

Abstract
Proteins with multiple binding sites exhibit a complex behavior that depends on the intrinsic affinities for each site and the energetic communication between the sites. The contributions from intrinsic affinity and cooperativity are difficult to deconvolute using conventional binding experiments that lack information about the occupancies of individual sites. Here, we report the concerted use of NMR and isothermal titration calorimetry to determine the intrinsic and cooperative binding free energies for a ligand–protein complex. The NMR measurements provided the site-specific information necessary to resolve the binding parameters. Using this approach, we observed that human ileal bile acid binding protein binds two molecules of glycocholic acid with low intrinsic affinity but an extraordinarily high degree of positive cooperativity. The highly cooperative nature of the binding provides insights into the protein's biological mechanism. With ongoing improvements in sensitivity and resolution, NMR methods are becoming more amenable to dissecting the complex binding energetics of multisite systems.
Proteins that bind more than one ligand molecule place special demands on the experimental analysis of the energetics (1–3). Consider a protein with two binding sites as in Fig. 1. The binding of a ligand to one site when the other site is unoccupied is represented by the intrinsic association constants Ki, where i = 1 or 2. When one site is occupied, the binding of the second ligand is given by the product of Ki and the cooperativity factor c12. The latter provides a measure of the energetic coupling between the sites. Values of c12 > 1 or ΔGc° < 0 indicate microscopic positive cooperativity, where the binding of a ligand to one site enhances the affinity for the second.
Figure 1.

Site-specific (microscopic) model for two-site binding.
Cooperativity and allostery are prominent regulatory mechanisms in biological systems (4–6), but are difficult to quantify. The site-specific model in Fig. 1 is characterized by three independent equilibria or binding parameters. Therefore, the experimental analysis of this two-site system demands at least three independent measurables. In general, a site-specific analysis of the binding energetics for a protein containing n sites requires 2n – 1 measurables, a daunting task even for modest values of n. To circumvent this problem, experimental binding data are often fit to models of reduced complexity. For example, the stepwise binding model contains n independent equilibria and parameters. For n = 2, the observed stepwise association constants K1obs and K2obs reflect the binding of the first and second moles of ligand (L) per mole protein (P), irrespective of site. These macroscopic constants provide a crude description of the binding energetics and may distort the magnitude or sign of the cooperativity. For example, microscopic positive cooperativity can be masked by macroscopic negative cooperativity (3). The analysis of binding curves in the case of positive cooperativity is particularly difficult, because the singly ligated intermediates are poorly populated. The relatively featureless binding curve is dominated by the unligated and doubly ligated states.
NMR is one of the few experimental methods with the resolving power to monitor the occupancies of individual binding sites on proteins. For this reason, NMR has untapped potential for analyzing the energetics of multisite systems. Here we illustrate for the first time how NMR can be used to measure the intrinsic and cooperative binding free energies for a ligand–protein complex. We applied NMR along with isothermal titration calorimetry (7) to investigate the binding properties of a protein with two sterol binding sites. Human ileal bile acid binding protein (I-BABP) is a member of the intracellular lipid binding protein family and is thought to help regulate the enterohepatic circulation of bile acids (8, 9). We chemically synthesized 13C- and 15N-enriched isotopomers of glycocholic acid, an abundant bile acid abundant in humans. Using these isotope-enriched ligands and heteronuclear two-dimensional (2D) NMR methods, we resolved and quantitated the ligand occupancies for each site as well as the unbound pool. The data were fit to a site-specific binding model, yielding the parameters defined in Fig. 1.
Materials and Methods
Protein Biosynthesis and Purification.
