

Abstract
A method for optimizing potential-energy functions of proteins is proposed. The method assumes a hierarchical structure of the energy landscape, which means that the energy decreases as the number of native-like elements in a structure increases, being lowest for structures from the native family and highest for structures with no native-like element. A level of the hierarchy is defined as a family of structures with the same number of native-like elements (or degree of native likeness). Optimization of a potential-energy function is aimed at achieving such a hierarchical structure of the energy landscape by forcing appropriate free-energy gaps between hierarchy levels to place their energies in ascending order. This procedure is different from methods developed thus far, in which the energy gap and/or the Z score between the native structure and all non-native structures are maximized, regardless of the degree of native likeness of the non-native structures. The advantage of this approach lies in reducing the number of structures with decreasing energy, which should ensure the searchability of the potential. The method was tested on two proteins, PDB ID codes 1FSD and 1IGD, with an off-lattice united-residue force field. For 1FSD, the search of the conformational space with the use of the conformational space annealing method and the newly optimized potential-energy function found the native structure very quickly, as opposed to the potential-energy functions obtained by former optimization methods. After even incomplete optimization, the force field obtained by using 1IGD located the native-like structures of two peptides, 1FSD and betanova (a designed three-stranded β-sheet peptide), as the lowest-energy conformations, whereas for the 46-residue N-terminal fragment of staphylococcal protein A, the native-like conformation was the second-lowest-energy conformation and had an energy 2 kcal/mol above that of the lowest-energy structure.
The prediction of protein structure solely on the basis of amino acid sequence and potential-energy function is one of the greatest challenges of contemporary computational biology and biophysics (1). This method is based on the physics of protein folding, namely on the thermo-dynamic hypothesis formulated by Anfinsen (2), according to which the native structure of a protein corresponds to the global minimum of its free energy under given conditions. Thus, protein structure prediction with ab initio methods is accomplished by a search for a conformation corresponding to the global minimum of an appropriate potential-energy function without the use of secondary structure prediction, homology modeling, threading, etc.
The necessary condition for this approach to work is that the potential-energy function must locate the native structure of a protein as the one of lowest energy. Crippen and coworkers (3, 4) designed a method that optimized the potential-energy function to locate the native structures of selected training proteins as the lowest-energy structures. However, the condition of the native structure being the lowest in energy is insufficient, because it does not ensure that this lowest-energy structure will be located by a random search. On the basis of simulation studies of a model lattice protein, Shakhnovich and coworkers (5) suggested that the sufficient condition is the presence of a sufficiently large energy gap between the native structure and the lowest-energy non-native structure. According to these workers (5), a large energy gap will provide a driving force to conduct the system to the global energy minimum. Substantial subsequent work on optimizing potential-energy functions has been carried out following this idea (6–11), with energy functions for both threading and de novo folding. Mathematically, the target function is the energy gap between the lowest-energy native-like structure and the lowest-energy non-native structure (ΔE) and/or the Z score (Z), defined as the difference between the mean energy of the native-like structures and the mean energy of the non-native structures divided by the standard deviation of the energy of the non-native structures (8–10):
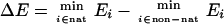 |
1 |
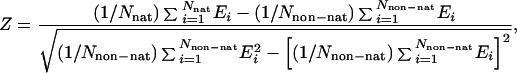 |
2 |
where nat and non-nat indicate the sets of native-like and non-native conformations, respectively [the criterion being the rms deviation (rmsd) from the experimental structure], and Nnat and Nnon-nat denote the number of native-like and non-native structures, respectively.
In our recent work (9, 10), we implemented this principle to optimize the united-residue (UNRES) force field (8, 12, 13) for off-lattice protein-structure prediction and simulation, initially using a set of decoys (8) from the Protein Data Bank (PDB) and, more recently (9, 10), a set that was generated by using the conformational space annealing (CSA) method (14).
