Epidemiology Statement
- TNM
Tumor, lymph node, metastasis staging system
- AUROC
The area under the receiver operating characteristic curve
- BMI
Body mass index
- IQR
Interquartile range
Author contributions
MH, HB, and TY were involved in the study concept and design. MH supervised this work. MH, HB, and TY had access to all the data. TY conducted all the analyses, designed the figures and wrote the first draft of the manuscript. KET, WR, BHM, AB, MK, HB, and MH were involved in the acquisition of data. All authors were involved in the revision of the manuscript for important intellectual content and approval of final version. The AI-assisted technology ChatGPT-3.5 was used by the first author to improve the readability and language of the first draft.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the German Research Council (BR 1704/6-1, BR 1704/6-3, BR 1704/6-4, CH 117/1-1, HO 5117/2-1, HO 5117/2-2, HE 5998/2-1, HE 5998/2-2, KL 2354/3-1, KL 2354 3-2, RO 2270/8-1, RO 2270/8-2, BR 1704/17-1, BR 1704/17-2); the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany; and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A, 01ER1505B, 01KD2104A).
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
The DACHS study was approved by the ethics committees of the Medical Faculty of Heidelberg University and the state medical boards of Baden-Wuerttemberg and Rhineland-Palatinate (Approval number: 310/2001). All participants provided informed consent to participate this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. 2023;73(3):233–54. [DOI] [PubMed] [Google Scholar]
- 2.Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16(12):713–32. [DOI] [PubMed] [Google Scholar]
- 3.Walter V, Jansen L, Hoffmeister M, Brenner H. Smoking and survival of colorectal cancer patients: systematic review and meta-analysis. Ann Oncol. 2014;25(8):1517–25. [DOI] [PubMed] [Google Scholar]
- 4.Liang PS, Chen TY, Giovannucci E. Cigarette smoking and colorectal cancer incidence and mortality: systematic review and meta-analysis. Int J Cancer. 2009;124(10):2406–15. [DOI] [PubMed] [Google Scholar]
- 5.Botteri E, Iodice S, Bagnardi V, Raimondi S, Lowenfels AB, Maisonneuve P. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765–78. [DOI] [PubMed] [Google Scholar]
- 6.Yang B, Jacobs EJ, Gapstur SM, Stevens V, Campbell PT. Active smoking and mortality among colorectal cancer survivors: the Cancer Prevention Study II nutrition cohort. J Clin Oncol. 2015;33(8):885–93. [DOI] [PubMed] [Google Scholar]
- 7.Alwers E, Carr PR, Banbury B, Walter V, Chang-Claude J, Jansen L, et al. Smoking behavior and prognosis after colorectal cancer diagnosis: a pooled analysis of 11 studies. JNCI Cancer Spectr. 2021;5(5):pkab077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang YM, Wei PL, Ho CH, Yeh CC. Cigarette smoking associated with colorectal cancer survival: a nationwide, population-based cohort study. J Clin Med. 2022;11(4):913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kukhareva PV, Caverly TJ, Li H, Katki HA, Cheung LC, Reese TJ, et al. Inaccuracies in electronic health records smoking data and a potential approach to address resulting underestimation in determining lung cancer screening eligibility. J Am Med Inform Assoc. 2022;29(5):779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor Gorber S, Schofield-Hurwitz S, Hardt J, Levasseur G, Tremblay M. The accuracy of self-reported smoking: a systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine Tob Res. 2009;11(1):12–24. [DOI] [PubMed] [Google Scholar]
- 11.Bollepalli S, Korhonen T, Kaprio J, Anders S, Ollikainen M. EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics. 2019;11(13):1469–86. [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain JD, Nusslé S, Chapatte L, Kinnaer C, Petrovic D, Pradervand S, et al. Blood DNA methylation signatures of lifestyle exposures: tobacco and alcohol consumption. Clin Epigenet. 2022;14(1):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenet. 2014;6(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCartney DL, Hillary RF, Stevenson AJ, Ritchie SJ, Walker RM, Zhang Q, et al. Epigenetic prediction of complex traits and death. Genome Biol. 2018;19(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Florath I, Saum KU, Brenner H. Self-reported smoking, serum cotinine, and blood DNA methylation. Environ Res. 2016;146:395–403. [DOI] [PubMed] [Google Scholar]
- 16.Liu C, Marioni RE, Hedman ÅK, Pfeiffer L, Tsai PC, Reynolds LM, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2018;23(2):422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner H, Chang-Claude J, Seiler CM, Rickert A, Hoffmeister M. Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med. 2011;154(1):22–30. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmeister M, Jansen L, Rudolph A, Toth C, Kloor M, Roth W, et al. Statin use and survival after colorectal cancer: the importance of comprehensive confounder adjustment. J Natl Cancer Inst. 2015;107(6):djv045. [DOI] [PubMed] [Google Scholar]
- 19.Sobin LH, Ch W. International Union Against Cancer (UICC). TNM Classification of Malignant Tumours, 6th ed. New York: Wiley; 2002.
