

Abstract
Drug design and development are central to clinical research, yet 90% of drugs fail to reach the clinic, often due to inappropriate selection of drug targets. Conventional methods for target identification lack precision and sensitivity. While various computational tools have been developed to predict the druggability of proteins, they often focus on limited subsets of the human proteome or rely solely on amino acid properties. Our study presents DrugProtAI, a tool developed by implementing a partitioning-based method and trained on the entire human protein set using both sequence- and non–sequence-derived properties. The partitioned method was evaluated using popular machine learning algorithms, of which Random Forest and XGBoost performed the best. A comprehensive analysis of 183 features, encompassing biophysical, sequence-, and non–sequence-derived properties, achieved a median Area Under Precision-Recall Curve (AUC) of 0.87 in target prediction. The model was further tested on a blinded validation set comprising recently approved drug targets. The key predictors were also identified, which we believe will help users in selecting appropriate drug targets. We believe that these insights are poised to significantly advance drug development. This version of the tool provides the probability of druggability for human proteins. The tool is freely accessible at https://drugprotai.pythonanywhere.com/.
Keywords: drug discovery, druggable targets, machine learning, ensemble-based methods, feature selection
Introduction
Drug design and development have been arduous tasks, yet a staggering 90% of drugs fail to reach the clinic [1]. The ability to predict whether a protein can effectively bind to small molecules or drugs has long been a key focus of interest for various structural and computational biologists [2]. The sole dependence on the 3D structure of proteins and the limited structure availability always hindered the process of accurate target identification [3]. The advent of deep learning models integrated into accurate identification of the 3D structure of proteins [4, 5] has bolstered the field to a great extent. Nevertheless, druggability prediction remains limited due to the failure to account for all associated physicochemical and biological properties. A substantial proportion of drugs fail to accomplish the goal of implementation in medical practice due to their targets being considered undruggable [6]. Druggability pertains to the therapeutic action of a drug whereas ligandability refers to the ability to bind to small molecules or ligands [7]. The assessment of the drug-likeness properties of the proteins [8, 9] and the association of the human genes encoding druggable proteins [7, 10] have remained subjects of continued scientific investigation.
The last decade has seen an unprecedented increase in studies that have employed machine learning (ML)–based computational approaches to identify druggable proteins. Shoombuatong et al. surveyed and highlighted different tools and algorithms and deployed webservers spanning this space, yet the full realization of maximal accuracy in uncovering the druggability potential of protein targets remains elusive [11]. Major concerns of the developed tools and algorithms toward the identification of druggable proteins have been raised and rightly pointed out by Charoenkwan et al. which led to the development of a new tool, SPIDER [12]. The study by Charoenkwan et al. [12] beautifully articulated the functionalities of the contemporary tools. The major limitations of most existing tools include the reliance on a limited set of sequence-derived features, lack of any blind validation, and training/testing on relatively small datasets of druggable and non-druggable proteins. Lin et al. emphasized the advantage of using bagging methods to enhance the predictability of druggable proteins [13], but did not propose any method for mitigating dataset bias. Moreover, most existing methods lack a web-based tool, thereby limiting accessibility for researchers. SPIDER also suffers from certain limitations, such as the training set that comprises only 2543 proteins. Additionally, the model is trained solely on features derived from amino acid sequences and their properties. The approach also lacks reference to key biophysical predictors that are fundamental in predicting protein druggability. Another tool, DrugnomeAI, which is based on a stochastic semi-supervised machine learning framework predicts the druggability of protein-coding genes by integrating gene-level data resources, but it lacks emphasis on features specific to proteins [7]. Furthermore, these tools fail to provide comprehensive information related to recent literature regarding proteins as candidate drug targets for various diseases. Another recent tool, DrugTar [14], represents a related effort that utilizes both structural and sequence-derived properties, incorporating features extracted using large language models (LLMs), along with Gene Ontology terms, to predict the druggability potential of proteins. However, a key limitation observed with DrugTar is its suboptimal performance when evaluated on a blind validation set, raising concerns about the model’s generalizability to unseen data.
To address key limitations in existing druggability prediction tools, we developed DrugProtAI, an advanced computational framework designed to overcome class imbalance and the narrow focus on amino acid properties. Unlike other tools restricted to protein subsets, DrugProtAI models nearly the entire human proteome. A partitioning method ensures balanced representation of druggable and non-druggable proteins during training, followed by validation on independent datasets. The framework integrates gradient boosting and random forest algorithms with partitioning to enhance predictive performance. Moreover, SHAP scores have been utilized to delineate the principal predictors and provide interpretable insights. We introduce two distinct modeling approaches, Partition Ensemble Classifier (PEC) and Partition Leave-One-Out Ensemble Classifier (PLOEC), with PLOEC uniquely nullifying the influence of the partition containing the test protein during training, ensuring unbiased assessment.
The advent of high-throughput omics technologies can pave the way for the identification of a large number of drug targets [15], and DrugProtAI can be used as a complementary tool in the drug discovery process. The search for recent literature on investigational targets is often cumbersome; therefore, DrugProtAI has integrated a feature for retrieving publications that provide supporting evidence for the protein as a drug target. We believe that DrugProtAI’s user-friendly web interface represents a step toward reducing uncertainties around druggable proteins, with the potential to substantially save time and resources in future drug discovery efforts.
