

Abstract
Toxoplasma gondii is an intracellular parasite that proliferates within most nucleated cells, an important human pathogen, and a model for the study of human and veterinary parasitic infections. We used a stable yellow fluorescent protein-α-tubulin transgenic line to determine the structure of the microtubule cytoskeleton in T. gondii. Imaging of living yellow fluorescent protein-α-tubulin parasites by laser-scanning confocal microscopy (LSCM) failed to resolve the 22 subpellicular microtubules characteristic of the parasite cytoskeleton. To understand this result, we analyzed sources of noise in the LSCM and identified illumination fluctuations on time scales from microseconds to hours that introduce significant amounts of noise. We confirmed that weakly fluorescent structures could not be imaged in LSCM by using fluorescent bead standards. By contrast, wide-field microscopy (WFM) did visualize weak fluorescent standards and the individual microtubules of the parasite cytoskeleton. We therefore measured the fluorescence per unit length of microtubule by using WFM and used this information to estimate the tubulin content of the conoid (a structure important for T. gondii infection) and in the mitotic spindle pole. The conoid contains sufficient tubulin for ≈10 microtubule segments of 0.5-μm length, indicating that tubulin forms the structural core of the organelle. We also show that the T. gondii mitotic spindle contains ≈1 microtubule per chromosome. This analysis expands the understanding of structures used for invasion and intracellular proliferation by an important human pathogen and shows the advantage of WFM combined with image deconvolution over LSCM for quantitative studies of weakly fluorescent structures in moderately thin living cells.
Toxoplasma gondii is an obligate intracellular parasite that is able to proliferate within virtually any nucleated cell including human tissues (1). Many members of the phylum Apicomplexa such as Toxoplasma, Plasmodium, Eimeria, and Cryptosporidia are important human and veterinary pathogens. Their ability to cause disease critically depends on both the initial invasion and later escape from host cells, processes that require active movements generated by the parasite cytoskeleton (2–4).
T. gondii encodes only one copy each of α- and β-tubulin in its genome (5), yet it contains several distinct kinds of microtubules including 22 subpellicular microtubules (6–8) located just beneath the cell's complex surface membrane, centrioles, and spindle poles (9–13). Based on morphological studies, the subpellicular microtubules are thought to help maintain the integrity of the cell surface as well as play a role in active shape changes during invasion (2, 7, 8, 14). The apical region of most apicomplexan parasites contains a specialized cytoskeletal structure, the conoid (15, 16), that is actively protruded and retracted during the invasion process (14, 17). In electron microscopy (EM) studies, the conoid appears as a tightly coiled spiral of 10–12 fibrous elements of unknown composition (6–8). The identity and structure of these fibers have been the subject of much speculation fueled by suggestive evidence that the active movements of the conoid may correlate with changes in packing of the fibers (18). Several lines of evidence have suggested that this structure is not composed of microtubules: the organelle fails to stain with antitubulin antibodies; protofilaments are not visible in the fibers, nor do they have a tubular cross-section; and the fibers are more tightly coiled (radius ≈ 0.2 μm) than is typical for microtubules.
Fluorescence microscopy is a ubiquitous technique for determining the molecular constitution of organelles, cells, and tissues. The introduction of fluorescently tagged proteins (19–21) in T. gondii parasites (12, 22) has facilitated studies in living cells greatly. For high-resolution three-dimensional (3D) fluorescence imaging, two techniques have been developed that use different methods to generate an image of the fluorescence distribution in cells or tissues. A laser-scanning confocal microscope scans the sample with a laser beam focused to a diffraction-limited point. In addition, a pinhole is installed in the emission light path to prevent out-of-focus light from reaching the detector. Together, these two elements ensure that light arising from regions outside the diffraction-limited focal spot never reaches the detector. By contrast, a wide-field microscope collects the light from all points in the focal plane at once plus all the light from illuminated regions of the sample that are above and below the focal plane. In wide-field microscopy (WFM), contrast is reduced by the blurred light from out-of-focus regions. Computational methods known as deconvolution algorithms can reverse the loss of contrast effectively, compensating for the blurring effects of defocus and reconstructing the 3D distribution of fluorescent molecules in the sample (23–25).
T. gondii is only 2 μm wide and 7 μm long, yet it has a full complement of all the organelles found in much larger eukaryotic cells. Thus, resolving subcellular structures within these parasites pushes the limits of performance of light microscopy. We report here our observations by digital fluorescence microscopy on the T. gondii cytoskeleton, including unsatisfactory results using laser-scanning confocal microscopy (LSCM), and successful studies by WFM/deconvolution. An unexpected distribution of yellow fluorescent protein (YFP)-tagged tubulin motivated quantitative analysis of tubulin content in the apical region and spindle pole of the parasite, providing new insights into parasite biology. The results serve to clarify the relative merits of LSCM and WFM in the determination of cell structure by fluorescence microscopy.
Methods
Specimens.
Transgenic T. gondii parasites expressing YFP-α-tubulin (ptubYFP-α-tub/sag CAT) were generously provided by Boris Striepen (12) and maintained as described (22). For short-term imaging of live parasites, small chambers were made by sealing coverslips of cultured cells on top of thin spacers.
