

Abstract
Strategies for C–H amination with sulfonamides have been dominated by stoichiometric strategies or Rh/Cu catalysis using strong oxidants, elevated temperatures, or excess equivalents of C–H partner. We developed a photocatalytic C–H amination with a bench-stable N-methoxypyridinium salt that is amenable to late-stage functionalization efforts. We discovered a competitive proton-coupled electron transfer pathway between sulfonamide and a pyridine byproduct that inhibits the reaction. Catalysis was re-established with the introduction of ZnBr2.
Graphical Abstract

Sulfonamides have long held a privileged role in small molecule drug discovery. In particular, the N-benzyl sulfonamide moiety is present in a wide range of small molecule therapies targeting cancer, Alzheimer’s disease, SARS-CoV-2, etc.1,2 When synthesizing N-functionalized sulfonamides, the classical strategy is to make a retrosynthetic disconnection at the N–S bond to utilize amine nucleophiles and highly reactive sulfonyl electrophiles. Sulfonyl chlorides are often challenging to handle due to their instability and can result in low yields or multiple sulfonylations.3,4 In contrast, a more direct approach to alkyl sulfonamide synthesis would be C–N formation from a sulfonamide and C–H partner. Recent efforts to modernize stoichiometric methods using this strategy demonstrate a broad substrate scope with mild conditions.5,6 Conversely, catalytic methods for intermolecular C–H sulfonamidation reveal that some practical limitations still exist, including strong oxidants, elevated temperatures, and/or excess amounts of C–H partner.6–8
Recently, our group reported on the use of photoredox catalysis to perform a benzylic C–H azolation with a range of heterocycles.9 Key to the success of the transformation was the use of N-methoxypyridinium salts (pyr-1) as an oxidant and a convenient source of electrophilic alkoxy radicals. Our lab and others have found N-alkoxypyridinium salts to be highly attractive reagents that are bench stable and easily prepared in a single step from commercial pyridine N-oxides, producing benign pyridine byproducts.10 Oxidative quench of an excited state photocatalyst, Ir(III)*, by pyr-1 generates Ir(IV), a neutral pyridine byproduct, and a methoxy radical capable of performing hydrogen-atom transfer (HAT) at benzylic C–H bonds. Subsequent oxidation of the resultant benzylic radical by Ir(IV) yields a carbocation, achieving formal hydride abstraction under mild catalytic conditions (Figure 1C). The ideal properties of this reagent led us to question whether the catalytic platform could facilitate the C–H functionalization of other valuable nucleophiles, specifically, sulfonamides. As a long-standing interest for the synthetic community, many intermolecular C–H sulfonamidation methods have been developed using Rh and Cu catalysis with a range of different oxidant types. Initially, electrophilic amination precursors such as organic azides, imidoiodinanes, hydroxylamines, and N-halogenated sulfonamides were used, but, by design, they lack modularity or a comprehensive scope (Figure 1A).11 In 2000, Che demonstrated that the oxidant could be separated from the sulfonamide with use of diacetoxyiodobenzene (PIDA) and a Rh catalyst, relying on an in situ generation of an imidoiodinane (Figure 1B).12 Du Bois and Bach demonstrated similar strategies using different hypervalent iodine reagents with trichloroethylsulfamate partners.13,14 Powell made use of a Cu catalyst with readily available tert-butoxy peroxybenzoate oxidants, albeit with an excess of the hydrocarbon partner.15 Emmert made use of N-fluorobenzenesulfonimide (NFSI) with Cu at elevated temperatures based on precedent from Stahl and Liu.16–18 More recently, Doyle and co-workers reported a photocatalytic C–H amination with amides using a custom N-sulfonate amide oxidant as the limiting reagent.19 A handful of azole and sulfonamide examples were included. We rationalized that pyr-1 could be a safe, bench-stable, and easily accessible alternative to prior oxidants.
Figure 1.