Results
We intended to discover and interpret key predictors contributing to protein druggability and numerically calculate the probability of a protein being druggable.
Feature extraction and engineering
We extracted comprehensive protein information from the UniProt [16] database and cross-referenced it with DrugBank [17], resulting in a broad classification of the human proteome into three categories, i.e. Druggable, Investigational, and Non-Druggable. The dataset presents a significant modeling challenge with only 10.93% druggable proteins and 85.73% non-druggable proteins (Fig. 1B). A total of 183 druggability-relevant extracted features were categorized into 10 classes (4 sequence based and 6 non-sequence based). The radar plot (Fig. 1A) depicts the distributions, with a complete feature list in Supplementary Table 1. The correlation analysis did not reveal much redundancy. A detailed description of how each feature was used in the model, along with the number of features extracted, is provided (Supporting Information).
Figure 1.

Illustration of the extracted dataset and druggability prediction model development workflow. (A) We utilized a comprehensive feature set that includes 10 diverse feature groups. These span across sequence-derived and annotation-derived (non-sequence) properties. The four sequence-derived properties include physicochemical features, Grouped Dipeptide Composition (GDPC) encodings, flexibility properties, and latent values. The six non-sequence properties include Protein-Protein Interaction (PPI) and PPI network properties, Post-translational Modification (PTM) and glycosylation counts, and subcellular locations and domains, forming a diverse robust input for druggability prediction. (B) The dataset, consisting of 20 434 proteins, is categorized into 85.73% non-druggable, 10.93% with approved drugs (termed druggable), and 3.34% drugs pending approval (termed investigational). (C) The model development workflow involves training multiple ensemble classifiers, including Random Forest and XGBoost, on balanced subsets of non-druggable partitions and druggable train sets. Each model undergoes validation and testing on an independent protein sample set for blind validation, ensuring robust and accurate predictions. The final Druggability Index and metrics are calculated by averaging predictive scores from each of the partition models, leading to reliable classification outcomes.
In addition to the above features, we performed a comparison to deep learning–based features from a protein encoder model. We incorporated embeddings from a state-of-the-art protein model, ESM-2-650M [18]. This model encodes protein sequences into dense numerical representations. For each protein, we used the 1280-dimensional output embedding corresponding to the [CLS] token, which provided a global summary of the input sequence. The detailed representation is provided in the Supporting Information.
Evaluation of the partition method and choosing the model for druggability prediction
To evaluate the partition-based modeling approach (Fig. 1C), the majority class (non-druggable) of the train set was then divided into nine partitions (~1897 proteins each). Each partition was trained against the full druggable set (1919 proteins), reducing class imbalance, and resulting in 9 trained partition models. Accuracy, sensitivity, and specificity have been used to assess the performance of the partition-based models. The performance metrics for each individual model, as well as the overall PEC model, are provided for both XGBoost and Random Forest (Supplementary Table 2a–b). The evaluation was performed using 20 distinct random seed configurations (comparable across models), simulating a cross-validation scenario. The reported metrics are presented as mean values along with their variances across these simulations. Notably, the XGBoost and Random Forest PEC model’s overall accuracy was ~2 percentage points higher than the average accuracy of the individual partition models. This supported our hypothesis that the collective performance of multiple models yields better results than any single partition alone, thereby confirming that our partitioning strategy functions as intended. We further tested other algorithms, including Logistic Regression, Support Vector Machine (with linear and RBF kernels), K-Nearest Neighbors, and Naive Bayes, to evaluate the best-performing model. The corresponding metrics are provided (Supplementary Table 3). Based on the metrics, it could be concluded that XGBoost and Random Forest performed well with overall accuracy 78.06 ± 2.03 and 75.94 ± 1.55, respectively, and was chosen further.
We applied the same PEC methodology using XGBoost on the ESM-2-650M–derived embeddings, following the identical experimental setup described previously. This model achieved improved performance across all three evaluation metrics, with an overall accuracy of 81.47 ± 1.42% (Supplementary Table 4). Although this model yielded higher predictive scores than our feature set, they came at the cost of interpretability. Deep learning embeddings do not allow biological insight into feature importance, which was a key motivation for our feature engineering approach.
We also applied a Genetic Algorithm (GA) with Roulette Wheel Selection for feature selection on the XGBoost classifier (Supplementary Fig. 1). It reduced the number of features to 85 across the 9 partitions, each containing 1919 proteins in the druggable training set. The provided metrics (Supplementary Table 2c) demonstrate an overall accuracy of 76.42%, which is comparable to the performance of the other models. The average score (Supplementary Table 5) and variance (Supplementary Table 6) across the generations are reported. PEC modeling thus demonstrated a robust and reliable strategy for classifying proteins based on druggability.
Feature improvement metrics and interpretability of partitioning models
Feature scores were calculated by averaging the individual importance scores from each model in the partition method to identify key predictors (Supplementary Table 7). Averaging ensures that consistently significant features are appropriately weighted while minimizing partition-specific biases. SHAP values provided interpretable insights by computing feature contributions across samples and partitions, then averaging them to capture both local and global effects (Fig. 2A). The relationship between test accuracy and the number of top features (K = 1 to 183) is shown (Fig. 2B), revealing a general trend of improved accuracy with increasing number of top features for both XGBoost and Random Forest (Supplementary Table 8a–8b). XGBoost exhibits higher variance across partitions due to its sensitivity to data subsets, while Random Forest remains more stable through ensemble averaging.