Five-day-old quail embryos were removed from eggs, dissected free of yolk and membranes, fixed in 4% formaldehyde in PBS with gentle agitation for 48 h, and then stained with 10 μM 4′,6-diamidino-2-phenylindole and 1.2 μM Alexa488-phalloidin (Molecular Probes) for 72 h. The embryos were mounted in the enzymatic oxygen-scavenging mixture (see Supporting Methods, which is published as supporting information on the PNAS web site, www.pnas.org) under a No. 1.5 coverslip supported by a spacer to avoid compression.
Preparation and Imaging of Fluorescent Beads in LSCM and WFM.
See Supporting Methods.
Microscopy.
LSCM.
LSCM images of parasites were acquired on a Zeiss LSM510 with an ×63 C Apochromat 1.2 numerical aperture (NA) water immersion lens with correction collar adjusted to minimize spherical aberration, 488-nm argon laser excitation, LP505 emission, and pinhole diameter set to 1.0 Airy disk, which was determined empirically to be optimal for this sample (26). For living cells, photomultiplier tube gain was set to maximum, and laser intensity then was set such that the brightest regions used the full 8-bit dynamic range of the detector. With this setting, 50% photobleaching required ≈200 scans.
LSCM images of quail embryo were recorded by using the same lens, argon 364- and 488-nm excitation, bandpass 390–470 nm for 4′,6-diamidino-2-phenylindole emission and longpass 515 nm for Alexa488 phalloidin, with pinhole set to 1.0 Airy disk.
WFM.
WFM images of parasites were recorded by using an ×100/1.4 NA oil immersion objective lens on a DeltaVision restoration microscope (Applied Precision, Issaquah, WA) built around an Olympus IX70 microscope equipped with mercury-arc illumination, a fluorescein filter set, and a Kodak 1401E charge-coupled device (CCD) mounted in a CH350 camera (Roper, Tucson, AZ). The effective pixel size was 0.067 μm. 3D images (37 optical sections, 0.25 μm per optical section) were corrected for variations in mercury-arc output by using a scaling factor determined by sampling 0.1% of the excitation beam during each exposure and then restored by constrained iterative deconvolution (23, 25).
For wide-field imaging of the fixed quail embryo, the slide used for LSCM was imaged on a DeltaVision restoration microscope built around a Zeiss Axiovert inverted microscope. Images were recorded by using an ×40/1.3 NA NeoFluar lens using an immersion oil that was found empirically to minimize spherical aberration 40–50 μm into the sample (laser liquid nD = 1.528, Cargille, Cedar Grove, NJ). Sixty-four optical sections spaced by 0.7 μm were collected by using an identical camera as described above. Each image measured 1,024 × 1,024, and effective image pixel size was 0.17 μm. Images were subsequently processed as described above. For presentation, the images were imported into Adobe PHOTOSHOP so that their full dynamic range was preserved.
Quantitative Measurements of YFP and Bead Fluorescence.
See Supporting Methods.
Measurement of LSCM Illumination Intensity Fluctuations.
Illumination intensity at the specimen plane was measured by using a flat, partially reflective mirror to reflect a portion of the laser light back through the objective lens to the photomultiplier tube detector. The surface of the mirror was positioned in the focal plane by maximizing the laser light reflected to the detector. A relatively low NA lens (×20, NA 0.5) was used with pinhole opened completely to eliminate variation caused by small changes in focus. The 488-nm line from a 25-mW argon laser was used at ≈10% full strength. For a few measurements, the beam from a small battery-powered diode laser (670 nm, 4.7 mW, LaserMax, Rochester, NY) was fed directly into the LSCM detector optics via an ×2.5 NA 0.12 objective. The signal to the detector from these test samples is extremely strong (at a photomultiplier tube voltage of less than 200 V the analog/digital converter became saturated), and thus the Poisson-distributed “shot noise” in the test images, proportional to the square root of the average number of photons detected per pixel, is expected to be insignificant in comparison with the real fluctuations in laser intensity. Indeed, we found that the root-mean-square noise hardly changed over a 6-fold range of laser intensities at constant dwell time. Real-space and Fourier-space (power spectrum) analyses of the intensity traces were carried out by using the SEMPER (Synoptics, Cambridge, U.K.) image-processing software.
Results
T. gondii is a highly successful protozoan parasite that grows well under laboratory conditions using fibroblasts in tissue culture as a host (Fig. 1A). The parasite is characterized by a complex array of cytoskeletal filaments thought to be involved in a variety of essential functions including host cell invasion, daughter parasite assembly, and maintaining the highly polarized organization of the cell. The extreme apical end of the parasite (the “stem” of this banana-shaped cell) contains the conoid, a somewhat mysterious organelle consisting of 10–12 tightly wound ribbon-like elements of unknown composition (refs. 7 and 8; Fig. 1B). Although sometimes assumed to consist of microtubules (15, 27), conoid filaments do not stain with antitubulin antibodies under most conditions, exhibit no clear subunit structure, appear much flatter than classical microtubules in EM images of cross-sections, and are bent into an unusually tight spiral (28) that seems incompatible with the structural rigidity of normal microtubules. Just below the conoid lies the inner apical polar ring, a probable microtubule organizing center (29). Twenty-two longitudinally oriented microtubules extend from this ring (Fig. 1B) and underlie the parasite's pellicle, a scaffolding that defines the parasite cell shape (6–8).
Figure 1.