Recombinant human I-BABP was biosynthesized in Escherichia coli and purified to homogeneity as follows. Bacteria harboring the pMON-hIBABP plasmid were grown at pH 7.2 in a New Brunswick Scientific Bioflow III high-density fermenter by using a nutrient-rich medium containing tryptone (10.8 g/l), yeast extract (22.5 g/l), potassium phosphate (0.1 M), magnesium sulfate (1–5 mM), calcium chloride (0.1–0.5 mM), as well as a trace amounts of iron sulfate and thiamine. Cells were grown to a final density of 55 as monitored by OD600. Protein expression, under control of the recA promoter, was induced in mid-log phase by adding nalidixic acid to 100 μg/ml. After harvesting, the protein was released from partially lysed cells by using a freeze–thaw protocol. The cells were suspended in Tris buffer, pH 8.1, containing a broad-spectrum protease inhibitor mixture (Roche Molecular Biochemicals) and were frozen in ethanol/dry ice and thawed. The freeze–thaw cycle was repeated five times. Further protein purification was achieved using extensive dialysis against 20 mM potassium phosphate, pH 7.4, followed by gel filtration chromatography using a 140 × 5 cm column of Sephadex G-50. The protein was delipidated by passing it over a column of lipophilic Sephadex type VI (Sigma, #H-6258) at 37°C. Protein purity, as assessed by overloaded Coomassie-stained SDS/PAGE gels, was >98%. The final yield of purified protein from a 4-l fermentation was ≈5 grams. Protein concentrations were determined spectrophotometrically. As calibrated by quantitative amino acid analysis, a 1 mg/ml solution of human I-BABP in water corresponds to an OD280 value of 0.846.
Isothermal Titration Calorimetry.
The calorimetry experiments were performed using a Microcal (Amherst, MA) OMEGA differential titration calorimeter. One hundred fifteen injections of 4-μl aliquots were followed by 35 injections of 7-μl aliquots of 6.24 mM potassium glycocholate into a reaction cell containing 1.4 ml of 0.19 mM human I-BABP. In most experiments, the final ligand:protein mole ratio at the end of the titration was ≈5:1. The protein and ligand were dissolved in identical buffers containing 20 mM potassium phosphate, 135 mM KCl, and 10 mM NaCl (pH 7.2). Separate isothermal titrations were carried out at a series of temperatures from 10–45°C in 5°C increments with the 35°C increment replaced by 37°C. Each titration series was repeated at least three times, with the 20°C increment repeated five times. The heats of injection were corrected for the heat of dilution of the ligand into buffer and normalized to the amount of glycocholate injected. The integrated peak intensities were fit to a two-step binding model, as outlined in Fig. 2, using the nonlinear least squares analysis program SCIENTIST (Scientific Instrument Services, Ringoes, NJ). The stepwise binding equations used to fit the calorimetry data are provided in Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org.
Figure 2.

Stepwise (macroscopic) model for the biding of two ligands, L, to protein, P.
Bayesian Analysis.
The fitting of the isothermal calorimetry data to the stepwise binding model, without the restraints from the NMR data, was assessed using a Bayseian analysis (10). A Markov chain Monte Carlo method was used to sample 10,000 points from the posterior probability distribution of the parameters Kd1obs and Kd2obs given the calorimetry data. The last nine thousand points were used to obtain the averages and uncertainties of the parameter space. The Markov Chain Monte Carlo sampler was adopted from Radford Neal's software package available at http://www.cs.toronto.edu/∼radford/fbm.software.html.
NMR Sample Preparation.
Glycocholic acid, uniformly 13C or 15N enriched in the glycine moiety, was synthesized and purified using previously reported methodology (11). Crystalline 13C- or 15N-enriched glycocholic acid was dissolved in tetrahydrofuran, and the concentrations of the stock solutions were determined by measuring dry weights using a Perkin–Elmer AD-4 microbalance. Gas-tight Hamilton syringes were used to aliquot appropriate amounts of the stock solution of bile acid, and the solvent was evaporated under a stream of nitrogen. The bile acid was solubilized using 1.1 equivalents of 1 M KOH, and each aliquot was brought up to a solution volume of 60 μl by using D2O. Then 540 μl of protein solution was added to bring the total volume of the NMR sample to 600 μl. The final protein NMR samples contained the following buffer conditions: 1.85 mM protein, 20 mM potassium phosphate, 135 mM KCl, and 10 mM NaCl (pH 7.2), in 90% H2O/10% D2O. Where necessary, 0.5-μl increments of 1 M KOH or HCl were added to correct the pH to 7.20 ± 0.05. To minimize pipetting errors and avoid sample losses, each point on the x axis of the NMR binding isotherm—i.e., each ligand:protein mole ratio—was represented by one or more separate NMR samples.
NMR Data Collection and Peak Volume Analysis.