Our recent algorithm for optimizing a potential-energy function involves iterations consisting of the following three steps (9, 10): (i) updating the decoy set by a global CSA search using the current weights; (ii) a local CSA search in the neighborhood of the experimental structure with the current weights, to locate the lowest-energy native-like structure corresponding to the current set of parameters of the energy function; and (iii) determination of new weights by making ΔE and Z as negative as possible. Steps i–iii are iterated until the global CSA search finds the native-like structure as the lowest-energy structure. The native likeness was defined in terms of the (Cα) rmsd from the experimental structure. Both the set of decoys and the set of native-like structures are updated in each iteration of the procedure. The algorithm can be implemented by using a single or more than one protein as a training set. However, although it works well for proteins with simple topologies, we found it difficult to optimize the energy function for proteins with more complicated folds, for which the native-like structures are far from low-energy conformations found with initial-guess parameters. Therefore, in this report, we propose a different method for optimizing a potential function based not only on energy ranking of native-like structures with respect to non-native structures but also on energy ranking of all structures, depending on native likeness. In this report, we describe the algorithm and preliminary tests with it on two proteins. More detailed results will be reported elsewhere. It should be noted that the hierarchy introduced here to optimize a potential function is different from the hierarchy (15) that is used to predict protein conformation by a combination of UNRES and all-atom models.
Methods
Force Field.
In the UNRES model (12), a polypeptide chain is represented by a sequence of α carbon (Cα) atoms linked by virtual bonds with attached united side chains (SC) and united peptide groups (p). Each united peptide group is located in the middle of two consecutive α carbons. Only these united peptide groups and the united side chains serve as interaction sites, the α carbons serving only to define the chain geometry (see figure 1 of ref. 12). All virtual bond lengths (i.e., Cα—Cα and Cα—SC) are fixed; the distance between neighboring Cαs is 3.8 Å corresponding to trans peptide groups, whereas the side-chain (αSC and βSC), virtual-bond (θ), and dihedral (γ) angles can vary. The energy of the virtual-bond chain is expressed by Eq. 3.
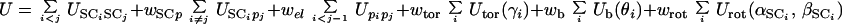 |
3 |
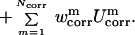 |
The term USCiSCj represents the mean free energy of the hydrophobic (hydrophilic) interactions between the side chains, which implicitly contains the contributions from the interactions of the side chain with the solvent. The term USCipj denotes the excluded-volume potential of the side-chain–peptide-group interactions. The peptide-group interaction potential (Upipj) accounts mainly for the electrostatic interactions (i.e., the tendency to form backbone hydrogen bonds) between peptide groups pi and pj. Utor, Ub, and Urot represent the energies of virtual-dihedral angle torsions, virtual-bond angle bending, and side-chain rotamers; these terms account for the local propensities of the polypeptide chain. Finally, the terms U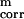 , m = 1, 2, … Ncorr are the correlation or multibody contributions that we derived (13) from a generalized-cumulant expansion (16) of the restricted free energy (RFE), and the ws are the weights of the energy terms. The multibody terms are indispensable for reproduction of regular α-helical and β-sheet structures. The internal parameters of the individual Us were derived by fitting the analytical expressions to the RFE surfaces of model systems (13) or by fitting the calculated distribution functions to those determined from the PDB (8), whereas the ws were calculated by optimization of ΔE and Z of the training proteins (8–10) (see the Introduction for a brief description of the procedure used thus far). The force field is now able to predict the structures of proteins containing both α-helical and β-sheet structures with a reasonable degree of accuracy, as assessed by tests on model proteins (15) as well as in the CASP3 (17) and CASP4 (15) blind prediction experiments.
, m = 1, 2, … Ncorr are the correlation or multibody contributions that we derived (13) from a generalized-cumulant expansion (16) of the restricted free energy (RFE), and the ws are the weights of the energy terms. The multibody terms are indispensable for reproduction of regular α-helical and β-sheet structures. The internal parameters of the individual Us were derived by fitting the analytical expressions to the RFE surfaces of model systems (13) or by fitting the calculated distribution functions to those determined from the PDB (8), whereas the ws were calculated by optimization of ΔE and Z of the training proteins (8–10) (see the Introduction for a brief description of the procedure used thus far). The force field is now able to predict the structures of proteins containing both α-helical and β-sheet structures with a reasonable degree of accuracy, as assessed by tests on model proteins (15) as well as in the CASP3 (17) and CASP4 (15) blind prediction experiments.