LSCM imaging of living T. gondii expressing YFP-α-tubulin. (A) Combined differential interference contrast and YFP fluorescence LSCM image of T. gondii-infected fibroblasts. (B Upper) Diagram of T. gondii with two developing daughters. ER, endoplasmic reticulum. (B Lower) Major cytoskeletal elements including conoid, apical polar rings (pr), central pair MT, and subpellicular MT (adapted from ref. 28). (C) LSCM images of YFP fluorescence in optical sections of live parasites. (Upper) A section containing the plasma membrane. Faintly visible longitudinal striations (arrows) arise from subpellicular MT. (Lower) optical section through the middle of a different group of parasites. The bright apical spot marks the conoid region. Internal spots include conoids and spindle pole bodies of developing daughters, not well resolved in this image. (Scale bars: A, 10 μm; C, 5 μm.) (D) Changes in the intensity of illumination in the LSCM on three different time scales. (Top) Single-pixel intensities 3.56 μsec per pixel. (Middle) Average intensity of 10,000 pixels (3.56 μsec per pixel) over a 15-min period. (Bottom) Average of 10,000 pixels over a 12-h period.
To study parasite replication in living cells, we have exploited stable transgenic parasite lines expressing various YFP-tagged cytoskeletal proteins by using fluorescence microscopy to observe the 3D distribution and dynamic rearrangements of YFP-α-tubulin and other cytoskeletal elements. In LSCM images of YFP-α-tubulin transgenics, the cytoplasm appears uniformly fluorescent (Fig. 1 A and C), suggesting the presence of a significant amount of unpolymerized YFP-α-tubulin, presumably as a soluble heterodimer with endogenous β-tubulin. In addition, a small dot of intense YFP fluorescence is observed at the extreme apical end of the parasite in the region occupied by the conoid (Fig. 1C). A bright dot is seen also at the apical region of daughter parasites developing within the mother. However, the individual subpellicular microtubules known to be present from EM studies (6–8) are visualized very poorly in LSCM images of either living or fixed cells. Direct measurements using the same objective lens showed that we could just resolve the lines in a 0.22-μm test grating, which is consistent with the performance expected for 488-nm wavelength illumination and objective lens NA of 1.2. This result indicated that our problem was not caused by poor resolution but primarily was caused by a low SNR in living YFP-α-tubulin parasites. Indeed, after detergent extraction of YFP-α-tubulin transgenic parasites, the 22 subpellicular microtubules are clearly visible by LSCM, as they are by immunofluorescence of fixed and extracted nontransgenic cells (data not shown), suggesting that unpolymerized YFP-α-tubulin masks the tubulin polymer in living parasites.
Clearly the low intensity of the fluorescence emission from intact parasites limits SNR. Increasing the fluorescence signal brightness by increasing laser power caused severe photobleaching and photodamage before complete 3D data could be collected. In rare images from the laser-scanning confocal microscope, we could faintly discern stripes that might correspond to individual microtubules (Fig. 1C), but the visibility of these stripes seemed to fluctuate erratically even in fixed cells. This variability suggested that random noise was reducing the contrast in our confocal images. We used the reflection from a mirror placed in the sample plane to measure the illumination delivered to the sample. Fig. 1D shows that the intensity of illumination was fluctuating significantly on time scales ranging from microseconds to hours. Approximately equivalent fluctuations in illumination intensity were measured on seven other laser-scanning confocal microscopes in five different locations including instruments from every major manufacturer (data not shown). By comparing measurements using the normal LSCM gas lasers and using a battery-driven diode laser we found evidence for at least three sources of noise: the gas lasers themselves, the LSCM detector electronics, and the fiber optic coupling of laser to microscope (see Discussion).
To determine whether it was possible to use LSCM for quantitative measurements of the YFP-α-tubulin in our cells or in any other weakly fluorescent specimen, we carried out a series of measurements with standard well characterized test objects. We recorded images of a mixture of 2.5-μm beads containing seven known relative fluorophore densities (manufacturer's specifications: 100, 30, 10, 3, 1, and 0.3% and nonfluorescent; Fig. 2A). We then determined how well LSCM measured the relative fluorescence content of the beads (see Methods) and compared this to the bead specifications and to a similar determination recorded by fluorescence-activated cell sorter (FACS). (Signal levels in YFP-α-tubulin parasites are similar to those in the weakly fluorescent beads.) Fig. 2B shows the distribution of summed fluorescence present in 250 individual beads measured by LSCM displayed on a logarithmic scale so that the peaks are spaced evenly. We observed five distinct populations of fluorescence densities that formed apparent normal distributions, the means of which increase in approximate geometric progression, paralleling the relative fluorescence recorded by FACS (Fig. 2B, arrows). The sixth and dimmest fluorescent bead group was observed as a scattered nonnormal distribution at the bottom of the scale. The relative intensity of this dimmest population measured by FACS was slightly lower than specified by the manufacturer.
Figure 2.

WFM and LSCM imaging of calibrated fluorescent standards. (A) Combined differential interference contrast and fluorescence image of a mixture of 2.5 μm polystyrene beads with six different fluorophore densities, appearing here as orange (brightly fluorescent) to gray (weakly fluorescent). (Scale bar, 5 μm.) (B–D) Total fluorescence in 3D images of beads (see Methods). The plots show the distribution of fluorescence measured by LSCM (B), WFM (C), and WFM after image deconvolution (D). After deconvolution, the absolute fluorescence rises substantially as out-of-focus information is shifted to an in-focus position, but the relative intensities do not change. The arrows indicate the median fluorescence of each bead population measured by FACS (see Supporting Methods).