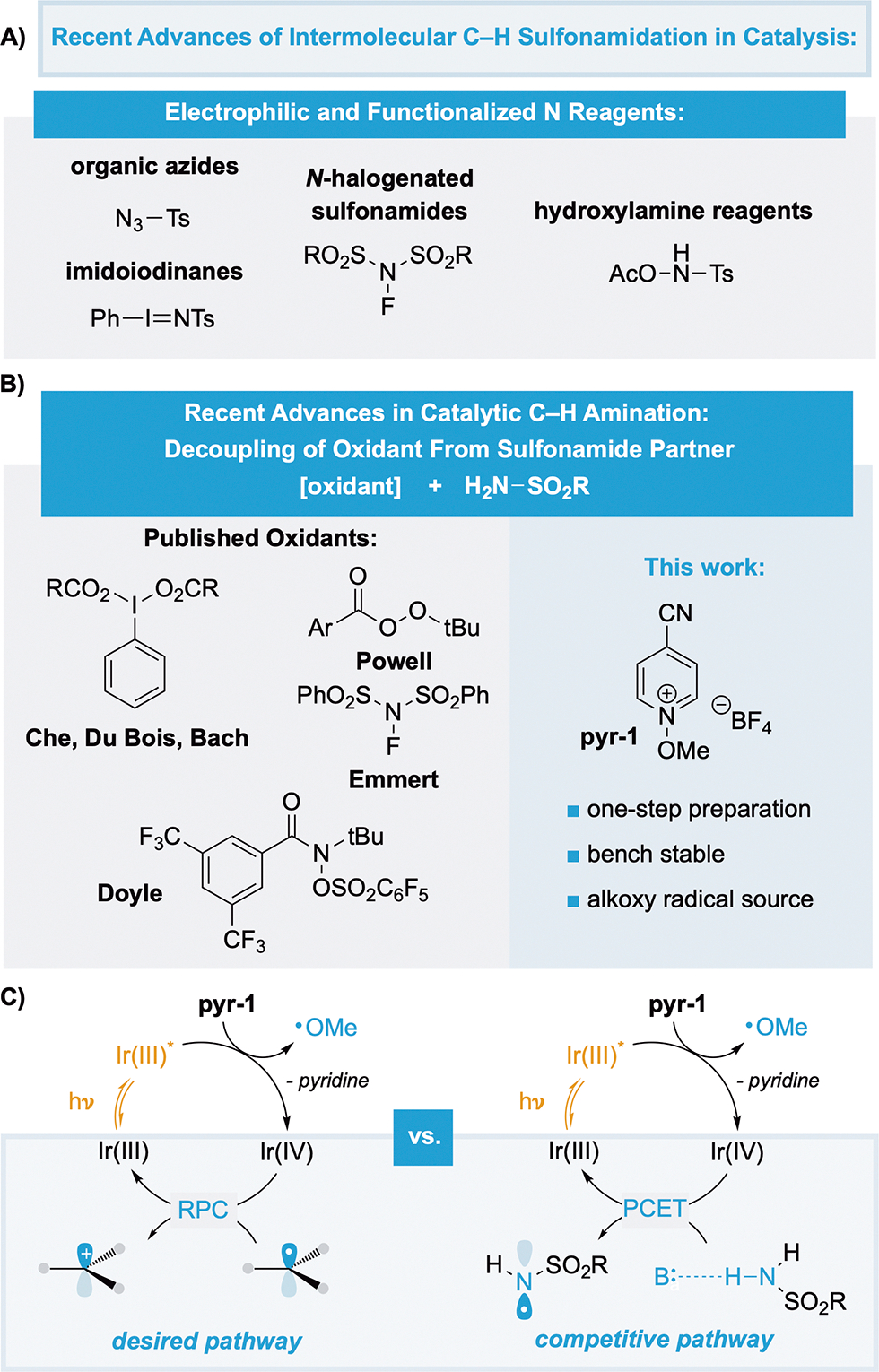
A) Recent advances of intermolecular C–H sulfonamidation in catalysis. B) Advances that separate oxidant from sulfonamide partner. C) Proposed competitive pathways in the photocatalysis cycle.