Figure 2.

Feature scores, feature improvement metrics, and distribution plots of top features. (A) SHAP summary plots illustrating the top 10 features selected using partition average feature importance scores for XGBoost and Random Forest models. SHAP values are computed on a representative subset of the training data to interpret feature contributions to protein druggability. Features toward the right of origin indicate a positive influence on druggability predictions, while those toward the left indicate a negative influence. Color represents feature magnitude highlighting how feature intensity affects model output. (B) Left axis—partition average feature scores for both XGBoost and RF models in decreasing order. The curve also displays the variance in feature scores across individual partition models. Right axis—improvement in test accuracy as more top features (from K = 1 to K = 183) are included. Highlighted points on the curve show where the PEC method achieves the best performance. (C) Demonstrates the data distribution plots for some of the top features from our partition-based ML modeling framework.
The SHAP plots highlight key features such as kinase, modified residues, instability index, and secondary structure sheets underscoring their importance for both algorithms. Positive SHAP values indicate a higher likelihood of being druggable, while negative values suggest the opposite. The x-axis depicts the magnitude and direction of feature influence, with values color coded from blue (lower) to red (high). Notably, higher counts of kinases, secondary structure sheets, glycosylation, and modified residues contribute positively to druggability. In contrast, features like disulfide bonds and glutamine percentage negatively influence druggability at higher values. Although minor discrepancies exist between Random Forest and XGBoost, the latter’s results align well with existing literature and observed feature patterns. We also observed a considerable overlap between the top features identified through Recursive Feature Elimination (RFE) (Supplementary Table 9) and the top SHAP predictors, including secondary structure sheet, instability index, modified residue, Grand Average of Hydropathy (GRAVY), amino acid percent Q, and flexibility mean. The top features identified from GA across partitions also revealed a significant amount of overlap with SHAP scores (Supplementary Table 8c and Supporting Information).
The top contributing feature group was further assessed (Supplementary Fig. 2) and physicochemical properties emerged as top. Both models and RFE consistently identified secondary structure sheets, instability index, and GRAVY as key features. Their distinct distribution patterns (Fig. 2C; Supplementary Fig. 3) further support their reliability in partition-based modeling.
Druggability index and blinded validation
The model deployed in the tool available at https://drugprotai.pythonanywhere.com/ was trained on the entirety of 2219 druggable and 17 377 non-druggable proteins, excluding investigational proteins from the training phase. To achieve this, the non-druggable set was divided into eight partitions 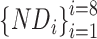 , and eight ensemble models
, and eight ensemble models  were trained, each using a partition of the non-druggable set (~2172 proteins) against the entire druggable set (|D| = 2219 proteins).
were trained, each using a partition of the non-druggable set (~2172 proteins) against the entire druggable set (|D| = 2219 proteins).
We calculated the Druggability Index (DI) for the 677 investigational proteins, which were unseen during the training, using the PEC model (Supplementary Table 10a–10b). We also calculated DI for 17 377 non-druggable proteins, using PLOEC model (Supplementary Table 11a–11b). The DI values can be considered a measure of how strongly a protein exhibit druggable tendencies. Our categorization was based on drug information available in DrugBank version 5.1.12 (dated 14 March 2024). Some protein targets have received FDA-approved drugs as per the updated data in version 5.1.13 (dated 2 January 2025) (Supplementary Table 12). A total of 81 new proteins now have FDA-approved drugs, reflecting the rapid pace of drug discovery. A few proteins were downgraded to investigational or non-druggable status, likely due to annotation inconsistencies. These rare cases were excluded from analysis due to their negligible impact.
The XGBoost-based PEC/PLOEC models achieved higher accuracy than Random Forest, correctly identifying 61 out of 81 newly druggable proteins. This blind validation yielded 75.31% accuracy for XGBoost and 74.07% for Random Forest. We also benchmarked our method against existing tools (Fig. 3A). Notably, the PEC model trained on ESM-2-650M embeddings using XGBoost performed poorly on blind validation, with an accuracy of only 65.43%. This suggests that while sequence-based features performed well in in-sample evaluations, they lacked robustness when applied to entirely new proteins. This underscores the importance of incorporating complementary non-sequence-derived features to achieve more reliable and generalizable druggability predictions.
Figure 3.

Comparison of our partition models (XGB and RF) against other tools. (A) Number of correctly predicted newly druggable proteins by DrugProtAI’s PEC and PLOEC models using XGBoost (XGB) and Random Forest (RF) consisting of 31 investigational and 50 non-druggable proteins, compared against SPIDER and DrugTar. (B) Bar plot depicting the probabilities of newly approved druggable targets, compared with other tools. The higher the probability of these targets, the better calibrated the particular method is.
Additionally, we assessed model calibration, defined as the average predicted probability (confidence score) assigned to newly druggable samples. Among correctly predicted proteins, models assigning higher probabilities are considered better calibrated. As shown in Fig. 3B, our feature selection approach yields superior calibration, indicating that the model is not only accurate but also more confident in its predictions. Our XGBoost PEC model predicted a DI ≥ 0.99 for 14 proteins in the investigational set (Supplementary Table 10a), two of which have since been approved. Illustrative examples across investigational, druggable, and non-druggable proteins are shown (Fig. 4A–B).