The Algorithm.
The algorithm described in the Introduction was successful when the training proteins were small proteins with relatively simple topology; we used betanova (18), the 10-58 part of the N-terminal domain of staphylococcal protein A [PDB ID code 1BDD (19)] (hereafter referred to as protein A) and the tissue-type plasminogen activator [PDB ID code 1TPM (20)]. The optimized parameters were energy-term weights of Eq. 3. Although CSA did not produce any native-like structure with the initial weights, the native-like structures started to appear after a few iterations and finally were located as the lowest-energy structures. However, this was not the case when trying to optimize the UNRES energy function for the Src-homology 3 (SH3) domain [PDB ID code 1BK2 (21)] (a β protein) or the third IgG-binding domain from streptococcal protein G, a 61-residue protein [PDB ID code 1IGD (22)] (an α/β protein), which have more complicated topologies. In the first case, the native-like structure was never found by a global CSA search, whereas for 1IGD we found some higher-energy native-like structures. However, even in the case of 1IGD, successive iterations did not lower the energies of native-like structures. For proteins with more complicated topology, we achieved some success only in the case of the DNA-binding POU-specific domain [PDB ID code 1POU (23)] (an α-helical protein); however, even in this case, the final set of weights produced the native-like structure as the third-lowest-energy family of structures in the global CSA search (10), and further improvement could not be obtained.
There seem to be two major reasons for limited success with the procedure for training proteins with complicated topologies. First, we attempt to interpolate between two remote points in conformational space: the native-like structures obtained by perturbing the experimental structure and the decoys obtained with weights (in general, parameters of the potential-energy function) that are far from the optimum parameters. Second, all non-native structures obtained in a global search are pushed high in energy regardless of their degree of native likeness. For example, in the case of 1IGD, which consists of two β hairpins and an α-helix in the middle, a conformation with a partially native-like secondary structure (e.g., with one of the hairpins and the α-helix in place) should be preferred to a structure with no native segment, but it is equally non-native according to the current procedure. For the search algorithm to proceed quickly, it would be logical to energy rank all structures according to the degree of native likeness (and not distinguish only the complete native-like from non-native structures). Then lowering the energy would also imply narrowing down the available conformational space to structures with an increasing degree of native likeness, in which the native structure would be found more easily, as illustrated for 1FSD [a designed 28-residue peptide that contains the minimal α/β fold (24)] in Fig. 1. This observation has been demonstrated by a recent lattice-simulation study of Tiana and Broglia (25): these authors found that the search proceeds quickly if stable intermediates with clusters of native contacts are formed during the simulated folding process. Such ranking of energy levels also enables us to design a procedure that would not rely on interpolation between remote points: although it is not probable to find native-like structures in the pool of conformations generated by a global-minimum search with arbitrary starting weights, usually conformations with some native-like elements are obtained with any reasonable parameters of the energy function.
Figure 1.

Schematic illustration of the energy levels, which is the goal of the algorithm for optimizing the potential function proposed in this work, using the 1FSD peptide (24) as an example. The energies of the conformations should decrease with their increasing “native likeness.” The highest energy level (Level 0) is occupied by structures with either no or non-native secondary structure. The next level (Level 1) is occupied by the structures with one native secondary structure element (the N-terminal β-hairpin or the C-terminal α-helix; the native-like structure fragments are indicated by thicker lines). Yet lower energy (Level 2) has structures with both α-helix and β-hairpin but no or incorrect packing of these two substructures and/or shifted turn in the β-hairpin. Finally, the native-like structures, with α-helix and β-hairpin packed correctly, occupy the lowest energy level (Level 3). Because the number of structures with more and more defined native-like elements decreases, such ordering of structures leads to diminishing conformational entropy following the energy decrease, which is highly desirable to find the native structure quickly in a spontaneous energy-driven search of the conformational space.
On the basis of the above considerations, we now propose the following method for optimizing the potential-energy function. The goal of this procedure is to obtain the best set of the parameters of the UNRES energy function (Eq. 3):
- 1.