Despite the platform’s prior success with azole nucleophiles, initial attempts using pyr-1 with ethylbenzene as the C–H substrate, p-tosylamine as the nucleophile, and [Ir(dF(CF3)-ppy)2(dtbbpy)](PF6) as the photocatalyst yielded only 20% of the desired product (Figure 2, entry 1). Extensive investigation of standard reaction parameters (i.e., solvent additives, temperature, photocatalyst identity, catalyst loading, etc.), unfortunately, proved ineffective. From the outset, we were cognizant that sulfonamides have a strong tendency to engage in hydrogen bonding (pKBHX = 1.0).20 Sulfonamides are particularly effective hydrogen bond donors, capable of hydrogen bonding with themselves despite being poor acceptors. The Knowles lab has exploited this feature to develop hydroamination reactions of sulfonamides using proton-coupled electron transfer (PCET) to generate openshell N-centered sulfonamidyl intermediates.21 Moreover, mechanistic studies by Berry on the Du Bois rhodium system suggest that a key step in metal nitrene formation involves a PCET process from a metal–sulfonamide complex.22 These precedents led us to hypothesize that an undesired oxidative PCET process could be occurring in our reaction. Upon complexation of the sulfonamide with an appropriate base, either the excited state or the ground state of the photocatalyst could facilitate an oxidative PCET. If the former, then reductive quenching of the excited state photocatalyst would give an Ir(II) species, which is catalytically unproductive. If the latter, then the oxidative PCET would be competitive with the desired radical-polar crossover (RPC) step needed for carbocation formation, which is mediated by the ground state Ir(IV) catalyst (Figure 1C). To this end, we proposed that addition of Lewis acid additives could disrupt a potential hydrogen-bonding complex and re-establish the desired catalysis. Gratifyingly, the hypothesis was corroborated with addition of 1 equiv of zinc bromide resulting in an increase in yield to 78%. Notably, reaction times were reduced from 24 to 6 h. With the optimized conditions in hand, we proceeded to investigate both the breadth of substrates and the nature of the competitive oxidative PCET.
Figure 2.

Optimization of photocatalytic C–H amination by addition of chelating additives.
We found that the optimized photocatalytic conditions accommodate a broad range of sulfonamide and hydrocarbon synthons (Figure 3). Examination of the benzylic C–H partners found that para (2–7, 48–77% yield), ortho (8, 49% yield), and meta substitutions (9 and 10, 42% and 65% yield, respectively) on the ring were tolerated. N-Benzyl sulfonamide products were obtained when the alkyl chain was extended at the benzylic position (11 and 12, 55% and 63% yield, respectively). Benzo-fused five- and six-membered rings gave 60% and 70% yield, respectively (13 and 14). The success of diphenylmethane product 15 led us to test phenyltoloxamine as a substrate. Gratifyingly, a 39% yield of aminated phenyltoloxamine (16) was obtained through the direct use of the maleic acid salt. The importance of diarylmethane substrates has been highlighted in medicinal chemistry programs.23–25 Additionally, late-stage amination of celestolide, 17, was achieved in 44% yield.
Figure 3.

Scope for C–H and sulfonamide partners. Reactions were run on a 0.5 mmol scale. DCE = 1,2-dichloroethane. HFIP = hexafluoroisopropanol. a0.2 mmol scale. bNo ZnBr2, 1.5 equiv C–H partner, 1 equiv sulfonamide, 1.5 equiv abstractor, 9:1 DCE:HFIP. cReaction time of 24 h. d1.5 equiv sulfonamide.
An array of aryl and alkyl sulfonamides were successful under the reaction conditions. Both electron-rich and electron-deficient functional groups were tolerated on the aryl sulfonamide at para (18–22, 45–72% yield), meta (23–25, 60–66% yield), and ortho positions (26, 66% yield). Sizable tert-butyl groups on the sulfonamide were tolerated to give 27 in 59% yield, as well as smaller motifs such as cyclopropyl and methyl (28 and 29, 66% and 67% yield, respectively). Secondary sulfonamides were also viable nucleophilic partners in the coupling (30, 62% yield). Importantly, pendant alkenes were tolerated in the reaction, despite the competitive allylic site for C–H functionalization (31, 23% yield). Addition of ZnBr2 was critical to prevent the generation of sulfonamidyl radical, which can undergo intramolecular cyclization. Lastly, heterocyclic arenes were also tolerated on the sulfonamide partner (32, 34% yield).