Figure 4.

Protein druggability status and predictions using PEC and PLOEC models and the layout of our tool DrugProtAI. The figure illustrates the druggability status of randomly proteins based on DrugBank data (v5.1.13, dtd. 2 January 2025) and our model predictions trained on druggability data (v5.1.12, dtd. 14 March 2024). (A) Six randomly chosen proteins from the investigational category under older version. Top: three proteins now approved, with scores predicted by PEC using XGBoost (left) and Random Forest (right). Bottom: three proteins still pending approval, with two predicted as druggable by our model, indicating potential for future drug approval. (B) Randomly chosen six proteins categorized as non-druggable under older version. Top: two proteins now approved and one pending approval. Bottom: three remain non-druggable, but PLOEC identifies potential druggability for two. (C) Depiction of the complete layout of our tool DrugProtAI.
Overview of DrugProtAI
DrugProtAI is a web-based tool, freely accessible for assessing protein druggability by integrating data from UniProt [16], DrugBank [17], PubMed [19], and AlphaFold [4], combined with advanced machine learning models. It predicts druggability using Partition-Based Ensemble Models with Random Forest and XGBoost, trained on 183 key biological features. Interactive pie charts display top-ranking features, while the DI aids in prioritizing therapeutic targets. Additionally, DrugProtAI offers quick access to ongoing research on specific drug targets, serving as a unified platform for early-stage drug discovery A schematic representation of DrugProtAI’s functionality is provided (Fig. 4C).
Discussion
We have sought to streamline target identification by equipping researchers to better interpret suitable drug candidates, thereby saving considerable time and effort in drug discovery. We present DrugProtAI, an easy-to-use web interface designed to facilitate target selection. Built on the PEC and PLOEC, DrugProtAI enables robust, unbiased, and accurate predictions of protein druggability, even with highly skewed data. Data from DrugBank [17] indicate that ~86% of proteins lack approved drugs, underscoring the challenge of imbalanced datasets in druggability prediction, as noted in prior studies [7, 9, 12, 20–23]. Our model employs a novel partitioning strategy, evaluated through accuracy across multiple random seeds. Achieving ~75% accuracy in blinded validation on newly approved drugs, it outperforms existing tools. To our knowledge, we are the first to identify key biophysical features distinguishing druggable from non-druggable proteins, offering valuable guidance for target selection in drug discovery.
Kinases are established drug targets, and the FDA approval of imatinib (DB00619) has spurred the development of multiple kinase inhibitors in oncology [24]. SHAP and feature importance scores indicate that kinase proteins might be one of the prime targets for inhibitor design and disease management. It has also emerged as top feature in RFE-based feature selection. Additionally, hydrogen bonds play a crucial role in determining the stability of the proteins [25] and shaping the structure of the proteins [26]. The secondary structure sheet, governed by hydrogen bonds [27], exhibits distinct distribution patterns between druggable and non-druggable proteins in our analysis. Glycosylation emerges as a top classifier in the XGBoost partitioning model, demonstrating strong concordance with findings reported in the literature [28–31]. Our results highlight the instability index as a key determinant of protein druggability from both RFE and SHAP-based top predictors, reflecting the importance of structural stability for effective drug binding. Aromaticity also emerges as a critical feature, influencing protein–drug interactions and binding specificity [32]. Disulfide bonds, which are known to stabilize proteins and are involved in regulating redox activity [33], might negatively impact druggability by reducing target flexibility, introducing redox sensitivity, and obscuring potential binding sites, particularly in the case of intracellular targets. All these factors highlight the key features responsible for determining the druggability of proteins, which may be overlooked if only the sequence-specific properties of amino acids are considered, as is done in other contemporary studies. The top predictors identified by RFE and SHAP were largely in concordance, with only a few features found to be unique to each method. The impact of subcellular localization was well captured during RFE-based feature selection using the XGBoost method, again, highlighting the significance of non–sequence-based properties in our feature set. Thus, we posit that it may function as a valuable guide for investigators in the selection of specific drug targets. However, we leave the majority of the interpretation to the users when selecting a particular drug target. The promising results from deep learning–based pre-trained protein models open new avenues. We aim to integrate these models with non–sequence-based features and offer users the option to choose between latent values or embeddings, enabling more informed decisions for assessing protein druggability, particularly in assessing the impact of mutation-driven changes on protein druggability. Such advancements may significantly accelerate target prioritization in precision medicine and rational drug design.
To avoid bias when a protein appears in the training set, we developed PLOEC, which excludes models trained on that protein from druggability scoring. This approach outperforms existing tools when tested on recently approved targets. For instance, the recently approved drug targeting the protein Activin A Receptor IIA, for the management of pulmonary arterial hypertension [34], has been assessed as druggable with exceptional ease using PEC method. Cantharidin, a well-known natural toxin inhibiting serine/threonine protein phosphatases, has been approved for treating skin-related disorders [35, 36]. Our PEC method effectively predicted druggability, as demonstrated by glycogen phosphorylase and Gamma-secretase subunit PEN2, both identified as druggable and now approved targets for pyridoxal phosphate and nirogacestat, respectively [37]. Similar results were observed for GTP cyclohydrolase I and phosphoenolpyruvate carboxykinases. PEC and PLOEC predict the druggability of investigational targets, identifying GTPase NRas, thioredoxin, and laminin subunit beta-1 as druggable, and spermine oxidase as non-druggable based on physicochemical properties. The tool thus facilitates target selection for diseases with multiple candidate proteins. Testing on an independent set, such as Intraflagellar transport protein 56 [38], which mediates ciliary transport, also fails to secure a majority vote as druggable, indicating its low therapeutic potential. To guide target selection, we provide druggability indices from both Random Forest and XGBoost models, allowing users to exercise discretion. The employment of novel partition-based methods has led to outperformance of recently developed tools [7, 11, 12], including those deploying advanced LLM strategies [14], in blinded validation using recently approved druggable targets, thereby highlighting the utility of our tool. However, researchers are advised to exercise caution, as some proteins may not be identified as druggable, and further analysis of physicochemical properties and experimental validation is recommended.