All NMR spectra were recorded on a Varian Unity 500 three-channel NMR spectrometer equipped with a Nalorac Cryogenics (Martinez, CA) 5-mm indirect triple resonance gradient probe. Two-dimensional HCACO spectra were collected using an in-house, gradient-enhanced version of the original pulse sequence (12). All HCACO spectra were acquired with a 1H spectral width of 6,500 Hz and 2,048 complex points, zero-filled to a total of 4,096 points. In the indirectly detected dimension, the carbonyl 13C spectral width was 1,250 Hz, 84 hypercomplex increments were acquired and zero-filled to a total of 256 points. Pseudoecho weighting was applied in the F2 dimension, using an exponential factor of −3 Hz and a Gaussian factor of 0.03. In F1, Gaussian weighting was used with a factor of 0.03. The gradient- and sensitivity-enhanced 1H/15N heteronuclear correlation spectra were collected using the pulse sequence of Kay and coworkers (13). All heteronuclear sequential quantum correlation (HSQC) specra were acquired with a 1H spectral width of 6,500 Hz and 2,048 complex points, zero-filled to a total of 4,096 points. In the 15N dimension, 40 hypercomplex increments were collected and zero-filled to a total of 256 points. The apodization functions were the same as for HCACO.
All of the 1H/15N correlation spectra used to construct the NMR isotherm isotherm were recorded under identical conditions except for the number of transients. To keep a consistent signal-to-noise ratio throughout the binding isotherm, the number of transients was scaled using the relationship that the signal-to-noise ratio increases proportionally with the square root of the number of transients. For example, an increase in the total ligand concentration of 2-fold resulted in a decrease in the number of transients by 4-fold. Peak volume integrations were carried out using the ll2d algorithm in the Varian VNMR software. The error bars in the peak volumes shown in the NMR isotherm represent twice the standard deviation of three separate spectra of the same sample accumulated under identical conditions. These are the errors in peak volume determination. In addition, at 1:1 and 3:1 ligand:protein, three separate samples were prepared at each mole ratio to assess the total errors (pipetting plus peak volume determination). The difference represents the pipetting errors, which were negligible compared with the peak volume errors.
The proportionality between NMR peak volumes and binding site occupancies can be complicated by the following issues. Differential spin-lattice relaxation of the NMR resonances can distort the relative peak intensities and site occupancies. However, the amide 1H T1 values for the three resonances were not significantly different in this case: 0.6 ± 0.2, 0.5 ± 0.1, and 0.4 ± 0.1 s for peaks U, 1, and 2, respectively. As a cross check, we collected 1H/15N correlation spectra with relaxation delays of 1, 2, and 3 seconds at the beginning of the pulse sequence. The relative volumes of peaks U, 1, and 2 varied by merely 2%, 3%, and 2% over this range of relaxation delays, well within the experimental uncertainties. A saturation transfer experiment was performed by collecting spectra with and without a selective 2-s, 20-Hz presaturation pulse on the water resonance to assess the possibility of ligand amide hydrogen exchange with solvent water. The presaturation of peaks 1, 2, and U produced an equivalent attenuation of 16% ± 3%, 14% ± 2%, and 16% ± 2%, respectively, ruling out differential hydrogen exchange. Moreover, the gradient and sensitivity-enhanced HSQC pulse sequence used for the quantitative analysis did not contain water presaturation pulses and was designed to minimally perturb the water resonance. Therefore, none of these potential artifacts affected the quantitative proportionality between NMR peak volumes and relative binding site occupancies.
The equations for the site-specific binding model used for fitting the NMR isotherms are provided in Supporting Text, which is published as supporting information on the PNAS web site.