Concurrent with the scope, we conducted mechanistic studies to 1) probe the feasibility of a PCET interaction, 2) better understand the role of ZnBr2, and 3) identify what catalyst state can facilitate the undesired PCET. First, we sought to identify the base involved. While previous sulfonamide PCET systems in photoredox catalysis use strong phosphate bases that are particularly good hydrogen bond acceptors, our system does not contain a base of similar strength. We considered the possibility that 4-cyanopyridine, a byproduct of pyr-1, could form a hydrogen bond with the N–H of p-tosylamine. Pyridines are relatively weak hydrogen bond acceptors, with a β value of 7.0, which is half that of phosphate (β = 14.3).26,27 However, despite the low hydrogen bond acceptor value, an equilibrium value for a hydrogen bond complex between p-tosylamine and pyridine was calculated to be K = 5.68, still favoring the complex formation (see Supporting Information). Indeed, the addition of 1 equiv of 4-cyanopyridine to the conditions reported in Figure 2, entry 1, decreased the yield from 20% to 6%. Figure 4A shows the decrease in product yield as a function of added 4-cyanopyridine.
Figure 4.

Mechanistic experiments to elucidate the competitive oxidative PCET. A) Pyridine inhibition study. B) Proposed illustration of the PCET pathway. C) Proposed illustration of ZnBr2 inhibiting the PCET pathway. D) 1H NMR experiments showing hydrogen bonding interactions. E) Cyclic voltammograms of the PCET pathway in MeCN vs SCE. Ewe = working electrode potential.
Next, we sought to detect the existence of the hydrogen bond. Figure 4D shows a 1H NMR experiment depicting the interactions between p-tosylamine and 4-cyanopyridine. As 4-cyanopyridine is added to a solution of sulfonamide, the N–H peak shifts downfield from 4.85 to 5.15 ppm, consistent with a hydrogen bonding interaction. Additionally, as zinc bromide is added in increasing equivalents, the N–H peak shifts back upfield to 4.95 ppm, thus demonstrating that the zinc bromide can disrupt the hydrogen bond interaction and prevent the competitive oxidative PCET.
Lastly, we turned our attention to identify the photocatalyst state that facilities PCET. One possibility is a reductive quench of the Ir(III)* species. This would result in the opposite order of electron transfers, as is required for the photocatalyst to generate a carbocation. Alternatively, the PCET could be facilitated by a ground state Ir(IV) catalyst, which would be competitive with the oxidative RPC step in the desired mechanistic sequence. To elucidate which catalyst state is involved in the undesired PCET, we carried out Stern–Volmer experiments. We observed that the excited state of the photocatalyst was not quenched by a mixture of p-tosylamine and 4-cyanopyridine (see Supporting Information). This stands in contrast to previous sulfonamide PCET systems which operate via reductive quench of the photocatalyst excited state with a strong phosphate base. We rationalize that this difference could be due to the use of 4-cyanopyridine, which would have a maximum pKa of 12.5 (pyridine in MeCN), resulting in a potential bond dissociation free energy (BDFE) of 88.9 kcal/mol that the photocatalyst excited state can access.28 To target the 105 kcal/mol BDFE of primary sulfonamide N–H bonds, the energetic difference would need to be compensated for with a more oxidizing photocatalyst state.21 The ground state Ir(IV) is more oxidizing than the Ir(III)* species by nearly half a volt (0.48 V) and has a target BDFE of 99.9 kcal/mol. Hence, we turned to cyclic voltammetry (CV) to probe the interaction of sulfonamide and pyridine with the ground state Ir(IV) species. As shown in Figure 4E, CV of the Ir photocatalyst exhibits a reversible oxidation peak. Addition of excess p-tosylamine and 4-cyanopyridine leads to an irreversible oxidation peak with an increase in current for the oxidative sweep. The behavior is characteristic of electrochemical catalysis where a soluble catalyst’s oxidation state is regenerated by substrate addition, effectively reversing the oxidation at the electrode. Control experiments verify that the oxidation of p-tosylamine is out of the photocatalyst’s range and that 4-cyanopyridine is not involved in any oxidative electron transfer process (see Supporting Information). Through these studies, we can conclude that there is a hydrogen bonding interaction between p-tosylamine and 4-cyanopyridine and that an oxidative PCET event likely occurs with an Ir(IV) catalyst.