Most druggability studies lack accessible tools for experimental scientists and focus narrowly on sequence properties. To address this, we developed an intuitive web interface that predicts druggability and highlights key contributing features, enabling researchers to prioritize targets and enhance the success of drug design efforts. The recent pandemic has underscored the potential of drug repurposing [39], and we are confident that it will substantially advance this strategy, ultimately conserving both time and financial resources. We provide researchers access to publications related to each drug-targeted protein.
DrugProtAI represents a pivotal step toward comprehensive druggability prediction. We aim to extend its application to other species and to responses involving antibodies, immunotherapies, antimicrobials, and drug resistance. With expanding datasets and advances in generative AI, we plan to develop more robust solutions in the future.
Methods
Data extraction and preprocessing
The dataset was curated manually, comprising 20 434 human proteins, with 183 features derived from the UniProt [16]. Proteins longer than 3000 amino acids were excluded for computational resources, resulting in a final set of 20 273 proteins. Feature extraction details are provided in the Supporting Information. Feature redundancy and collinearity analyses were performed. The correlation heatmap (Supplementary Fig. 4) indicated minimal correlations among features, except for latent values and a few physicochemical properties. The features were grouped into 10 well-defined categories encompassing biological, physicochemical, and sequence-derived properties. Additionally, we compared these with deep learning–based embeddings from the ESM-2-650M protein model [18]. Detailed methods are included in the Supporting Information.
Protein datasets were categorized as druggable, investigational, or non-druggable based on the presence of approved drugs, investigational drugs, or absence of drug in DrugBank (v5.1.12, dated 14 March 2024). This version listed 677 investigational target proteins. All were evaluated using DrugProtAI to assess druggability. As of DrugBank v5.1.13 (dated 2 January 2025), 31 of the investigational proteins had received drug approval, along with an additional 50 proteins approved for new drugs from non-druggable set (Supplementary Table 12). These proteins were used in a blinded validation to evaluate model performance. Details on protein stratification are provided in the Supporting Information.
Machine learning workflows and the partition method employed to build a robust model to predict druggability of proteins
The ML techniques were employed to decipher the probable therapeutic action of a target protein using 183 extracted features. A metric has been devised, namely, the ‘Druggability Index (DI)’ to determine the protein’s capacity to be druggable. In total, 677 investigational proteins were excluded from our training dataset. The remaining dataset, comprising 19 596 proteins, resembled a skewed distribution (2219 druggable and 17 377 non-druggable). Model performance was evaluated on a held-out test set comprising 300 proteins from each of the druggable and non-druggable categories, ensuring fair evaluation. Consequently, the training dataset consisted of 18 996 proteins (1919 druggable and 17 077 non-druggable).
It is crucial to handle the substantial imbalance in the dataset before training ML models. The popular approaches like oversampling have been highlighted in different literature for handling imbalances [22]. Approaches such as synthetic oversampling like SMOTE [40] and ADASYN [41] are used to generate artificial samples of the minority class to balance the dataset. However, these methods lack transparency that the synthetic samples generated belong to true distribution of the minority class [42]. Another commonly used method undersampling may lead to loss of critical datapoints [42].
We then proposed a Partition Ensemble Classifier (PEC), which trains multiple ensemble models on different partitions of training data, while retaining the same feature set across partitions. The PEC has been designed to mitigate the class imbalance. A total of 2219 druggable and 17 377 non-druggable proteins were partitioned into 8 models, with each partition comprising 2172 non-druggable proteins paired against the entire set of 2219 druggable proteins. Each partition was evaluated using 20 random seeds. The rationale for adopting the PEC design is detailed in the Supporting Information.
We have used Random Forests [43] (Bagging or Bootstrap Aggregating) and XGBoost [44] (Boosting), the two popular ensemble learning methods. It has been extensively used to handle imbalances in biological datasets and is increasingly being used to tackle this complicated issue of class imbalance in biological datasets [45, 46], thereby avoiding performance degradation. Mechanisms involved in bagging and boosting classifiers are depicted (Fig. 1C). The detailed rationale for choosing the two methods and the parameters used are provided in the Supporting Information.
We have also utilized a genetic algorithm [47] (GA) (Supplementary Fig. 1), a stochastic optimization methodology that boosts the selection of key features for druggability score prediction. Three key genetic operations, crossover, mutation, and selection, are repeated until stopping criteria are met. We have provided the details of the feature selection process and the metrics involved in the GA process in the Supporting Information.
Once it was established that the PEC method functioned as intended, we compared XGBoost and Random Forest with other ML models to identify the best-performing model for predicting protein druggability. The details are provided in the Supporting Information.