Results
The isothermal calorimetry results highlight the difficulty in analyzing binding data in the case of positive cooperativity. The data in Fig. 3 exhibited an atypical profile and did not fit well to binding models that assumed a single class of identical sites or multiple classes of noninteracting sites. Therefore, the calorimetric isotherms were fit to the equations for the stepwise binding model in Fig. 2, which assumed nothing about the presence or absence of cooperativity. Excellent fits were obtained with the stepwise model using least squares minimization. However, the best fit parameters were not unique in that divergent sets of stepwise binding constants gave comparably good χ2 values. To map the statistically relevant parameter space, we performed a Bayesian analysis to assess the probability distribution for different combinations of the observed binding constants (10). As illustrated in Fig. 3D, the calorimetric results are consistent with pairs of stepwise dissociation constants that vary over several orders of magnitude. In all cases, the affinity of the second step was greater than that of the first, indicative of macroscopic positive cooperativity. However, the magnitude of the cooperative free energy could not be determined. The stepwise parameters were inherently insufficient for deconvoluting the intrinsic and cooperative free energies, as discussed above. In addition, a unique set of stepwise binding constants could not be determined in this case because of the positive cooperativity. The calorimetry data did not have enough information content to establish even a reduced, macroscopic description of the binding energetics.
Figure 3.

Isothermal titration calorimetry results for glycocholic acid binding to human I-BABP. The raw data collected at 37°C (A) and 20°C (B) are shown as injection profiles. The discontinuity at an x axis value of ≈60 min represents a change in the injection volume from 4 μl to 7 μl. C displays the calorimetric binding isotherm for the data collected at 20°C. The y axis is the normalized heat released or absorbed for each injection of ligand. The curve through the points represents a least squares fit to the two-step binding model. The fitting equations are defined in Supporting Text, which is published as supporting information on the PNAS web site. D displays the Bayesian analysis of the probability distribution of the fitted stepwise dissociation constants for the data at 20°C.
By contrast, the NMR results provided three experimental measurables as well as site-specific information about binding site occupancies. Heteronuclear 2D NMR spectra of samples containing isotopically enriched ligand and unenriched protein revealed three sets of NMR resonances, as shown in Fig. 4. Peak U represented unbound glycocholic acid, as assigned by spectra of otherwise identical control samples lacking protein. The other peaks represented glycocholic acid bound to two distinct environments on the protein, arbitrarily designated sites 1 and 2. In 2D gradient-enhanced HCACO spectra as in Fig. 4A, the [13C]glycine-enriched bile acid bound to each site gave rise to a pair of 1H resonances at the same carboxyl 13C chemical shift. As shown in Fig. 4B, the 2D 1H/15N correlation spectra for samples containing [15N]glycocholate and I-BABP contained three resonances corresponding to bile acids bound to each site and the unbound bile acid. All three resonances were observed at each ligand/protein mole ratio examined between 0.5 and 6. The stacked plot in Fig. 4C illustrates the artifact-free baseline and good signal-to-noise necessary for accurate volume integration. Whereas the 2D 1H/13C correlation spectra had similarly clean baselines, the sensitivity and resolution of the 1H/15N spectra were somewhat better. Therefore, we selected the latter as a basis for the quantitative analysis.
Figure 4.

NMR spectra and site-specific binding isotherms for glycocholic acid binding to human I-BABP. (A) Two-dimensional gradient-enhanced HCACO spectrum of a sample containing a 3:1 mole ratio of [1′,2′-13C]glycocholate to unenriched I-BABP. The resonances corresponding to the unbound bile acid and the bile acid bound to sites 1 and 2 are designated U, 1, and 2, respectively. (B) Gradient- and sensitivity-enhanced 1H/15N heteronuclear sequential quantum correlation (HSQC) spectrum (contour plot) of 15N labeled glycocholate for a sample containing a 3:1 mole ratio of [15N]glycocholate to I-BABP. (C) A stacked plot representation of the spectrum in B. The small peak at 1H = 3.5 ppm and 15N = 123 ppm resulted from chemical exchange of ligand between site 2 and the unbound pool. (D) Site-specific NMR isotherms for the binding of [15N]glycocholate to I-BABP. The spectra at each mole ratio were recorded in triplicate by using separate samples for each mole ratio. The average ±95% confidence limits are reported for the unbound (squares), site 1 (triangles), and site 2 (open circles) resonances. The scaled peak volumes for site 2 include the measured volumes of peak 2 plus the volumes for the small exchange cross peak as seen in C.