We next compared this distribution with data obtained from an equivalent sample of 290 beads on a wide-field CCD-based microscope (see Methods; Fig. 2C). 3D images of individual beads were recorded and fluorescence content was measured as described above. We again observed a series of normal distributions representing each of the bead populations. However, the weakest bead population was a discrete normal distribution in this measurement, suggesting that the intrinsic noise level was lower in this WFM than in our LSCM. The WFM relative intensities closely approximate the manufacturer's specifications and are similar to the data obtained by FACS (Fig. 2C, arrows) with the exception that the FACS again tends to slightly underestimate the relative intensity of the dimmest population of beads (see Fig. 2 B and C). The range of the distribution within each group measured by WFM was narrower than by LSCM, and the coefficients of variation for each group were also smaller than in the LSCM data (Table 1). We concluded that the WFM produces a more accurate measurement of sample fluorescence than the LSCM.
Table 1.
Statistical analysis of noise in measurements of fluorescent beads
| Beads* | LSCM | FACS† | WFM/Raw | WFM/Decon | FACS‡ |
|---|---|---|---|---|---|
| .3 | .44 | .09 | .35 | .24 | .20 |
| 1 | .15 | .07 | .09 | .10 | .10 |
| 3 | .18 | .05 | .08 | .08 | .08 |
| 10 | .37 | .06 | .09 | .08 | .07 |
| 30 | .24 | .06 | .07 | .08 | .06 |
| 100 | .13 | .05 | .06 | .07 | .06 |
Values are coefficients of variation (standard deviation/arithmetic mean) for each of the bead populations measured in Fig. 2. Between 30 and 45 beads were used for each bead population assayed by microscopy. 1200–1500 beads from each population were used for each FACS assay (see Methods).
Nominal relative bead fluorophore density as specified by manufacturer.
Coefficients of variation for beads used for LSCM measured by FACS.
Coefficients of variation for beads used for WFM measured by FACS.
A major difference between LSCM and WFM lies in the amount of out-of-focus information present in WFM images. In WFM images of living parasites, a large amount of out-of-focus light degrades the contrast, interfering with quantitative measurements of YFP-tubulin fluorescence. Constrained deconvolution is a computational algorithm that restores out-of-focus light from its recorded position back to the object from which it emanated (23–25). To determine the effect of removing out-of-focus light on quantitative measurements, we processed WFM bead images by constrained deconvolution (Fig. 2D). As expected, the absolute bead intensities were increased substantially after deconvolution, because the “misplaced” out-of-focus light had been restored, but the distribution of relative bead intensities before and after deconvolution were similar (Fig. 2D). The increase in contrast after deconvolution had a significant effect on the SNR in measurement of the weakest beads, as shown by the decreased coefficient of variation compared with unprocessed WFM data for this population (Table 1). Thus combining WFM with a CCD detector and image deconvolution generated the most accurate measurement of sample fluorescence for weakly fluorescent objects and therefore might be used for quantitative analysis of images of living cells with limited signal levels.
We repeated our earlier attempt to image and measure microtubules in living T. gondii expressing YFP-α-tubulin, this time by WFM instead of LSCM. As predicted by the results with imaging of weakly fluorescent beads, images of living YFP-α-tubulin transgenic parasites acquired with a sensitive CCD camera on a wide-field microscope were much better than our confocal images and clearly showed the individual subpellicular microtubules even before deconvolution (Fig. 3A). Constrained iterative deconvolution of the WFM images removed the blurring caused by the objective lens and reduced the background caused by fluorescence from out-of-focus regions (Fig. 3B). This markedly improved the visibility of the individual microtubules, increasing contrast to 10–20% vs. 2–3% in the raw images (Fig. 3C). Based on the finding that image restoration by deconvolution faithfully maintains the relative intensities of fluorescent beads, we used deconvolved images to quantitate the localization of tubulin polymer in living T. gondii parasites.
Figure 3.

WFM imaging of T. gondii expressing YFP-α-tubulin. (A) Raw WFM YFP fluorescence image of several living parasites with the focal plane near the plasma membrane. (B) Same as A, after processing of the 3D stack by constrained iterative deconvolution. Microtubules are clearly visible as bright striations. Arrows in A and B point to a single microtubule. Compare these with LSCM images in Fig. 1. (C) Plot of the change in intensity along the dashed line in B and the corresponding pixels in A. (D) Cartoon (Lower left) and deconvolved image showing a single focal plane through the interior of a group of parasites revealing conoid, spindle pole, and centriole. Developing daughters are revealed by the U-shaped outline of their growing subpellicular microtubules, capped by the bright conoid. The cartoon of the lowermost parasite in the image shows the substructures within the parasite (see also Fig. 1B). Arrows indicate conoids of mother and daughter parasites. The subpellicular microtubules of the mother (bright perimeter originating at the conoid) extend half the length of the parasite. The distal portion of the cartoon parasite is outlined by the dotted lines. Distal to the daughter conoid, the two dots indicate the location of the centriole and spindle pole, in that order. (Scale bar in A, B, and D, 3 μm.)
