

The extended family of major histocompatibility complex (MHC) class I-related proteins serves a variety of immunological functions, among which those of the polymorphic MHC class I molecules are paramount. These heterodimers of a membrane-anchored HLA-A, -B, or -C chain and soluble β2-microglobulin (β2m) present intracellularly processed peptides to circulating CD8+ T cells with αβ T cell antigen receptors (TCRs), which are thus enabled to detect and destroy pathogen-infected cells (1, 2). Activation of these effector T cells can be modulated by interactions of MHC class I molecules with inhibitory or activating receptors that were originally identified as natural killer (NK) cell receptors (NKRs) but are variably expressed on T cells. Best known in humans are the inhibitory or activating isoforms of the killer cell Ig-like receptors (KIRs), which bind to HLA-A, -B, or -C, and the C-type lectin-like inhibitory CD94-NKG2A and activating CD94-NKG2C heterodimers, which interact with HLA-E (3, 4). There is substantial evidence that engagement of the inhibitory receptors increases TCR-dependent activation thresholds, which may limit chronic T cell activation (5, 6). By contrast, much less is known about whether and how signals from the activating receptors are functionally integrated. However, distant relatives of MHC class I molecules, MICA and MICB, which have no role in antigen presentation but function as signals of cellular distress, interact with a distinct activating receptor complex, NKG2D–DAP10, which triggers NK cells, potently augments CD8+ αβ T cell and γδ T cell effector functions, and costimulates T cell proliferation and cytokine production (7–9). DAP10 signals similar to the CD28 T cell costimulatory receptor, by activation of phosphatidylinositol 3-kinase (PI3K) upon phosphorylation of a YXXM motif in its cytoplasmic domain (10).
There is substantial evidence that engagement of the inhibitory receptors increases TCR-dependent activation thresholds.
MICA and MICB are encoded by genes near HLA-B in the human MHC but share only about 30% amino acid sequence identity in the extracellular α1α2α3 domains with conventional class I chains (11). Sequences orthologous to MIC are conserved in the genomes of most mammals except rodents. MIC proteins are highly glycosylated and lack association with β2m and peptides, which is reflected by restructured domain interfaces and the presence of only a shallow remnant of a peptide-binding groove (12, 13). In the receptor-ligand complex structure, the upper sides of the MICA α1α2 platform domain defined by the two canonical antiparallel α-helices interface extensively with NKG2D homodimers (14). Because of the potent functions of NKG2D and its broad distribution on most CD8+ αβ T cells, γδ T cells, and NK cells, the arguably most interesting aspects in the immunobiology of MIC are related to gene and protein regulation and expression. Unlike the ubiquitous MHC class I molecules, MIC are expressed almost exclusively on fibroblast and epithelial cell lines. They are not regulated by interferons but can be induced by heat shock, similar to heat shock protein 70 (hsp70), presumably because of the presence of putative heat shock elements (HSEs) in the 5′-flanking regions of the MICA and MICB genes (12, 15). In accord with the modus of hsp70 regulation, MIC are expressed in significant amounts on rapidly proliferating epithelial cells but are scarce on quiescent cells, on which surface MIC becomes about 10-fold amplified after heat shock induction. In healthy individuals, expression of MIC is restricted to variable areas of intestinal epithelium (12). It has remained unclear, however, whether this tissue distribution is the result of permanent cellular stress or whether it may be induced by environmental factors. This topic has been explored by Tieng and colleagues (16), who now report their intriguing observation that adhesion of diarrheagenic Escherichia coli strains of the diffusely adherent pathogenic group to the intestinal epithelial Caco-2 and cervical carcinoma HeLa cell lines causes a rapid increase of MICA expression. This effect is induced by a specific interaction of the bacterial AfaE-III adhesin with the cellular CD55 receptor, a glycosyl-phosphatidylinositol (GPI)-anchored protein that is expressed on most human cells. It is otherwise known as decay-accelerating factor, which inhibits complement C3b deposition by C3 convertase inactivation. The experimental evidence is conclusive, as Tieng et al. show that adhesion and MICA induction could be conferred to noninvasive Shigella flexneri strains by gene transfer of AfaE-III, but not of another candidate adhesin of the same gene family, and that these effects were mimicked by AfaE-III-coated polystyrene beads. Antibody blocking experiments confirmed the specific involvement of CD55 (16). As could be expected, the induced expression of MICA was sufficient to trigger INF-γ secretion by an NK cell line. It should be noted, however, that NK cells are virtually absent among intestinal intraepithelial lymphocytes (IELs).