In summary, we have developed a catalytic platform for the synthesis of a broad array of N-benzylic sulfonamides directly from C–H partners with a bench-stable oxidant. Additionally, during the optimization process, we uncovered an unproductive competitive PCET pathway that involves a hydrogen bonding complex between the sulfonamide reagent and the 4-cyanopyridine byproduct. NMR experiments corroborate that the addition of zinc bromide can disrupt hydrogen bonding through coordination to the 4-cyanopyridine. Stern–Volmer and CV experiments elucidated the involvement of the ground state Ir(IV) in this process. Such competitive PCET processes with weaker hydrogen bonding participants may be unknowingly present in other redox methods. We hope these findings may assist others in their exploration and use of redox reactions.
Supplementary Material
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.5c01637.
Additional experimental details, characterization data, optimization, and relevant spectra (PDF)
ACKNOWLEDGMENTS
N.A.F., A.M.H. and P.Z.M. would like to thank Pfizer, Inc. for funding this collaborative project. We would like to thank Matthew Crawley at the University at Buffalo for assistance with NMR, and the University at Buffalo’s Chemistry Instrumentation Center for HRMS assistance. Support for UB’s NMR is from NSF MRI grant CHE-2018160. We also would like to thank Dr. Ryan Evans for helpful discussions.
Funding
Research reported in this publication was supported by a research grant from Pfizer, Inc., startup funds from The State University of New York at Buffalo and the National Institute of Health (R35GM147021).
Footnotes
The authors declare no competing financial interest.
Contributor Information
Nicholas A. Fitzpatrick, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, United States.
Anna M. Howarth, Natural Sciences Complex, The State University of New York at Buffalo, Buffalo, New York 14260, United States.
Grzegorz J. Skrzypek, Natural Sciences Complex, The State University of New York at Buffalo, Buffalo, New York 14260, United States; Medicine Design, Pfizer Research and Development, Groton, Connecticut 06340, United States
Jisun Lee, Medicine Design, Pfizer Research and Development, Groton, Connecticut 06340, United States.
Hatice G. Yayla, Medicine Design, Pfizer Research and Development, Groton, Connecticut 06340, United States
Patricia Z. Musacchio, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, United States; Natural Sciences Complex, The State University of New York at Buffalo, Buffalo, New York 14260, United States
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- (1).Zhao X; Xu H; Huang X; Zhou JS Asymmetric Stepwise Reductive Amination of Sulfonamides, Sulfamates, and a Phosphinamide by Nickel Catalysis. Angew. Chem., Int. Ed. 2019, 58, 292–296. [DOI] [PubMed] [Google Scholar]
- (2).Shen Z; Ratia K; Cooper L; Kong D; Lee H; Kwon Y; Li Y; Alqarni S; Huang F; Dubrovskyi O; Rong L; Thatcher GRJ; Xiong R Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mukherjee P; Woroch CP; Cleary L; Rusznak M; Franzese RW; Reese MR; Tucker JW; Humphrey JM; Etuk SM; Kwan SC; am Ende CW; Ball ND Sulfonamide Synthesis via Calcium Triflimide Activation of Sulfonyl Fluorides. Org. Lett. 2018, 20, 3943–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).DeForest JC; Winters KR; Sach NW; Bernier L; Sutton SC Silyl Triflate-Promoted Sulfonylations. Org. Lett. 2025, 27, 1276–1280. [DOI] [PubMed] [Google Scholar]