By measuring YFP-α-tubulin fluorescence along microtubules and subtracting the cytoplasmic (i.e., nonpolymer) background fluorescence, we determined the YFP-α-tubulin fluorescence per unit length on subpellicular microtubules. The average of these values then was used to determine the tubulin polymer content in the apical region of the parasites, the centriole, and the spindle pole. We found that the conoid in interphase cells corresponds to 4.7 μm (SEM = 0.61 μm) of microtubule. This result is compatible with a conoid composed of 10–11 microtubule segments (or other polymeric structure of similar tubulin content), each ≈0.5 μm long. This large amount of YFP fluorescence could not arise from the two central microtubules within the conoid, because their combined length is only ≈0.7 μm (Fig. 1B), nor is the fluorescence likely to be accounted for by tubulin in the apical polar rings, which are not long enough to contain this amount of tubulin polymer (≈1.0-μm circumference). We also measured tubulin content in the conoids of mother and daughter in parasites undergoing cell division (Fig. 3D). The daughters' conoids contain significantly more tubulin polymer than do the maternal conoids in the same cell (maternal, 5.1 μm, SEM = 0.37 μm; daughter, 7.1 μm, SEM = 0.3 μm), suggesting that either the maternal conoid begins disassembling relatively early during cell division or the daughter conoids undergo some maturation step after their initial assembly.
In addition to the apical conoid region and subpellicular microtubules, YFP-α-tubulin also highlights two other microtubule structures in T. gondii transgenics, the centriole and spindle poles, during cell division (Fig. 3D). The spindle pole has been observed in electron micrographs (11) as two cone-shaped electron-dense regions localized on the surface of the intact nucleus during cell division. From EM images, the length of the spindle pole was measured to be 0.12–0.15 μm. We measured YFP fluorescence in the spindle pole of parasites in several different vacuoles containing populations of cells that were in early and midstages of cell division by using the same deconvolved images as used for the conoid measurements. There was some tendency for the tubulin content to be larger in the cells at earlier stages of cell division, but the uncertainty in these measurements was too large for us to be sure of its significance. Ignoring the difference in stage of cell cycle and pooling all measurements together yield an average tubulin content corresponding to 1.26 μm (SEM = 0.22 μm) of subpellicular microtubule or ≈10 individual microtubules for a spindle pole that is 0.12 μm long. T. gondii has 11 chromosomes (30, 31), and thus our measurement of tubulin polymer content is consistent with a mitotic spindle consisting of ≈1 microtubule per chromosome. The disposition of kinetochore and nonkinetochore microtubules in this organism will require further analysis by EM.
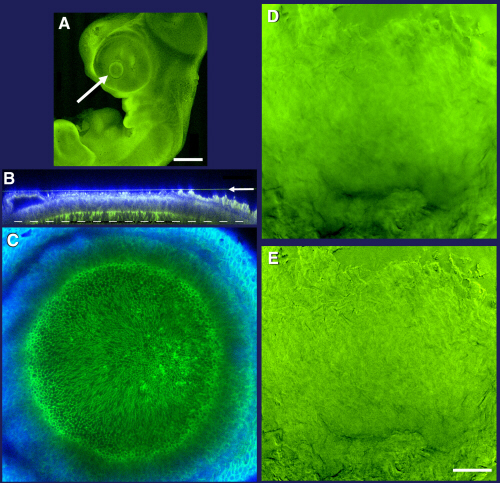
Given the disappointing performance of our LSCM in quantitative measurements of 3D fluorescence distribution, should all 3D fluorescence microscopy be done on a wide-field microscope? Fig. 4 shows a different low-contrast specimen, for which the laser-scanning confocal microscope outperformed the wide-field microscope. A whole-mount fixed quail embryo was stained with 4′,6-diamidino-2-phenylindole to highlight nuclei and a fluorescent derivative of phalloidin to label actin filaments (see Supporting Methods). An optical section from an LSCM 3D data set clearly shows actin outlining the cortices of individual cells in the developing lens. A WFM optical section recorded in the same place in the same sample is completely uninformative (Fig. 4B and Fig. 5, which is published as supporting information on the PNAS web site), and deconvolution is unable to reveal any more detail (see Supporting Methods). In relatively thick and densely stained samples, the SNR in WFM images is degraded by out-of-focus light, whereas the LSCM minimizes out-of-focus light reaching the detector. For such samples, LSCM should be used.
Figure 4.

Comparison of LSCM and WFM performance in imaging of a thick tissue. A 5-day-old quail embryo fixed in formaldehyde and stained with Alexa488-phalloidin is shown. See Supporting Methods for more details. (A) A single LSCM optical section ≈50 μm below the exterior surface of the embryonic eye. Concentrations of actin at cell cortices are visible. (B) The same location in the embryo imaged by WFM recorded with a CCD camera. (Scale bar, 50 μm.)
Discussion
Living T. gondii parasites stably expressing YFP-α-tubulin (12) contain a high concentration of tubulin in the conoid region, a finding of great importance in trying to understand the structure and function of this intriguing organelle. Previous workers have speculated that the ribbon-like fibers of the conoid might be microtubules (15, 27), but no structural, biochemical, or immunological evidence supports this speculation (32). In EM images of thin sections, the conoidal fibers are not tubular, whereas the 22 microtubules located just under the pellicle look entirely normal (28). Nevertheless, our quantitative measurements of YFP fluorescence in the apical region, calibrated by measuring the fluorescence of individual subpellicular microtubules, reveal that the apical fluorescent spot contains as much tubulin as ≈5 μm of microtubule. This result agrees well with the total length of the spirally wound ribbon-like elements visible in the conoid by EM, suggesting that they are in fact some form of polymeric tubulin. Quantitative measurements rule out the central pair of microtubules and the apical polar rings as the source of YFP fluorescence in this region, leaving the conoid itself as the only plausible site. Careful EM analysis will be necessary to determine how the ultrastructural arrangement of this tubulin is related to the lattice arrangement of typical microtubules or other tubulin polymers (33, 34). Because the conoid protrudes during parasite invasion (14, 17), it will be interesting to determine whether a microtubule-based motor protein of the dynein or kinesin family is present. Our measurements also suggest that the amount of tubulin in the conoid decreases as daughters mature into adults.