Define minimal integral fragments of the native structure. We define these fragments in terms of regular hydrogen-bonded patterns encountered in α-helices and β-sheets. For example, in the case of 1IGD, there are three native fragments: the N-terminal β-hairpin, middle α-helix, and C-terminal βhairpin.
- 2.
Define the hierarchy levels of the protein. For a protein with n native fragments, the minimal choice is to define n + 2 levels numbered from 0 to n + 1. Levels 0, 1, … , n consist of conformations containing 0, 1, … , n native fragments, respectively, without yet taking care of their proper packing. A fragment is considered native-like if it contains no less than half of the hydrogen-bonded contacts that occur in the corresponding fragment of the experimental structure. It is also allowed that the contacts are shifted by up to three amino acid residues: e.g., if a native β-hairpin that extends from residue 7 to residue 26 contains a turn at residues 16 and 17, we will also consider conformations that contain a turn at residues 13 and 14. Level n + 1 consists of the actual native-like structures (i.e., all of the constituent native fragments are properly arranged and packed). The division into hierarchy levels is illustrated in Fig. 1 for the example of 1FSD (24).
- 3.
Carry out the global conformational search of the protein with current parameters of the energy function.
- 4.
Assign conformations obtained during the search to structural levels, as described in step 2.
- 5.
Add those of the resulting conformations that are different from conformations found previously to the database of conformations used to optimize the energy function. If no new conformations were found, terminate.
- 6.
- Check whether the free-energy relationships (Eqs. 4 and 5) between structural levels are satisfied. If they are, the procedure ends. If not, adjust the parameters of the potential-energy function to satisfy the following relationships between configurational free energies of the subsets of the database of conformations:
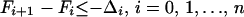
4 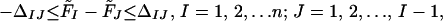
5
- where Fi is the configurational free energy of the ensemble consisting of the conformations of the structural class i, F̃I is the configurational free energy of the conformations within any structural class containing native-like fragment I, and the Δs are target gaps between the free energies. The gaps ΔI should be of the order of a few kcal/mol (we use 1 or 2 kcal/mol), whereas the gaps Δi depend on what structural levels are separated. From our experience, it follows that a larger gap should be assigned between level 0 (completely unfolded structures) and level 1, and then the gaps should decrease. Typically, we used 10, 5, and 2 kcal/mol for gaps between level 0 and level 1, level 1 and level 2, and level 2 and level 3, respectively (see Results and Discussion). The free energies of Eqs. 4 and 5 are defined by Eqs. 6 and 7, respectively:
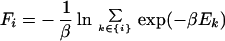
6 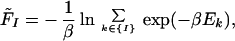
7
with {i} and {I} denoting the set of conformations of the ith structural level and those containing native-like fragment I, respectively. β can be identified with 1/RT, R being the gas constant and T being the absolute temperature, or treated as a parameter of the method. With large β (low temperature), Eqs. 4 and 5 approach energy gaps, as that in Eq. 1. The purpose of introducing the requirements in Eq. 4 is to push conformations with higher native likeness lower and lower in free energy, whereas the requirements in Eq. 5 cause all structural segments to be found with comparable probability.
Iterate steps 3–6.
To accomplish the task of step 6, we minimize the following target function:
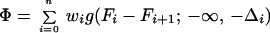 |
8 |
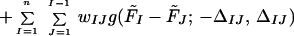 |
with
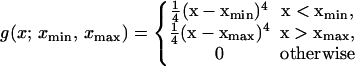 |
9 |
and the ws being the weights of the respective terms in the target function (it should be noted that they are different from the energy-term weights of Eq. 3). The Fs are defined by Eqs. 6 and 7, respectively, whereas the Δs are defined in the text following Eqs. 4 and 5, respectively. The components of the first sum in Eq. 8 correspond to the inequality relations defined by Eq. 4 and those of the second sum to the inequality relations defined by Eq. 5. Each component is zero, if the corresponding inequality (Eqs. 4 and 5) is satisfied. If Φ is zero, all inequalities are satisfied, which is the case of a perfect solution. A nonzero solution means that the free-energy differences exceed the target gaps Δ; i.e., there is a compromise between contradictory requirements to satisfy the respective inequalities. By assigning different ws to different free-energy differences in Eq. 8, we can guide the optimization in favor of selected inequalities; however, at present, we usually set all the ws to the same value. The adjustable parameters are, first of all, the energy-term weights in Eq. 3 and also other parameters of the UNRES energy function. In this work, we optimized some of the parameters of the potential of SC–SC interaction (see Results and Discussion), in addition to optimizing energy-term weights.