These findings may mark the beginning of a more exhaustive search for other enteric pathogens that may induce MIC expression through interactions with the intestinal mucosa, including bacterial toxins. However, some smaller issues remain to be addressed. For one, it is difficult to rationalize how CD55 engagement results in MIC induction. Tieng et al. (16) propose that it may depend on a conformational change, because antibody crosslinking of CD55 produced no effect. Regardless, a transcriptional induction of the heat shock response, which is regulated by cytoplasmic protein overload and misfolding, seems implausible (17). Perhaps, CD55 might directly or indirectly accelerate the flow of endosomal recycling of endocytosed MIC polypeptides and/or interfere with their degradation. Second, experimental evidence from polarized epithelial cells is desirable because basolaterally induced MIC expression has not been shown and the intracellular MIC polypeptide sequences lack a discernible cytoplasmic sorting signal. Previous immunohistochemical images have not been informative with regard to polarized MIC localization (12).
Other tissue sites with induced expression of MIC are associated with disease conditions. Among these are many epithelial tumors, including breast, lung, gastric, renal, colon, ovarian, and prostate carcinomas (18). Moreover, MIC are strongly induced by cytomegalovirus (CMV) and mycobacterial infection and probably by at least some other pathogens (8, 9). Interestingly, a CMV membrane glycoprotein, UL16, binds to MICB and a second set of NKG2D ligands termed ULBPs (19) The regulation and expression of these molecules are undefined, but they presumably correspond to the mouse retinoic acid-early inducible (RAE-1) family of NKG2D ligands (20). Common to all of these proteins is an MHC class I-like α1α2 platform domain, lack of an α3 domain, and a GPI membrane anchor. Expression of UL16 in CMV-infected cells and an inhibitory effect on the function of NKG2D have not been demonstrated. However, it remains an intriguing possibility that UL16 may have evolved to promote viral evasion from T cell control, analogous to the many well-known strategies of viral interference with host defense (21). This would highlight the significance of NKG2D in immune responses against CMV, which in all likelihood extends to other infections as well. Hence, the report by Tieng et al. (16) reinforces the notion that the MIC–NKG2D system, with its ability to rapidly mobilize targeted T cell and NK cell responses, functions as an emergency defense against infectious agents and hazardous conditions that cause cellular distress.
Expression of MIC effectively endows large areas of intestinal epithelium with costimulatory capacity. This activity is conceptually opposed to the immunobiology of the CD80–CD86 ligands of the CD28 costimulatory receptor, which are restricted to professional antigen-presenting cells (22). Intestinal IELs include mostly CD8+ αβ T cells, many of which express CD8αα homodimers that are virtually absent from circulating T cells, smaller populations of the uncommon CD4−CD8− αβ T cells, and γδ T cells. Characteristic of most IELs are memory phenotypes and potent cytolytic and immunoregulatory capacities (23). Typical of most of the γδ T cells is expression of Vδ1 γδ TCRs—a phenotype that is relatively rare among γδ T cells in the circulation (24). Many of these T cells respond against diverse transfected or untransfected target cell types expressing MIC. Their activation requires triggering of TCRs and NKG2D (7, 15). It remains unresolved, however, whether their TCRs recognize MIC or some unidentified ubiquitous surface components. Confirmation of the former alternative would validate the previously postulated idea that Vδ1 γδ T cells recognize self antigens that may be stress induced (25)—a model that was originally proposed for mouse intraepithelial γδ T cells with invariant TCRs (26). Because of the enormous surface area of intestinal epithelium and the dense population of its IELs (about one IEL per five enterocytes), these represent the largest lymphocyte pool (23). Many IELs are continuously exposed to MIC and thus, conceptually, are in a state of alert. Hence, it is not surprising that IELs express low levels of NKG2D, which may be down-modulated to prevent chronic T cell stimulation and to limit autoreactive bystander T cell activation (27). This regulation would be analogous to the ligand-induced endocytosis of CD28 and KIR, which serves to control T cell responsiveness (28, 29). However, expression of NKG2D is induced by interleukin (IL)-15 (27), which exerts stimulatory effects similar to those of IL-2 and shares the same receptor. IL-15 is produced by intestinal epithelial cells upon external stimuli and infection (30). Thus, through induced expression of MIC and secretion of IL-15, intestinal epithelial cells are able to regulate the response capacity of IELs.