We were able also to estimate the tubulin content of the T. gondii spindle pole. Mitosis in this parasite proceeds without breakdown of the nuclear membrane. The mitotic spindle has not been seen by light microscopy and is also difficult to observe by EM. Consequently, the structure of the spindle and spindle pole is not well known. The estimates of tubulin content reported here will be useful in further analysis of the structures used for mitosis in T. gondii.
Our LM measurements of tubulin content will be invaluable in interpreting future EM studies of conoid ultrastructure, just as gel-based measurements have been crucial in interpreting past EM studies (see ref. 35 for review). Our estimates assume that the ratio of YFP-α-tubulin to endogenous tubulin incorporated into subpellicular microtubules is the same as into other structures. This assumption might be incorrect for two reasons. First, YFP-α-tubulin might be an imperfect mimic of α-tubulin, encountering obstacles that do not affect the smaller endogenous protein. In yeast, green fluorescent protein-α-tubulin appears to be incorporated normally into all microtubules but nevertheless cannot complement mutations of the endogenous α-tubulin gene (36), suggesting imperfect mimicry. Second, endogenous α-tubulin itself may be used to a different extent in different organelles. T. gondii, similar to most eukaryotes, has multiple tubulins (including α, β, and γ isoforms), and it is clear from other organisms that the different tubulin isoforms are not used equally for all purposes (37–40).
To calibrate our measurement of tubulin in the apical spot of YFP-α-tubulin parasites, we tried to visualize individual subpellicular microtubules within living cells by confocal microscopy but failed despite careful optimization of instrument parameters (26), imaging conditions, and different objective lenses. Attempts to understand this failure highlight the differences in performance between LSCM and WFM and uncovered surprisingly large artifactual pixel-to-pixel intensity fluctuations in LSCM images, especially at low signal levels. Further investigation revealed significant intensity fluctuations in every confocal microscope examined. Our own measurements and discussions with the instrument manufacturers identified two major sources of this intensity fluctuation: the laser and the optical fiber. The output of gas lasers used in commercial confocal microscopes fluctuates significantly over short time periods. During normal operation of beam-scanning confocal microscopes, the beam dwells on each pixel for 1–20 μsec. Averaged over 4 μsec (as in Fig. 1D) the laser intensity fluctuates by 2–3%, but fluctuations up to 10–15% are observed with submicrosecond dwell times. When averaged over a much longer time (msec to sec), the amplitude of the fluctuation is small, in the range of 0.5–1%. However, on this time scale another source of noise becomes significant. Most modern instruments bring the laser beam to the microscope via an optical fiber, typically a single-mode polarization-preserving fiber. Both the intensity and plane of polarization of the fiber output fluctuate on time scales from seconds to hours in response to imperceptible environmental changes such as gentle breezes, temperature oscillations, etc. Sensitivity to the environment can be reduced by painstaking alignment of one axis of birefringence of the fiber with the plane of polarization of the laser, but even with the most skillful alignment the residual fluctuations are ≥5%. Without frequent realignment, fluctuations in the range of 15% are routine. Finally, we note that comparison of power spectra from images acquired by using LSCM gas lasers vs. a battery-driven diode laser revealed an additional noise contribution that we presume must arise from the LSCM electronics. All these noise sources seem likely to stem from problems in implementation rather than fundamental physical limitations.
Why are subpellicular microtubules not visible in LSCM images of T. gondii parasites? WFM images revealed that the contrast between the microtubules and the surrounding cytoplasm is very small, of the order of 2–3%. This is the expected order of magnitude for the contrast of a microtubule immersed in cytoplasm containing 20–30 μM nonpolymerized tubulin and imaged by using a high NA lens. If the random intensity fluctuations in the image are much greater than 2–3%, individual microtubules will be difficult to see. Thus the available contrast caused by small differences in fluorescence between the microtubules and the surrounding cytoplasm is lost in the LSCM images, simply buried in noise.
WFM images are much less noisy for three reasons. First, illumination is delivered in parallel to all regions of the sample simultaneously, eliminating pixel-to-pixel differences in delivered illumination. Second, the CCD has ≈4-fold higher detection efficiency than the photomultiplier tubes of the laser-scanning confocal microscope, increasing the number of detected photons by 4-fold, thus reducing the Poisson shot noise by a factor of 2 for the same total illumination (which is limited in these living samples by photobleaching and phototoxicity). Finally, the CCD read-out electronics are adding less noise to the signal than the LSCM electronic circuitry.
LSCM images, particularly images of fixed samples protected from photobleaching, often are less noisy than our measurements on test samples (e.g., Fig. 1D). A common artifact of LSCM, ground-state depletion of the fluorophore, leads to substantial noise reduction. When using point-scanned laser illumination, it is very easy to achieve illumination levels that promote most of the fluorophores in the focal spot into the excited state. When this occurs, the amount of fluorescent light emitted by the sample becomes independent of modest changes in illumination intensity. Note that even with complete ground-state depletion, the image remains a faithful quantitative representation of spatial variations in fluorophore concentration; only the representation of illumination intensity changes is hidden. Changes in illumination intensity are revealed only when a large fraction of the molecules remains in the ground state (i.e., very low laser power), as in live-cell imaging, for which one uses the absolute minimum illumination intensity to minimize photodamage to the specimen.