In contrast to our previous procedure (9, 10), the algorithm described above does not require interpolation between the perturbed native-like conformations and diverse conformations obtained in the global search. It should be sufficient that only representatives of structural level 1 (i.e., with each having a single native-like element) but representing all native-like elements be found in the global search with the initial parameters, which, in our experience, is usually the case. Then conformations containing gradually more and more native-like elements (i.e., representing higher structural levels) should emerge in subsequent cycles, as conformations of level 1 will start to prevail over conformations of level 0 (which contain no native-like elements). Because the inequalities of Eq. 5 are satisfied, the probability of finding conformations with each of the native-like elements will tend to be equal, which in turn will enable them to assemble to give conformations containing more native-like elements.
A procedure that weighted the target energy gap of a conformation with respect to the energy of the experimental structure depending on rmsd from the native structure was described earlier by Maiorov and Crippen (26). However, rmsd is a measure that is unable to distinguish conformations very far from the native structure, whereas our procedure can find native-like elements in conformations very different from the native structure in terms of rmsd. These workers also implemented continuous weighting of energy gaps depending on rmsd rather than energy levels. Therefore, the procedure described in this report is substantially different from that of Maiorov and Crippen (26).
Results and Discussion
In these preliminary studies, we wanted to determine how much can be achieved by potential-function optimization by requiring only that all correct native-like elements be present in the target structures after potential-function optimization. We therefore weakened the condition for the highest structural level (step 2 of the procedure described in Methods), which requires that the rmsd from the native structure be within a chosen cutoff limit, to require that all elements have correct hydrogen-bonded contacts (especially that the turns in β hairpins are not shifted).
The first system was 1FSD (24). The structural levels of this peptide are illustrated in Fig. 1. We started from the parameters of the UNRES force field that we used in the CASP4 experiment (10, 15), except that we replaced the torsional parameters with those obtained from MP2/6-31G* ab initio energy surfaces of model blocked amino acids, and we also included double-torsional energy terms calculated in our recent work.‡‡ Thus, the initial energy function was not optimized. The parameter β in Eq. 6 was equal to 10, which effectively means optimizing the difference between the lowest energies of the conformations belonging to the structural levels occurring in the corresponding inequality. Only the energy-term weights (the ws in Eq. 3) were optimized. The target gaps Δi were as follows: 10 kcal/mol between level 0 and level 1; 5 kcal/mol between level 1 and level 2; and 1 kcal/mol between level 2 and level 3. Because there were only two native-like segments, we did not implement the gaps between the free energies of structures with a defined native-like element (Eq. 5).
The evolution of the energy levels during the course of optimization is shown in Fig. 2. For the starting parameters (iteration 0), the conformations with one native element (level 1) are the lowest in energy (Fig. 2a). After the first iteration, conformations containing both α-helix and β-hairpin become the lowest in energy; they differ from the correct native structure by the packing of these two elements and by the fact that the β-turn is shifted one residue forward. After the fourth iteration, the lowest-energy conformation had the topology of the native structure, with 2.8 Å Cα rmsd from the native structure. This result is remarkable in view of the fact that the rmsd was not used as a criterion of native likeness during the optimization; also, because of the weakening of the conditions for the highest-level conformations, we did not require native-like packing. The lowest-energy structure is superposed on the experimental structure in Fig. 3.
Figure 2.

Illustration of the progress of optimization of the UNRES energy function for 1FSD [a designed α/β peptide (24)]. The crosses represent conformations obtained in CSA runs after optimizing parameters in a given iteration: (a) initial parameters; (b) iteration 1; (c) iteration 4 (the last iteration). See text for description.
Figure 3.