Disturbance of this regulatory balance could exacerbate autoimmune conditions such as inflammatory bowel diseases in genetically susceptible individuals. Tieng et al. (16) show that in Crohn's disease, which is associated with frequent occurrences of diffusely adherent diarrheagenic E. coli strains and high levels of inflammatory cytokines, MICA expression is markedly increased (see Fig. 1). Thus, together with events triggering deregulation of cytokine networks, MICA could represent a pathogenicity factor. This possibility has been investigated for a number of diseases with autoimmune etiologies, such as ankylosing spondylitis, Behçet's disease, psoriasis, Addison's disease, Kawasaki's disease, and ulcerative colitis, because MICA (and to a lesser extent MICB) is polymorphic (31). So far, however, no primary allelic association has been found. There are about 50 alleles of MICA with fairly randomly distributed biallelic amino acid substitutions in its extracellular domains (31). As to their potential significance, the locations of allelic positions and their analysis in the context of the MICA structure have yielded no insights. However, a single amino acid substitution in the α2 domain is associated with large differences in the strength of NKG2D binding, which may significantly affect thresholds of T cell activation, especially under conditions of suboptimal expression of MIC (32). The physiological significance of this allelic bimorphism and its potential relevance to the findings of Tieng et al. remain to be explored.
Figure 1.
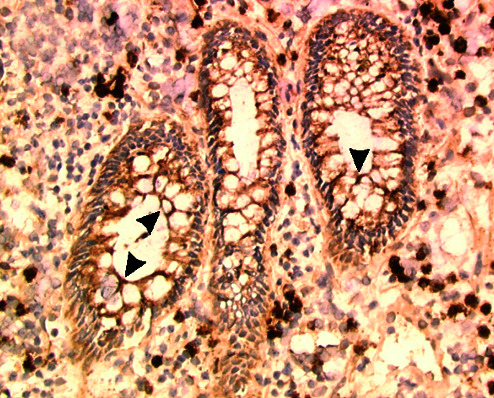
Indirect immunoperoxidase staining on cryocut sections shows MICA expression (arrowheads) on colonic glandular epithelia cells from patients with Crohn's disease. See article by Tieng et al. (16), from which this micrograph is reproduced.
Footnotes
See companion article on page 2977.
References
- 1.Bjorkman P J, Parham P. Annu Rev Biochem. 1990;90:253–288. doi: 10.1146/annurev.bi.59.070190.001345. [DOI] [PubMed] [Google Scholar]
- 2.Davis M M, Boniface J J, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 3.Lanier L L. Annu Rev Immunol. 1998;16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 4.Long E O. Annu Rev Immunol. 1999;17:875–904. doi: 10.1146/annurev.immunol.17.1.875. [DOI] [PubMed] [Google Scholar]
- 5.Carena I, Shamshiev A, Donda A, Colonna M, De Libero G. J Exp Med. 1997;186:1769–1774. doi: 10.1084/jem.186.10.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakker A B H, Phillips J H, Figdor C G, Lanier L L. J Immunol. 1998;160:5239–5245. [PubMed] [Google Scholar]
- 7.Bauer S, Groh V, Wu J, Steinle A, Phillips J H, Lanier L L, Spies T. Science. 1999;285:727–729. [PubMed] [Google Scholar]
- 8.Das H, Groh V, Kuiji C, Sugita M, Morita C T, Spies T, Bukowski J F. Immunity. 2001;15:83–93. doi: 10.1016/s1074-7613(01)00168-6. [DOI] [PubMed] [Google Scholar]