The two samples examined in this paper demonstrate that it is not possible to use a single type of microscope for 3D imaging of all specimens. As a rule of thumb, the deciding factor in choosing between LSCM and WFM/deconvolution will be the thickness of the layer of fluorescent material in the specimen. With thin specimens (including thin layers of fluorescence within thick specimens), the SNR of WFM images nearly always will be superior to LSCM. As the fluorescing layer increases in thickness, deconvolution will be useful to restore image contrast until, above some limiting thickness, the SNR in the raw image data drops to such low values that the algorithm fails, as happened for WFM imaging of the embryo shown in Fig. 4. The contrast in the LSCM image of the embryo was acceptable, because the out-of-focus light was mostly removed by the pinhole.
How can the thickness beyond which WFM/deconvolution is inappropriate be estimated? Consider a thick specimen in which a fluorophore is uniformly distributed laterally over an area that is larger than the field of view. In a microscope, fluorescence is excited in conical regions above and below the plane of focus. Assuming for the moment that absorption of the excitation beam is small, then the same number of photons passes through every horizontal plane within these conical regions. Imagine a thin horizontal layer somewhere within this cone of illumination. The total amount of fluorescence arising from each such layer will be the same no matter where it is positioned vertically in the sample. If we choose the thickness of this horizontal layer to be the z-axis resolution, then we see that the contrast in a WFM image (ratio of in-focus to out-of-focus fluorescent light) will be approximately the vertical resolution (i.e., thickness of the in-focus layer) divided by the thickness of the specimen. We can expect trouble with WFM/deconvolution when this ratio approaches the minimum SNR in the raw image data that the deconvolution algorithm can accept. If the deconvolution algorithm begins to fail when the contrast in the raw image data falls to below 1%, for example, then samples with a (fluorescent layer) thickness greater than 100 times the vertical resolution will be inappropriate, providing an upper bound on the thickness. If the fluorophore concentration or light scattering is high enough to cause significant attenuation of the excitation light, then the thickness limit will be much smaller. Applying this guideline to a common situation, cells growing in tissue culture (thickness typically less than 30 μm; vertical resolution typically ≈0.5 μm) nearly always will yield better images with WFM plus deconvolution than with LSCM.
Supplementary Material
Acknowledgments
Some of the data used for this paper were collected on an Applied Precision DeltaVision workstation installed at the Marine Biological Laboratory (Woods Hole, MA) for the 2000 Optical Microscopy and Imaging in the Biological Sciences course. We are grateful to Applied Precision, Inc., the Marine Biological Laboratory, and the course organizers for the opportunity to use this equipment. We thank Dr. Boris Striepen for the YFP-α-tubulin transgenic parasites and Sarah Blackwood and Simon Powis for help with FACS analysis. This research was supported by National Institutes of Health Grants R01 A149301 (to J.M.M. and D.S.R.) and R21 GM60216 (to J.M.M.) and Wellcome Trust Career Development Fellowship 054333 (to J.R.S.). D.S.R. is a Burroughs Wellcome Scholar in Molecular Parasitology.
Abbreviations
- 3D
three-dimensional
- LSCM
laser-scanning confocal microscopy
- WFM
wide-field microscopy
- YFP
yellow fluorescent protein
- NA
numerical aperture
- CCD
charge-coupled device
- SNR
signal-to-noise ratio
- EM
electron microscopy
- FACS
fluorescence-activated cell sorter
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Frenkel J K. Toxoplasmosis: Parasite Life Cycle, Pathology, and Immunology. Baltimore: University Park; 1973. [Google Scholar]
- 2.Dobrowolski J, Sibley L D. Behring Inst. Mitt. 1997. 90–96. [PubMed] [Google Scholar]
- 3.Morisaki J H, Heuser J E, Sibley L D. J Cell Sci. 1995;108:2457–2464. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- 4.Sibley L D, Hakansson S, Carruthers V B. Curr Biol. 1998;8:R12–R14. doi: 10.1016/s0960-9822(98)70008-9. [DOI] [PubMed] [Google Scholar]
- 5.Nagel S D, Boothroyd J C. Mol Biochem Parasitol. 1988;29:261–273. doi: 10.1016/0166-6851(88)90081-3. [DOI] [PubMed] [Google Scholar]