Feature importance and interpretability in PEC model
The major limitation faced by contemporary tools is the lack of interpretable features depicting druggability. In any classifier algorithm, the features used to train the model are considered to be a true representation of the real-world scenario. However, this is not always the case, as noise in the data can also be inadvertently learned. Random Forest uses the mean decrease in Gini impurity to determine the important features [43]. On the other hand, XGBoost Classifier uses the Gain measure for feature scoring [44]. Our PEC model involved eight models trained on eight different non-druggable partitions. We reported the average feature importance scores for each model as the final feature score, ensuring a balanced consideration of their impacts. Though these built-in methods are well suited for achieving higher performance metrics, they do not provide insight into how each feature affects the class predictions. Hence, we used SHAP (SHapley Additive exPlanations) values to gain better insights about impact on classes by different features [12, 48]. This technique tends to ensure a fair distribution of credit (in this case, the prediction) among features [49]. The detailed methods related to SHAP score calculations are provided in the Supporting Information. We also tried to estimate the feature importance using Recursive Feature Elimination (RFE) method, a computationally less expensive method than SHAP. However, it has got its limitations, as it does not account for complex feature interactions or nonlinear dependencies as effectively as SHAP. The detailed information and metrics are provided in Supporting Information.
Quantifying druggable tendency by Druggability Index
Given a predictive model, the Druggability Index (DI) of a protein is defined as the probability of the given protein predicted by the model as druggable. The DI scores were separately obtained from the XGBoost and Random Forest models.
We adopted separate strategies to calculate DI, depending on protein class.
For proteins belonging to the investigational class, the predictions were generated using the PEC (Partition Ensemble Classifier) model. The PEC model calculates DI value as the average of the prediction outputs of individual models trained on each partition of the dataset as proposed in the Partition method earlier. The prediction given by a PEC model in
 terms of prediction probabilities of individual models
terms of prediction probabilities of individual models  where
where  is the input feature vector corresponding to an investigational protein, is given by the equation:
is the input feature vector corresponding to an investigational protein, is given by the equation: 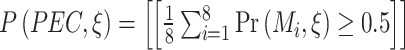 where
where 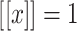 iff x is true.
iff x is true.-
For proteins belonging to the non-druggable class, the predictions were generated using the PLOEC (Partition Leave-One-Out Ensemble Classifier) model. Every non-druggable protein is present in the training set in exactly one of the partitions. In the PLOEC model, we do not consider the partition containing this protein. The PLOEC model evaluates DI as the average of the prediction outputs of individual models trained on each of the remaining partitions. The prediction given by a PLOEC model
 in terms of prediction probabilities of individual models
in terms of prediction probabilities of individual models  , where
, where 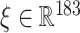 is the input feature vector corresponding to a non-druggable protein, is given by the equation:
is the input feature vector corresponding to a non-druggable protein, is given by the equation:  where
where  be such that
be such that and
and  iff x is true.
iff x is true.A flowchart depicting PEC and PLOEC model is provided (Supplementary Fig. 5).
Overview of our tool: DrugProtAI
DrugProtAI is powered by a Python-based Flask backend and features a dynamic application based on Javascript, while in-house styling was used for the layout of the webpage. Also, PostgreSQL was used for backend databasing and storing preloaded results. The interactive data visualization plots were created using the chart() JS function (https://www.jsdelivr.com/). The 3D protein rendering tool leveraged 3Dmol.js (https://3dmol.csb.pitt.edu/). The current version of DrugProtAI features a dynamic website that integrates data from popular knowledgebases with in-house computed DI derived from ML models and is freely accessible. The detailed functionalities of the DrugProtAI is provided in the supporting information.
Key Points
We tried to address the critical challenge of high drug attrition rates by introducing a robust machine learning framework for accurate target identification.
Proposed a novel partitioning method–based Ensemble classifier to handle class imbalance, evaluating the druggability of 20 273 human proteins using 183 biophysical and sequence-derived features.
Demonstrated the superior performance of XGBoost-based PEC and PLOEC models by correctly predicting 61 out of 81 newly approved drug targets and utilized SHAP analysis to reveal key features influencing protein druggability.
We believe the key predictors identified will help users in deciding on a particular drug target.
Introducing DrugProtAI, a user-friendly platform for druggability prediction of proteins, integrating target-linked publications and approved drugs, and validated to outperform existing tools on recent drug targets.
Supplementary Material
.jpeg){kind=link}
{kind=link}
.jpeg){kind=link}
{kind=link}
.jpeg){kind=link}
{kind=link}
Acknowledgements
We are grateful to the repositories and databases that have been instrumental in supporting this study. We acknowledge Nirjhar Banerjee and Suhisna Dutta for their assistance in designing the DrugProtAI UI, and Harshit Patil for his help in drafting the manuscript.
Dr Srivastava’s laboratory focuses on utilizing multi-omics approaches to delineate the pathophysiology of various tumors and infectious diseases, aiming to identify novel druggable targets.
Contributor Information
Ankit Halder, Department of Biosciences and Bioengineering, Indian Institute of Technology Bombay, Powai, Mumbai 400076, Maharashtra, India.
Sabyasachi Samantaray, Department of Computer Science and Engineering, Indian Institute of Technology Bombay, Powai, Mumbai 400076, Maharashtra, India.