- (5).(a) Hou Z-W; Liu D-J; Xiong P; Lai X-L; Song J; Xu H-C Site-Selective Electrochemical Benzylic C–H Amination. Angew. Chem., Int. Ed. 2021, 60, 2943–2947. [DOI] [PubMed] [Google Scholar]; (b) Chen Y; Yang B; Li Q-Y; Lin Y-M; Gong L Selectfluor®-enabled photochemical selective C(sp3)–H(sulfonyl)amidation. Chem. Commun. 2022, 59, 118–121. [DOI] [PubMed] [Google Scholar]; (c) Wu F; Ariyarathna JP; Kaur N; Alom N-E; Kennell ML; Bassiouni OH; Li W Halogen-Bond-Induced Consecutive Csp3–H Aminations via Hydrogen Atom Transfer Relay Strategy. Org. Lett. 2020, 22, 2135–2140. [DOI] [PubMed] [Google Scholar]; (d) Fan R; Li W; Pu D; Zhang L Transition-Metal-Free Intermolecular Amination of sp3 C–H Bonds with Sulfonamides. Org. Lett. 2009, 11, 1425–1428. [DOI] [PubMed] [Google Scholar]
- (6).For a comprehensive review on the synthesis of sulfonamides, see: Mondal, S.; Malakar, S. Synthesis of Sulfonamide and Their Synthetic and Therapeutic Applications: Recent Advances. Tetrahedron 2020, 76, 131662. [Google Scholar]
- (7).For a review on transition metal-catalyzed C–H amination methods, see: Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- (8).Gephart RT III; Warren TH Copper-Catalyzed sp3 C–H Amination. Organometallics 2012, 31, 7728–7752. [Google Scholar]
- (9).Das M; Zamani L; Bratcher C; Musacchio PZ Azolation of Benzylic C–H Bonds via Photoredox-Catalyzed Carbocation Generation. J. Am. Chem. Soc. 2023, 145, 3861–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Chang L; An Q; Duan L; Feng K; Zuo Z Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev. 2022, 122, 2429–2486. [DOI] [PubMed] [Google Scholar]; (b) Capaldo L; Ravelli D Alkoxy radicals generation: facile photocatalytic reduction of N-alkoxyazinium or azolium salts. Chem. Commun. 2019, 55, 3029–3032. [DOI] [PubMed] [Google Scholar]; (c) Kim I; Park B; Kang G; Kim J; Jung H; Lee H; Baik M-H; Hong S Visible-Light-Induced Pyridylation of Remote C(sp3)–H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed. 2018, 57, 15517–15522. [DOI] [PubMed] [Google Scholar]; (d) Bao X; Wang Q; Zhu J Dual Photoredox/Copper Catalysis for the Remote C(sp3)–H Functionalization of Alcohols and Alkyl Halides by N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed. 2019, 58, 2139–2143. [DOI] [PubMed] [Google Scholar]; (e) Barthelemy A-L; Tuccio B; Magnier E; Dagousset G Alkoxyl Radicals Generated under Photoredox Catalysis: A Strategy for anti-Markovnikov Alkoxylation Reactions. Angew. Chem., Int. Ed. 2018, 57, 13790–13794. [DOI] [PubMed] [Google Scholar]; (f) Quint V; Chouchene N; Askri M; Lalevée J; Gaumont A-C; Lakhdar S Visible-light-mediated α-phosphorylation of N-aryl tertiary amines through the formation of electrondonor–acceptor complexes: synthetic and mechanistic studies. Org. Chem. Front. 2019, 6, 41–44. [Google Scholar]
- (11).(a) Suárez JR; Chiara JL Rhodium-Catalyzed Intermolecular C–H Amination of Simple Hydrocarbons Using the Shelf-Stable Nonafluorobutanesulfonyl Azide. Chem. Commun. 