- 6.Cintra W M, De Souza W. J Submicrosc Cytol. 1985;17:503–508. [PubMed] [Google Scholar]
- 7.Morrissette N S, Murray J M, Roos D S. J Cell Sci. 1997;110:35–42. doi: 10.1242/jcs.110.1.35. [DOI] [PubMed] [Google Scholar]
- 8.Nichols B A, Chiappino M L. J Protozool. 1987;34:217–226. doi: 10.1111/j.1550-7408.1987.tb03162.x. [DOI] [PubMed] [Google Scholar]
- 9.Perkins F O. J Cell Sci. 1975;18:327–346. doi: 10.1242/jcs.18.2.327. [DOI] [PubMed] [Google Scholar]
- 10.Musisi F L, Bird R G, Brown C G, Smith M. Z Parasitenkd. 1981;65:31–41. doi: 10.1007/BF00926551. [DOI] [PubMed] [Google Scholar]
- 11.Sheffield H G, Melton M L. J Parasitol. 1968;54:209–226. [PubMed] [Google Scholar]
- 12.Striepen B, Crawford M J, Shaw M K, Tilney L G, Seeber F, Roos D S. J Cell Biol. 2000;151:1423–1434. doi: 10.1083/jcb.151.7.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canning E U. BioSystems. 1988;21:333–340. doi: 10.1016/0303-2647(88)90030-5. [DOI] [PubMed] [Google Scholar]
- 14.Chiappino M L, Nichols B A, O'Connor G R. J Protozool. 1984;31:288–292. doi: 10.1111/j.1550-7408.1984.tb02963.x. [DOI] [PubMed] [Google Scholar]
- 15.de Souza W. Rev Inst Med Trop Sao Paulo. 1974;16:32–38. [PubMed] [Google Scholar]
- 16.McLaren D J, Paget G E. Parasitology. 1968;58:561–571. doi: 10.1017/s0031182000028869. [DOI] [PubMed] [Google Scholar]
- 17.Mondragon R, Frixione E. J Eukaryotic Microbiol. 1996;43:120–127. doi: 10.1111/j.1550-7408.1996.tb04491.x. [DOI] [PubMed] [Google Scholar]
- 18.Senaud J. Protistologica. 1967;3:167–232. [Google Scholar]
- 19.Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 20.Stearns T. Curr Biol. 1995;5:262–264. doi: 10.1016/s0960-9822(95)00056-x. [DOI] [PubMed] [Google Scholar]
- 21.Pepperkok R, Squire A, Geley S, Bastiaens P I. Curr Biol. 1999;9:269–272. doi: 10.1016/s0960-9822(99)80117-1. [DOI] [PubMed] [Google Scholar]
- 22.Striepen B, He C Y, Matrajt M, Soldati D, Roos D S. Mol Biochem Parasitol. 1998;92:325–338. doi: 10.1016/s0166-6851(98)00011-5. [DOI] [PubMed] [Google Scholar]
- 23.Swedlow J R, Sedat J W, Agard D A. In: Deconvolution of Images and Spectra. Jansson P A, editor. New York: Academic; 1997. pp. 284–309. [Google Scholar]
- 24.McNally J G, Karpova T, Cooper J, Conchello J A. Methods. 1999;19:373–385. doi: 10.1006/meth.1999.0873. [DOI] [PubMed] [Google Scholar]
- 25.Agard D A. Annu Rev Biophys Bioeng. 1984;13:191–219. doi: 10.1146/annurev.bb.13.060184.001203. [DOI] [PubMed] [Google Scholar]
- 26.Sandison D R, Piston D W, Williams R M, Webb W W. Appl Opt. 1995;34:3576–3588. doi: 10.1364/AO.34.003576. [DOI] [PubMed] [Google Scholar]
- 27.Dubey J P, Lindsay D S, Speer C A. Clin Microbiol Rev. 1998;11:267–299. doi: 10.1128/cmr.11.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrissette N S. Biology. Philadelphia: Univ. of Pennsylvania; 1995. p. 191. [Google Scholar]
- 29.Russell D G, Burns R G. J Cell Sci. 1984;65:193–207. doi: 10.1242/jcs.65.1.193. [DOI] [PubMed] [Google Scholar]
- 30.Sibley L D, Boothroyd J C. Mol Biochem Parasitol. 1992;51:291–300. doi: 10.1016/0166-6851(92)90079-y. [DOI] [PubMed] [Google Scholar]
- 31.Sibley L D, LeBlanc A J, Pfefferkorn E R, Boothroyd J C. Genetics. 1992;132:1003–1015. doi: 10.1093/genetics/132.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwartzman J D, Krug E C, Binder L I, Payne M R. J Protozool. 1985;32:747–749. doi: 10.1111/j.1550-7408.1985.tb03115.x. [DOI] [PubMed] [Google Scholar]
- 33.Dustin P. Microtubules. Berlin: Springer; 1984. [Google Scholar]
- 34.Amos L A, Klug A. J Cell Sci. 1974;14:523–549. doi: 10.1242/jcs.14.3.523. [DOI] [PubMed] [Google Scholar]
- 35.Murray J M. Int Rev Cytol. 1991;125:47–93. doi: 10.1016/s0074-7696(08)61216-4. [DOI] [PubMed] [Google Scholar]
- 36.Ding D Q, Chikashige Y, Haraguchi T, Hiraoka Y. J Cell Sci. 1998;111:701–712. doi: 10.1242/jcs.111.6.701. [DOI] [PubMed] [Google Scholar]
- 37.Marschall L G, Jeng R L, Mulholland J, Stearns T. J Cell Biol. 1996;134:443–454. doi: 10.1083/jcb.134.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mogensen M M, Mackie J B, Doxsey S J, Stearns T, Tucker J B. Cell Motil Cytoskeleton. 1997;36:276–290. doi: 10.1002/(SICI)1097-0169(1997)36:3<276::AID-CM8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 39.Stearns T, Evans L, Kirschner M. Cell. 1991;65:825–836. doi: 10.1016/0092-8674(91)90390-k. [DOI] [PubMed] [Google Scholar]
- 40.Chang P, Stearns T. Nat Cell Biol. 2000;2:30–35. doi: 10.1038/71350. [DOI] [PubMed] [Google Scholar]
- 41.Efron B, Tibshirani R. Science. 1991;253:390–395. doi: 10.1126/science.253.5018.390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}