- 20.Hoffmeister M, Jansen L, Stock C, Chang-Claude J, Brenner H. Smoking, lower gastrointestinal endoscopy, and risk for colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2014;23(3):525–33. [DOI] [PubMed] [Google Scholar]
- 21.Lash TL, Cole SR. Immortal person-time in studies of cancer outcomes. J Clin Oncol. 2009;27(23):e55–6. [DOI] [PubMed] [Google Scholar]
- 22.Dugué PA, Bodelon C, Chung FF, Brewer HR, Ambatipudi S, Sampson JN, et al. Methylation-based markers of aging and lifestyle-related factors and risk of breast cancer: a pooled analysis of four prospective studies. Breast Cancer Res. 2022;24(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dugué PA, Yu C, Hodge AM, Wong EM, Joo JE, Jung CH, et al. Methylation scores for smoking, alcohol consumption and body mass index and risk of seven types of cancer. Int J Cancer. 2023;153(3):489–98. [DOI] [PubMed] [Google Scholar]
- 24.Howard G, Wagenknecht LE, Burke GL, Diez-Roux A, Evans GW, McGovern P, et al. Cigarette smoking and progression of atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) study. JAMA. 1998;279(2):119–24. [DOI] [PubMed] [Google Scholar]
- 25.Kärkkäinen M, Kettunen HP, Nurmi H, Selander T, Purokivi M, Kaarteenaho R. Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res. 2017;18(1):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ordóñez-Mena JM, Walter V, Schöttker B, Jenab M, O’Doherty MG, Kee F, et al. Impact of prediagnostic smoking and smoking cessation on colorectal cancer prognosis: a meta-analysis of individual patient data from cohorts within the CHANCES consortium. Ann Oncol. 2018;29(2):472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren GW, Kasza KA, Reid ME, Cummings KM, Marshall JR. Smoking at diagnosis and survival in cancer patients. Int J Cancer. 2013;132(2):401–10. [DOI] [PubMed] [Google Scholar]
- 28.Boyle T, Fritschi L, Platell C, Heyworth J. Lifestyle factors associated with survival after colorectal cancer diagnosis. Br J Cancer. 2013;109(3):814–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogel CFA, Haarmann-Stemmann T. The aryl hydrocarbon receptor repressor - more than a simple feedback inhibitor of AhR signaling: clues for its role in inflammation and cancer. Curr Opin Toxicol. 2017;2:109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bojesen SE, Timpson N, Relton C, Davey Smith G, Nordestgaard BG. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax. 2017;72(7):646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuboi Y, Yamada H, Munetsuna E, Fujii R, Yamazaki M, Ando Y, et al. Increased risk of cancer mortality by smoking-induced aryl hydrocarbon receptor repressor DNA hypomethylation in Japanese population: a long-term cohort study. Cancer Epidemiol. 2022;78:102162. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Zhou SX, Gao L, Li XA. Regulation of CD40 signaling in colon cancer cells and its implications in clinical tissues. Cancer Immunol Immunother. 2016;65(8):919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Josephs L, Culliford D, Johnson M, Thomas M. Improved outcomes in ex-smokers with COPD: a UK primary care observational cohort study. Eur Respir J. 2017;49(5):1602114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carreras G, Pistelli F, Falcone F, Carrozzi L, Martini A, Viegi G, et al. Reduction of risk of dying from tobacco-related diseases after quitting smoking in Italy. Tumori. 2015;101(6):657–63. [DOI] [PubMed] [Google Scholar]
- 35.Tran B, Falster MO, Douglas K, Blyth F, Jorm LR. Smoking and potentially preventable hospitalisation: the benefit of smoking cessation in older ages. Drug Alcohol Depend. 2015;150:85–91. [DOI] [PubMed] [Google Scholar]
- 36.Inoue-Choi M, Ramirez Y, Fukunaga A, Matthews CE, Freedman ND. Association of adherence to healthy lifestyle recommendations with all-cause and cause-specific mortality among former smokers. JAMA Netw Open. 2022;5(9):e2232778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shang J, Nie X, Qi Y, Zhou J, Qi Y. Short-term smoking cessation leads to a universal decrease in whole blood genomic DNA methylation in patients with a smoking history. World J Surg Oncol. 2023;21(1):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Coorens T, Maughan EF, et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020;578(7794):266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Bustos C, Ramos E, Young JM, Tran RK, Menzel U, Langford CF, et al. Tissue-specific variation in DNA methylation levels along human chromosome 1. Epigenet Chromatin. 2009;2(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elisia I, Lam V, Cho B, Hay M, Li MY, Yeung M, et al. The effect of smoking on chronic inflammation, immune function and blood cell composition. Sci Rep. 2020;10(1):19480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caliri AW, Tommasi S, Besaratinia A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat Res Rev Mutat Res. 2021;787:108365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin Q, Zhou Y, Guo J, Chen Q, Tang W, Li Y, et al. Conserved methylation signatures associate with the tumor immune microenvironment and immunotherapy response. Genome Med. 2024;16(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu D, Zeng S, Su C, Li J, Xuan Y, Lin Y, et al. The interaction between DNA methylation and tumor immune microenvironment: from the laboratory to clinical applications. Clin Epigenet. 2024;16(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Z, Wen W, Cai Q, Long J, Wang Y, Lin W, et al. From tobacco smoking to cancer mutational signature: a mediation analysis strategy to explore the role of epigenetic changes. BMC Cancer. 2020;20(1):880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Govender P, Ghai M, Okpeku M. Sex-specific DNA methylation: impact on human health and development. Mol Genet Genomics. 2022;297(6):1451–66. [DOI] [PubMed] [Google Scholar]
- 46.Forsyth KS, Jiwrajka N, Lovell CD, Toothacre NE, Anguera MC. The conneXion between sex and immune responses. Nat Rev Immunol. 2024;24(7):487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiang D, Liu Y, Zhou S, Zhou E, Wang Y. Protective effects of estrogen on cardiovascular disease mediated by oxidative stress. Oxid Med Cell Longev. 2021;2021:5523516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013;22(R1):R7-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Solomon O, MacIsaac J, Quach H, Tindula G, Kobor MS, Huen K, et al. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics. 2018;13(6):655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.