Superposition of the lowest-energy structures (green) of 1FSD (Left) and 1IGD (Right) obtained with the force fields optimized on these proteins on their experimental structures (22, 24) (red). For 1FSD, residues 3–26 (which are well-defined in the NMR structure) are superposed. The Cα-rmsd over residues 3–26 is 1.8 Å, whereas the Cα-rmsd over all 28 residues is 2.8 Å. For 1IGD, residues 25–47 (the central α-helix and the second loop) are superposed.
It should also be noted that, although the energy gap between the structures of level 3 and those of level 2 is small, the native-like structures of 1FSD (level 3) started to appear in CSA runs as the lowest-energy structures very quickly (after less than 2,000 energy minimizations) with the optimized energy function (Fig. 3). In our previous approach to optimization, in which only the energy gap and Z score between the native-like and non-native structures were optimized, the native-like structures appeared after ≈10,000 local minimizations, although the energy gap between the native and non-native structure was more than 2 kcal/mol. It appears that proper energy ranking of the non-native structure allows the native structure to be found quickly. This improved efficiency seems to be a result of the hierarchical design of the energy landscape so that, with decreasing energy, the conformational-search method chooses the structure from more and more native-like conformations. We observed that, if only the energy gap between the native and non-native structures is optimized, the search for the native structure with such a potential is slowed down by low-energy non-native structures that are geometrically far from the native structure, even though the energy gap between the native and non-native structures is significant.
Last, we carried out a preliminary study on 1IGD, which is a 61-residue α/β protein with a more complex fold (22). With the old procedure (9, 10), we were unable to optimize the potential-energy function, because the native-like structures were too far away from any structure obtained in a global CSA search (A.L., J.P., C.C., J.L., and H.A.S., unpublished work). On the other hand, structures with a single or two native elements were found quite often.
The native structure of 1IGD consists of three secondary-structure elements: the N-terminal β-hairpin from residue 5 to residue 26 (I), the middle α-helix from residue 27 to residue 42 (II), and the C-terminal β-hairpin from residue 46 to residue 51 (III). This time, we included, in the target functions defined by Eq. 8, the free energies calculated with β from 0.05 to 1.6 to optimize both the gaps between the lowest energies and those between averages over reasonably low energies. The rationale for this is that, in the early stages of the search, conformations with higher energies are found, and the CSA method, being a genetic algorithm, gives a greater chance for survival to those that have lower energies. Thus, if the lowest-energy conformations belonging to a higher structural level had lower energy compared to those of a lower level, they could be eliminated in favor of less native-like conformations, if their predecessors had significantly higher energies than those of the less native-like conformations.
The optimizable parameters consisted of energy-term weights, as well as the well depths (ɛ°) in the Gay–Berne potential of side-chain–side-chain interaction (see equation 6 in ref. 12 for definition). With those parameters, conformations with a single native element II or III, as well as those containing I and III (i.e., full β-sheet conformations) were present; in other words, the set contained all “building blocks.” After the first iteration, conformations containing all three native elements (i.e., belonging to level 3) started to appear, and they became the lowest in energy after 10 iterations. The lowest-energy conformation is shown in Fig. 3. It can be seen that the packing of the three elements is not native like, although all three elements are present. At this point, conformations obtained by perturbing the crystal structure could be introduced into the procedure, as in our earlier procedure (9, 10), to complete the optimization. Unlike the simpler case of 1FSD, structures with native-like packing did not start to appear spontaneously.
Even with incomplete optimization for 1IGD, we found it interesting to check the capability of the force field obtained with this protein. 1IGD is a protein that is complex enough for one to hope that the force field optimized by using this molecule may be transferable to other proteins. We therefore carried out a global conformational search of 1FSD (24), betanova (22), and the 10–45 N-terminal fragment of staphylococcal protein A (19). For 1FSD and betanova, the lowest-energy structures turned out to be native-like, whereas, for protein A, the native-like three-helix bundle had an energy 2 kcal/mol higher than the global minimum (Fig. 4). It should be noted that, in our previous studies, we had to optimize the energy function simultaneously on two proteins (betanova and protein A) to obtain a force field that recognized both α- and β-folds. It can therefore be suggested that hierarchical optimization of the potential-energy function results in a better-transferable force field than those obtained by optimization of energy gap/Z score alone.