- 9.Groh V, Rhinehart R, Randolph-Habecker J, Topp M S, Riddell S R, Spies T. Nat Immunol. 2001;2:255–260. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 10.Wu J, Song Y, Bakker A B H, Bauer S, Spies T, Lanier L L, Phillips J H. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 11.Bahram S, Bresnahan M, Geraghty D E, Spies T. Proc Natl Acad Sci USA. 1994;91:6259–6263. doi: 10.1073/pnas.91.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Proc Natl Acad Sci USA. 1996;93:12445–12450. doi: 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li P, Willie S T, Bauer S, Morris D L, Spies T, Strong R K. Immunity. 1999;10:577–584. doi: 10.1016/s1074-7613(00)80057-6. [DOI] [PubMed] [Google Scholar]
- 14.Li P, Morris D L, Willcox B E, Steinle A, Spies T, Strong R K. Nat Immunol. 2001;2:443–451. doi: 10.1038/87757. [DOI] [PubMed] [Google Scholar]
- 15.Groh V, Steinle A, Bauer S, Spies T. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 16.Tieng V, Le Bouguenec C, du Merle L, Bertheau P, Desreumaux P, Janin A, Charron D, Toubert A. Proc Natl Acad Sci USA. 2002;99:2977–2982. doi: 10.1073/pnas.032668099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morimoto R I. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 18.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein K H, Spies T. Proc Natl Acad Sci USA. 1999;96:6879–6884. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosman D, Müllberg J, Sutherland C L, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny N J. Immunity. 2001;14:123–133. doi: 10.1016/s1074-7613(01)00095-4. [DOI] [PubMed] [Google Scholar]
- 20.Cerwenka A, Bakker A B H, McClanahan T, Wagner J, Wu J, Phillips J H, Lanier L L. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 21.Ploegh H L. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 22.Chambers C A, Allison J P. Curr Opin Cell Biol. 1999;11:203–210. doi: 10.1016/s0955-0674(99)80027-1. [DOI] [PubMed] [Google Scholar]
- 23.Hayday A, Theodoridis E, Ramsburg E, Shires J. Nat Immunol. 2001;2:997–1003. doi: 10.1038/ni1101-997. [DOI] [PubMed] [Google Scholar]
- 24.Deusch K, Luling F, Reich K, Classen M, Wagner H, Pfeffer K. Eur J Immunol. 1991;21:1053–1059. doi: 10.1002/eji.1830210429. [DOI] [PubMed] [Google Scholar]
- 25.Chowers Y, Holtmeier W, Harwood J, Morzycka-Wroblewska E, Kagnoff M F. J Exp Med. 1994;180:183–190. doi: 10.1084/jem.180.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Havran W L, Chien Y H, Allison J P. Science. 1991;252:1430–1432. doi: 10.1126/science.1828619. [DOI] [PubMed] [Google Scholar]
- 27.Roberts A I, Lee L, Schwarz E, Groh V, Spies T, Ebert E C, Jabri B. J Immunol. 2001;167:5527–5530. doi: 10.4049/jimmunol.167.10.5527. [DOI] [PubMed] [Google Scholar]
- 28.Linsley P S, Bradshaw J, Urnes M, Grosmaire L, Ledbetter J A. J Immunol. 1993;150:3161–3169. [PubMed] [Google Scholar]
- 29.Huard B, Karlsson L. Nature (London) 2000;403:325–328. doi: 10.1038/35002105. [DOI] [PubMed] [Google Scholar]
- 30.Fehniger T A, Caligiuri M A. Blood. 2001;97:14–32. doi: 10.1182/blood.v97.1.14. [DOI] [PubMed] [Google Scholar]
- 31.Stephens H A F. Trends Immunol. 2001;22:378–385. doi: 10.1016/s1471-4906(01)01960-3. [DOI] [PubMed] [Google Scholar]
- 32.Steinle A, Li P, Morris D L, Groh V, Lanier L L, Strong R K, Spies T. Immunogenetics. 2001;53:279–287. doi: 10.1007/s002510100325. [DOI] [PubMed] [Google Scholar]