Sahil Barbade, Department of Civil Engineering, Indian Institute of Technology Bombay, Powai, Mumbai 400076, Maharashtra, India.
Aditya Gupta, Department of Mechanical Engineering, Indian Institute of Technology Bombay, Powai, Mumbai 400076, Maharashtra, India.
Sanjeeva Srivastava, Department of Biosciences and Bioengineering, Indian Institute of Technology Bombay, Powai, Mumbai 400076, Maharashtra, India.
Author contributions
A.H., S.S.R., and S.S. conceived and designed the project. A.H., S.S.R., and S.B. performed data analysis. A.G. implemented the web tool. A.H., S.S.R., S.B., and A.G. drafted the manuscript. S.S. edited the manuscript. All authors have approved the final version of the manuscript.
Conflict of interest: None declared.
Funding
This study was supported by the MHRD-UAY Project (UCHHATAR AVISHKAR YOJANA), project #IITB_016 (2017) awarded to S.S., and by MASSFIITB (Mass Spectrometry Facility at IIT Bombay), funded by the Department of Biotechnology (BT/PR13114/INF/22/206/2015). We also thank MERCK-COE (DO/2021-MLSP) for their extended support. A.H. was supported by the Ministry of Education, India, through the PMRF program.
Data availability
All relevant code and data files for analysis can be found at https://github.com/Sachi-27/DrugProtAI.
References
- 1. Sun D, Gao W, Hu H, et al. Why 90% of clinical drug development fails and how to improve it? Acta Pharmaceutica Sinica B 2022;12:3049–62. 10.1016/j.apsb.2022.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hajduk PJ, Huth JR, Tse C. Predicting protein druggability. Drug Discov Today 2005;10:1675–82. 10.1016/S1359-6446(05)03624-X. [DOI] [PubMed] [Google Scholar]
- 3. Batool M, Ahmad B, Choi S. A structure-based drug discovery paradigm. IJMS 2019;20:2783. 10.3390/ijms20112783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583–9. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024;630:493–500. 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakharkar MK, Sakharkar KR, Pervaiz S. Druggability of human disease genes. Int J Biochem Cell Biol 2007;39:1156–64. 10.1016/j.biocel.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 7. Raies A, Tulodziecka E, Stainer J, et al. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun Biol 2022;5:1291. 10.1038/s42003-022-04245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bull SC, Doig AJ. Properties of protein drug target classes. PloS One 2015;10:e0117955. 10.1371/journal.pone.0117955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics 2009;25:451–7. 10.1093/bioinformatics/btp002. [DOI] [PubMed] [Google Scholar]
- 10. Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med 2017;9:eaag1166. 10.1126/scitranslmed.aag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shoombuatong W, Schaduangrat N, Nikom J. Empirical comparison and analysis of machine learning-based approaches for druggable protein identification. EXCLI J 2023;22:915–27. 10.17179/excli2023-6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Charoenkwan P, Schaduangrat N, Lio P, et al. Computational prediction and interpretation of druggable proteins using a stacked ensemble-learning framework. iScience 2022;25:104883. 10.1016/j.isci.2022.104883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin J, Chen H, Li S, et al. Accurate prediction of potential druggable proteins based on genetic algorithm and bagging-SVM ensemble classifier. Artif Intell Med 2019;98:35–47. 10.1016/j.artmed.2019.07.005. [DOI] [PubMed] [Google Scholar]
- 14. Borhani N, Izadi I, Motahharynia A. et al. DrugTar Improves Druggability Prediction by Integrating Large Language Models and Gene Ontologies. Bioinformatics 2025;btaf360. 10.1093/bioinformatics/btaf360. [DOI] [PMC free article] [PubMed]
- 15. Halder A, Verma A, Biswas D, et al. Recent advances in mass-spectrometry based proteomics software, tools and databases. Drug Discov Today Technol 2021;39:69–79. 10.1016/j.ddtec.2021.06.007. [DOI] [PubMed] [Google Scholar]
- 16. Apweiler R, Bairoch A, Wu CH, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2004;32:115D–9. 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Knox C, Wilson M, Klinger CM, et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res 2024;52:D1265–75. 10.1093/nar/gkad976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin Z, Akin H, Rao R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023;379:1123–30. 10.1126/science.ade2574. [DOI] [PubMed] [Google Scholar]
- 19. White J. PubMed 2.0. Med Ref Serv Q 2020;39:382–7. 10.1080/02763869.2020.1826228. [DOI] [PubMed] [Google Scholar]
- 20. Gong Y, Liao B, Wang P, et al. DrugHybrid_BS: using hybrid feature combined with bagging-SVM to predict potentially druggable proteins. Front Pharmacol 2021;12:771808. 10.3389/fphar.2021.771808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arif M, Fang G, Ghulam A, et al. DPI_CDF: druggable protein identifier using cascade deep forest. BMC Bioinformatics 2024;25:145. 10.1186/s12859-024-05744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jamali AA, Ferdousi R, Razzaghi S, et al. DrugMiner: comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Drug Discov Today 2016;21:718–24. 10.1016/j.drudis.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 23. Iraji MS, Tanha J, Habibinejad M. Druggable protein prediction using a multi-canal deep convolutional neural network based on autocovariance method. Comput Biol Med 2022;151:106276. 10.1016/j.compbiomed.2022.106276. [DOI] [PubMed] [Google Scholar]