Figure 4.

Superposition of the lowest-energy structure of betanova (18), 1FSD (24), and the second lowest in energy structure of protein A (19) (green) on the corresponding NMR structures (red), left to right. The Cα rmsds are 0.9, 2.9, and 3.1 Å, respectively.
Conclusion
We have proposed a method for optimizing a potential-energy function that is based on a hierarchical design of the energy landscape, with energy decreasing with increasing degree of native likeness. Preliminary tests on two systems, 1FSD and 1IGD, demonstrated that such a hierarchical design results in a force field with fast-folding properties because the set of accessible conformations is narrowed down, making them gradually closer to the native structure with decreasing energy. In this way, optimization also appears to result in better transferability of the force field, compared to the previous approaches, in which only the energy gap and/or Z score between the native and non-native structures were optimized, without taking into account energy ranking among partially unfolded and completely unfolded structures.
It should also be noted that the hierarchical optimization proposed in this work is very closely related to the form of real protein potential-energy landscapes, provided that folding is hierarchical. Here, we made an arbitrary assumption that native-like elements of structure should accumulate as folding progresses, which is not always the case [e.g., β-lactoglobulin, which is a β-sheet protein, has transition structures that contain a substantial amount of α-helical structure (27)]. We also assigned arbitrary energy gaps between the structural levels. In view of this problem, the use of experimental information from unfolding studies (such as the sequence of partially unfolded intermediates and their energy relations) should provide much more realistic potential-energy functions and extend their design beyond the features included here for hierarchical folding.
Acknowledgments
We thank Prof. Ron Elber and Dr. Jarosław Meller, Department of Computer Science, Cornell University, for helpful discussions. This research was supported by grants from the National Institutes of Health (GM-14312), the National Science Foundation (MCB00-03722), the Fogarty Foundation (R03 TW1064), the National Institutes of Health National Center for Research Resources (P41RR-04293), and the Polish State Committee for Scientific Research (KBN) (3 T09A 111 17 and 7 T09A 158 21). Support was also received from the National Foundation for Cancer Research. A large part of the computations in this work was carried out at: (i) the Cornell Theory Center, which receives funding from Cornell University, New York State, the National Center for Research Resources at the National Institutes of Health, and members of the Theory Center's Corporate Partnership Program; (ii) the computing resources provided by the National Partnership for Advanced Computational Infrastructure at the San Diego Supercomputer Center, supported in part by National Science Foundation cooperative agreement ACI-9619020; (iii) the resources of the Informatics Center of the Metropolitan Academic Network in Gdańsk; and (iv) the Interdisciplinary Center of Mathematical and Computer Modeling in Warsaw.
Abbreviations
- CSA
conformational space annealing
- PDB
Protein Data Bank
- rmsd
rms deviation
- UNRES
united residue
Footnotes
Ołdziej, S., Dobrzańska, U. & Liwo, A. (2001) in Seventh Electronic Computational Chemistry Conference (ECCC7), The Cooper Union for the Advancement of Science and Art, April 12–30, 2001, poster no. 41. (http://www.cooper.edu/engineering/chemechem/ECCC7)