The temperature dependence of the bile acid amide resonances is shown in Fig. 5. At the lowest temperature, the three resonances were narrow and in slow exchange on the NMR chemical shift time scale (kex ≪ 350 s−1). With increasing temperature, peaks 2 and U became selectively broadened and nearly disappeared by 35°C. This behavior was indicative of intermediate exchange between the bile acid bound to site 2 and the unbound pool (kex ≈ 350 s−1). Exchange cross peaks were also observed in isotope-directed NOESY spectra not shown here. At the higher temperatures, the exchange broadening obscured the site-specific information and complicated the analysis of binding site occupancies. Therefore, the quantitative site-specific analysis of binding energetics was performed only at 20°C in this study. A stepwise analysis of the calorimetry data alone at 37°C was consistent with two-step binding with macroscopic positive cooperativity, qualitatively similar to the stepwise analysis at 20°C. Of course, the binding and cooperativity constants are likely to be temperature dependent, and those changes may have contributed in part to the spectral changes observed in Fig. 5.
Figure 5.

Temperature dependence of bile acid amide 1H resonances. One-dimensional first increments of the 2D 1H/15N correlation spectra were collected at temperatures ranging from 15° to 40°C. All spectra were collected using a 3:1 mole ratio of [15N]glycocholate complexed to I-BABP with sample conditions identical, except for temperature, to that in Fig. 4 B and C.
The site-specific binding isotherms obtained by fitting the NMR peak volumes are displayed in Fig. 4D. The curves represent a solution that fits both the NMR and the calorimetry data at 20°C. The resulting site-specific and stepwise parameters are summarized in Table 1. The binding of glycocholate to human bile acid binding protein is characterized by two intrinsically weak binding sites, but strong positive cooperativity. The microscopic cooperativity factor c12 exceeds 1,000, corresponding to a standard state free energy change of approximately −17 kJ/mol. Hence, the binding of the ligand to one site dramatically increased its affinity for the second binding site.
Table 1.
Site-specific and stepwise binding parameters for the interactions of glycocholic acid with human ileal bile acid binding protein at 20°C
| Parameter type | Dissociation constants*, M | Binding free energies†, kJ/mol |
|---|---|---|
| Site-specific binding | ||
| Cooperativity | 8.6 (± 0.3) × 10−4 | −17.2 ± 0.1 |
| Site 1, intrinsic | 1.5 (± 0.1) × 10−3 | −15.8 ± 0.2 |
| Site 2, intrinsic | 2.1 (± 0.4) × 10−3 | −15.0 ± 0.5 |
| Stepwise binding‡ | ||
| Step 1 | 1.8 (± 0.3) × 10−3 | −15.4 ± 0.4 |
| Step 2 | 1.5 (± 0.9) × 10−6 | −32.6 ± 2.2 |
| Hill coefficient§ | 1.94 |
ΔG° = RTlnKd or RTln(1/c12), where the standard state is defined as 1 M.
The observed stepwise dissociation constants are related to the site-specific dissociation constants as follows: (K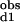 )−1 = (2Kd1)−1 + (2Kd2)−1 and K
)−1 = (2Kd1)−1 + (2Kd2)−1 and K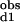 *K
*K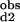 = Kd1*Kd2/c12.
= Kd1*Kd2/c12.
The Hill coefficient, a commonly used measure of macroscopic cooperativity, varies from limiting values of 0 to 2 for extremely negatively and positively cooperative systems, respectively (3). It assumes a value of 1 for a noncooperative system. The Hill coefficient at half saturation is related to the stepwise binding parameters as follows: nH = 2/[1 + (K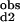 /K
/K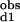 )1/2].
)1/2].
In principle, the three independent measurables obtained from NMR should have sufficed for obtaining the site-specific binding constants. However, we used all five measurable parameters: three from NMR and two from calorimetry. In this case, the very high degree of positive cooperativity precluded an analysis based solely on the NMR data. However, other types of two binding site systems that are less cooperative may be amenable to an NMR-only analysis.
Discussion
Macroscopic binding constants provide a global description of the energetics of multisite systems. This global description is crude and difficult to interpret. By contrast, site-specific binding constants provide a local description of the binding energetics. They yield the intrinsic binding free energies for each site as well as the cooperative free energies reflecting the interactions between all pairs of sites. Armed with the local description, the global binding properties of the system can be calculated, but the converse is not possible. Therefore, a site-specific analysis is the most informative approach for interpreting multisite binding data (1, 3). However, a site-specific analysis is much more demanding experimentally. This study shows how the resolving power of NMR can be used to meet those demands.