- 24. Attwood MM, Fabbro D, Sokolov AV, et al. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat Rev Drug Discov 2021;20:839–61. 10.1038/s41573-021-00252-y. [DOI] [PubMed] [Google Scholar]
- 25. Pace CN, Fu H, Lee Fryar K, et al. Contribution of hydrogen bonds to protein stability. Protein Sci 2014;23:652–61. 10.1002/pro.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oklejas V, Zong C, Papoian GA, et al. Protein structure prediction: do hydrogen bonding and water-mediated interactions suffice? Methods 2010;52:84–90. 10.1016/j.ymeth.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koch O, Bocola M, Klebe G. Cooperative effects in hydrogen-bonding of protein secondary structure elements: a systematic analysis of crystal data using Secbase. Proteins 2005;61:310–7. 10.1002/prot.20613. [DOI] [PubMed] [Google Scholar]
- 28. Solá RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs 2010;24:9–21. 10.2165/11530550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Costa AF, Campos D, Reis CA, et al. Targeting glycosylation: a new road for cancer drug discovery. Trends in Cancer 2020;6:757–66. 10.1016/j.trecan.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 30. Diniz F, Coelho P, Duarte HO, et al. Glycans as targets for drug delivery in cancer. Cancers 2022;14:911. 10.3390/cancers14040911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith BAH, Bertozzi CR. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat Rev Drug Discov 2021;20:217–43. 10.1038/s41573-020-00093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lanzarotti E, Defelipe LA, Marti MA, et al. Aromatic clusters in protein–protein and protein–drug complexes. J Chem 2020;12:30. 10.1186/s13321-020-00437-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bechtel TJ, Weerapana E. From structure to redox: the diverse functional roles of disulfides and implications in disease. Proteomics 2017;17:17. 10.1002/pmic.201600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Villanueva J, Wade J, Torres A, et al. Sotatercept: the first FDA-approved activin A receptor IIA inhibitor used in the management of pulmonary arterial hypertension. Am J Cardiovasc Drugs 2025;25:17–24. 10.1007/s40256-024-00694-w. [DOI] [PubMed] [Google Scholar]
- 35. Honkanen RE. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett 1993;330:283–6. 10.1016/0014-5793(93)80889-3. [DOI] [PubMed] [Google Scholar]
- 36. Keam SJ. Cantharidin topical solution 0.7%: first approval. Pediatr Drugs 2024;26:95–100. 10.1007/s40272-023-00600-y. [DOI] [PubMed] [Google Scholar]
- 37. Vincenzi B, Bui N, Dileo P, et al. Efficacy of nirogacestat in participants with poor prognostic factors for desmoid tumors: analyses from the randomized phase 3 DeFi study. JCO 2024;42:11556–6. 10.1200/JCO.2024.42.16_suppl.11556. [DOI] [Google Scholar]
- 38. Xin D, Christopher KJ, Zeng L, et al. IFT56 regulates vertebrate developmental patterning by maintaining IFTB complex integrity and ciliary microtubule architecture. Development 2017;144:1544–53. 10.1242/dev.143255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodrigues L, Bento Cunha R, Vassilevskaia T, et al. Drug repurposing for COVID-19: a review and a novel strategy to identify new targets and potential drug candidates. Molecules 2022;27:2723. 10.3390/molecules27092723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sowjanya AM, Mrudula O. Effective treatment of imbalanced datasets in health care using modified SMOTE coupled with stacked deep learning algorithms. Appl Nanosci 2023;13:1829–40. 10.1007/s13204-021-02063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Munshi RM. Novel ensemble learning approach with SVM-imputed ADASYN features for enhanced cervical cancer prediction. PloS One 2024;19:e0296107. 10.1371/journal.pone.0296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alkhawaldeh IM, Albalkhi I, Naswhan AJ. Challenges and limitations of synthetic minority oversampling techniques in machine learning. World J Methodol 2023;13:373–8. 10.5662/wjm.v13.i5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Breiman L. No title found. Machine Learning 2001;45:5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 44. Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System. KDD '16: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016, 785–94. 10.1145/2939672.2939785. [DOI]
- 45. Salunkhe UR, Mali SN. Classifier ensemble design for imbalanced data classification: a hybrid approach. Procedia Computer Science 2016;85:725–32. 10.1016/j.procs.2016.05.259. [DOI] [Google Scholar]
- 46. Liu L, Wu X, Li S, et al. Solving the class imbalance problem using ensemble algorithm: application of screening for aortic dissection. BMC Med Inform Decis Mak 2022;22:82. 10.1186/s12911-022-01821-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Katoch S, Chauhan SS, Kumar V. A review on genetic algorithm: past, present, and future. Multimed Tools Appl 2021;80:8091–126. 10.1007/s11042-020-10139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lundberg S, Lee S-I. A Unified Approach to Interpreting Model Predictions. NIPS'17: Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017, 4768–77. 10.48550/ARXIV.1705.07874. [DOI]
- 49. Sun MW, Moretti S, Paskov KM, et al. Game theoretic centrality: a novel approach to prioritize disease candidate genes by combining biological networks with the Shapley value. BMC Bioinformatics 2020;21:356. 10.1186/s12859-020-03693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant code and data files for analysis can be found at https://github.com/Sachi-27/DrugProtAI.