References
- 1.Vásquez M, Némethy G, Scheraga H A. Chem Rev. 1994;94:2183–2239. [Google Scholar]
- 2.Anfinsen C B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 3.Crippen G M, Snow E. Biopolymers. 1990;29:1479–1489. doi: 10.1002/bip.360291014. [DOI] [PubMed] [Google Scholar]
- 4.Seetharamulu P, Crippen G M. J Math Chem. 1991;6:91–110. [Google Scholar]
- 5.Sali A, Shakhnovich E I, Karplus M. Nature (London) 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 6.Godzik A, Koliński A, Skolnick J. J Comput Aid Mol Des. 1993;7:397–438. doi: 10.1007/BF02337559. [DOI] [PubMed] [Google Scholar]
- 7.Hao M-H, Scheraga H A. Curr Opin Struct Biol. 1999;9:184–188. doi: 10.1016/s0959-440x(99)80026-8. [DOI] [PubMed] [Google Scholar]
- 8.Liwo A, Pincus M R, Wawak R J, Rackovsky S, Ołdziej S, Scheraga H A. J Comput Chem. 1997;18:874–887. [Google Scholar]
- 9.Lee J, Ripoll D R, Czaplewski C, Pillardy J, Wedemeyer W J, Scheraga H A. J Phys Chem B. 2001;105:7291–7298. [Google Scholar]
- 10.Pillardy J, Czaplewski C, Liwo A, Wedemeyer W J, Lee J, Ripoll D R, Arłukowicz P, Ołdziej S, Arnautova Y A, Scheraga H A. J Phys Chem B. 2001;105:7299–7311. [Google Scholar]
- 11.Meller J, Elber R. Proteins. 2001;45:241–261. doi: 10.1002/prot.1145. [DOI] [PubMed] [Google Scholar]
- 12.Liwo A, Ołdziej S, Pincus M R, Wawak R J, Rackovsky S, Scheraga H A. J Comput Chem. 1997;18:849–873. [Google Scholar]
- 13.Liwo A, Czaplewski C, Pillardy J, Scheraga H A. J Chem Phys. 2001;115:2323–2347. [Google Scholar]
- 14.Lee J, Scheraga H A, Rackovsky S. J Comput Chem. 1997;18:1222–1232. [Google Scholar]
- 15.Pillardy J, Czaplewski C, Liwo A, Lee J, Ripoll D R, Kaźmierkiewicz R, Ołdziej S, Wedemeyer W J, Gibson K D, Arnautova Y A, et al. Proc Natl Acad Sci USA. 2001;98:2329–2333. doi: 10.1073/pnas.041609598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kubo R. J Phys Soc Japan. 1962;17:1100–1120. [Google Scholar]
- 17.Orengo C A, Bray J E, Hubbard T, Conte L L, Sillitoe I. Proteins Struct. Funct. Genet. 1999. Suppl. 3, 149–170. [Google Scholar]
- 18.Kortemme T, Ramirez-Alvarado M, Serrano L. Science. 1998;282:253–256. doi: 10.1126/science.281.5374.253. [DOI] [PubMed] [Google Scholar]
- 19.Gouda H, Torigoe H, Saito A, Sato M, Arata Y, Shimada I. Biochemistry. 1992;31:9665–9673. doi: 10.1021/bi00155a020. [DOI] [PubMed] [Google Scholar]
- 20.Downing A K, Driscoll P C, Harvey T S, Dudgeon T J, Smith B O, Baron M, Campbell I D. J Mol Biol. 1992;225:821–833. doi: 10.1016/0022-2836(92)90403-7. [DOI] [PubMed] [Google Scholar]
- 21.Musacchio A, Noble M, Pauptit R, Wierenga R, Saraste M. Nature (London) 1992;359:851–855. doi: 10.1038/359851a0. [DOI] [PubMed] [Google Scholar]
- 22.Derrick J P, Wigley D B. J Mol Biol. 1994;243:906–918. doi: 10.1006/jmbi.1994.1691. [DOI] [PubMed] [Google Scholar]
- 23.Assa-Munt N, Mortishire-Smith R J, Aurora R, Herr W, Wright P E. Cell. 1993;73:193–205. doi: 10.1016/0092-8674(93)90171-l. [DOI] [PubMed] [Google Scholar]
- 24.Dahiyat B I, Mayo S L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 25.Tiana G, Broglia R A. J Chem Phys. 2001;114:2503–2510. [Google Scholar]
- 26.Maiorov V N, Crippen G M. J Mol Biol. 1992;227:876–888. doi: 10.1016/0022-2836(92)90228-c. [DOI] [PubMed] [Google Scholar]
- 27.Forge V, Hoshino M, Kuwata K, Arai M, Kuwajima K, Batt C A, Goto Y. J Mol Biol. 2000;296:1039–1051. doi: 10.1006/jmbi.1999.3515. [DOI] [PubMed] [Google Scholar]