The system used to illustrate this approach, glycocholate binding to human ileal bile acid binding protein, revealed an extraordinarily high degree of microscopic and macroscopic positive cooperativity. More than one-third of the total binding free energy of −48 kJ/mol was derived from the microscopic cooperativity. To put the cooperativity in perspective, human I-BABP can be compared with human hemoglobin. The largest cooperativity factor in hemoglobin, which occurs with the binding of the fourth molecule of oxygen, is ≈400 (14). For glycocholate binding to human I-BABP, the cooperativity factor exceeds 1,000. A crude but commonly used measuring stick for macroscopic cooperativity is the Hill coefficient, which adopts a value of 1 for a noncooperative system and a maximum value of 2 for an extremely positively cooperative two-site system. The Hill coefficient in this case is 1.94. The microscopic and macroscopic cooperativity observed in this system may be the largest ever measured for a ligand–protein interaction.
The remarkable binding behavior of I-BABP has implications for its biological function. Most other members of the intracellular lipid binding protein family bind a single fatty acid or retinoid molecule with relatively high affinity (15). By contrast, human I-BABP has two glycocholate binding sites with weak intrinsic affinities and a strong positive cooperativity. This distinction suggests a fundamentally different role for I-BABP. This protein is abundant in the absorptive epithelial cells located in the distal small intestine and may help regulate the passage of bile acids through the cytoplasm. Because of the modest intrinsic affinity, a sizeable fraction of glycocholate could remain unbound at low ligand:protein ratios. This could permit a significant flux of bile acid through the enterocyte as unbound monomers. Compared with retinoids and fatty acids, bile acids have much higher monomer solubility and may not need a protein carrier to traverse the cytoplasm. However, because of the strong positive cooperativity, the fraction of unbound glycocholate decreases as the bile acid:protein ratio increases. This trend occurs up to the 2:1 stoichiometric binding capacity of the protein. A noncooperative system does not show this trend. By this mechanism, I-BABP could buffer ileal enterocytes against higher levels of free glycocholate that may trigger abnormal apoptosis or cytotoxicity (16, 17). The binding energetics of I-BABP suggest that it may function as a “regulated sponge”: permissive at low glycocholate concentrations but protective at higher levels.
The structural basis for the positive cooperativity is not yet known. Cooperativity could result from allostery, where the binding of the first ligand is energetically communicated to the second site through a conformational change in the protein. Alternatively, the binding of the first bile acid may provide a favorable binding surface for the second through a direct ligand–ligand interaction. A determination of the structural correlates of the cooperativity must await a careful comparison of the three-dimensional structures of the doubly ligated and unligated proteins. Curiously, solution-state NMR structures have been reported for the singly ligated forms of the porcine and human I-BABP (18, 19). Because of the cooperativity, the population of the singly ligated protein should not exceed a few percent. The apparent discrepancy between the reported structure(s) and the current binding results could possibly be explained by the temperature dependence of ligand exchange rates (Fig. 5). The structures were determined at temperatures above 30°C, where one of the two glycocholate binding sites may have been undetected because of NMR exchange broadening. The current study provides definitive evidence that the human protein contains two bile acid binding sites and also explains the “paradoxical” behavior seen with photoaffinity labeling experiments (20).
The energetics by NMR approach can be extended to systems with more than one type of ligand if unique isotope enrichment patterns are used for each ligand. In addition, it can be extended to systems with larger numbers of binding sites, especially if the site-specific NMR analysis is combined with macroscopic binding data from other techniques, as illustrated herein. The main limitation of NMR as a quantitative site-specific binding tool has been its relatively poor sensitivity and the corresponding requirements for high macromolecule concentrations. In turn, this requirement has restricted the method to relatively weak binding regimes or systems in rapid exchange where the site-specific information may be lost. However, the ongoing improvements in sensitivity and resolution resulting from improved pulse sequences, higher magnetic fields and cryogenic probes are pushing the method into regimes characterized by submicromolar dissociation constants and higher molecular weight. For these reasons, NMR holds great promise for dissecting the complex binding energetics of an increasingly wide range of macromolecules with multiple binding sites.
Supplementary Material
Acknowledgments
The source code for the NMR pulses sequences was kindly provided by Lewis Kay, and the cDNA for human I-BABP was a generous gift from Paul Dawson. We are indebted to Gary Ackers for his thoughtful insights and encouragement. This work was supported by Public Health Service Grants R01 DK48046 and P30 DK52574 (to D.P.C.). K.R. was a visiting student from the Biochemistry Diploma Program, University of Regensburg, Germany and supported by a fellowship from Deutscher Akademischer Austauschdienst.
Abbreviations
- I-BABP
human ileal bile acid binding protein
- 2D
two-dimensional
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ackers G K, Shea M A, Smith F R. J Mol Biol. 1983;170:223–242. doi: 10.1016/s0022-2836(83)80234-4. [DOI] [PubMed] [Google Scholar]
- 2.Wyman J, Gill S J. Binding and Linkage. Mill Valley, CA: Univ. Sci. Books; 1990. [Google Scholar]
- 3.Di Cera E. Thermodynamic Theory of Site-Specific Binding Processes in Biological Macromolecules. Cambridge, U.K.: Cambridge Univ. Press; 1995. [Google Scholar]
- 4.Monod J, Changeux J-P, Jacob F. J Mol Biol. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 5.Ackers G K, Doyle M L, Myers D, Daugherty M A. Science. 1992;255:54–63. doi: 10.1126/science.1553532. [DOI] [PubMed] [Google Scholar]
- 6.Ackers G K. Adv Protein Chem. 1998;51:185–253. doi: 10.1016/s0065-3233(08)60653-1. [DOI] [PubMed] [Google Scholar]
- 7.Wiseman T, Williston S, Brandts J F, Lin L N. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 8.Sacchettini J C, Hauft S M, Van Camp S L, Cistola D P, Gordon J I. J Biol Chem. 1990;265:19199–19207. [PubMed] [Google Scholar]
- 9.Lin M C, Kramer W, Wilson F A. J Biol Chem. 1990;265:14986–14995. [PubMed] [Google Scholar]
- 10.Berry D A. Statistics: A Bayesian Perspective. Belmont, CA: Duxbury Press; 1996. [Google Scholar]
- 11.Tserng K Y, Hachey D L, Klein P D. J Lipid Res. 1977;18:404–407. [PubMed] [Google Scholar]
- 12.Powers R, Gronenborn A M, Clore G M, Bax A. J Magn Reson. 1991;94:209–213. [Google Scholar]
- 13.Zhang O, Kay L E, Olivier J P, Forman-Kay J D. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 14.Ackers G K, Holt J M, Huang Y, Grinkova Y, Klinger A L, Denisov I. Proteins Struct Funct Genet. 2000;4:23–43. doi: 10.1002/1097-0134(2000)41:4+<23::aid-prot30>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 15.Glatz J F, van der Vusse G J. Prog Lipid Res. 1996;35:243–282. doi: 10.1016/s0163-7827(96)00006-9. [DOI] [PubMed] [Google Scholar]
- 16.Garewal H, Bernstein H, Bernstein C, Sampliner R, Payne C. Cancer Res. 1996;56:1480–1483. [PubMed] [Google Scholar]
- 17.Pritchard D M, Watson A J. Pharmacol Ther. 1996;72:149–169. doi: 10.1016/s0163-7258(96)00102-7. [DOI] [PubMed] [Google Scholar]
- 18.Lücke C, Zhang F, Hamilton J A, Sacchettini J C, Rüterjans H. Eur J Biochem. 2000;267:2929–2938. doi: 10.1046/j.1432-1327.2000.01307.x. [DOI] [PubMed] [Google Scholar]
- 19.Kramer W, Sauber K, Baringhaus K H, Kurz M, Stengelin S, Lange G, Corsiero D, Girbig F, Konig W, Weyland C. J Biol Chem. 2000;276:36020–36027. doi: 10.1074/jbc.M006877200. [DOI] [PubMed] [Google Scholar]
- 20.Kramer W, Corsiero D, Friedrich M, Girbig F, Stengelin S, Weyland C. Biochem J. 1998;333:335–341. doi: 10.1042/bj3330335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.