2013, 49, 9194–9196. [DOI] [PubMed] [Google Scholar]; (b) Bhuyan R; Nicholas KM Efficient Copper-Catalyzed Benzylic Amidation with Anhydrous Chloramine-T. Org. Lett. 2007, 9, 3957–3959. [DOI] [PubMed] [Google Scholar]; (c) Fructos MR; Trofimenko S; Díaz-Requejo MM; Pérez PJ Facile Amine Formation by Intermolecular Catalytic Amidation of Carbon–Hydrogen Bonds. J. Am. Chem. Soc. 2006, 128, 11784–11791. [DOI] [PubMed] [Google Scholar]; (d) Gephart III RT; Huang DL; Aguila MJB; Schmidt G; Shahu A; Warren TH Catalytic C–H Amination with Aromatic Amines. Angew. Chem., Int. Ed. 2012, 51, 6488–6492. [DOI] [PubMed] [Google Scholar]; (e) Lu H; Subbarayan V; Tao J; Zhang XP Cobalt(II)-Catalyzed Intermolecular Benzylic C–H Amination with 2,2,2-Trichloroethoxycarbonyl Azide (TrocN3). Organometallics 2010, 29, 389–393. [Google Scholar]; (f) Xiong T; Li Y; Lv Y; Zhang Q Remote Amide-Directed Palladium-Catalyzed Benzylic C–H Amination with N-Fluorobenzenesulfonimide. Chem. Commun. 2010, 46, 6831–6833. [DOI] [PubMed] [Google Scholar]; (g) Wang Z; Zhang Y; Fu H; Jiang Y; Zhao Y Efficient Intermolecular Iron-Catalyzed Amidation of C-H Bonds in the Presence of N-Bromosuccinimide. Org. Lett. 2008, 10, 1863–1866. [DOI] [PubMed] [Google Scholar]; (h) Wang A; Venditto NJ; Darcy JW; Emmert MH Nondirected, Cu-Catalyzed sp3 C–H Aminations with Hydroxylamine-Based Amination Reagents: Catalytic and Mechanistic Studies. Organometallics 2017, 36, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yu X-Q; Huang J-S; Zhou X-G; Che C-M Amidation of Saturated C–H Bonds Catalyzed by Electron-Deficient Ruthenium and Manganese Porphyrins. A Highly Catalytic Nitrogen Atom Transfer Process. Org. Lett. 2000, 2, 2233–2236. [DOI] [PubMed] [Google Scholar]
- (13).Fiori KW; Du Bois J Catalytic Intermolecular Amination of C–H Bonds: Method Development and Mechanistic Insights. J. Am. Chem. Soc. 2007, 129, 562–568. [DOI] [PubMed] [Google Scholar]
- (14).Nörder A; Herrmann P; Herdtweck E; Bach T Acyclic Stereocontrol in the Catalytic C–H Amination of Benzylic Methylene Groups. Org. Lett. 2010, 12, 3690–3692. [DOI] [PubMed] [Google Scholar]
- (15).Powell DA; Fan H Copper-Catalyzed Amination of Primary Benzylic C–H Bonds with Primary and Secondary Sulfonamides. J. Org. Chem. 2010, 75, 2726–2729. [DOI] [PubMed] [Google Scholar]
- (16).Wang A; DeOliveira CC; Emmert M Non-Directed, Copper Catalyzed Benzylic C–H Amination Avoiding Substrate Excess. ChemRxiv Preprint 2019, DOI: 10.26434/chemrxiv.8792243. [DOI] [Google Scholar]
- (17).Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hu H; Chen S-J; Mandal M; Pratik SM; Buss JA; Krska SW; Cramer CJ; Stahl SS Copper-catalysed benzylic C-H coupling with alcohols via radical relay enabled by redox buffering. Nat. Catal. 2020, 3, 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ruos ME; Kinney RG; Ring OT; Doyle AG A General Photocatalytic Strategy for Nucleophilic Amination of Primary and Secondary Benzylic C–H Bonds. J. Am. Chem. Soc. 2023, 145, 18487–18496. [DOI] [PubMed] [Google Scholar]
- (20).Laurence C; Brameld KA; Graton J; Le Questel J-Y; Renault E The pKBHX Database: Toward a Better Understanding of Hydrogen-Bond Basicity for Medicinal Chemists. J. Med. Chem. 2009, 52, 4073–4086. [DOI] [PubMed] [Google Scholar]
- (21).Zhu Q; Graff DE; Knowles RR Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2018, 140, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kornecki KP; Berry JF Evidence for a One-Electron Mechanistic Regime in Dirhodium-Catalyzed Intermolecular C–H Amination. Chem.–Eur. J. 2011, 17, 5827–5832. [DOI] [PubMed] [Google Scholar]
- (23).Glogowski MP; Cercizi N; Lynch-Colameta T; Ridgers LH; Phelan JP; Rowley AM; Rauch MP Utilization of High-Throughput Experimentation (HTE) and ChemBeads Toward the Development of an Aryl Bromide and Benzyl Bromide Photoredox Cross-Electrophile Coupling. Org. Lett. 2024, 26, 2420–2424. [DOI] [PubMed] [Google Scholar]
- (24).Gulati U; Gandhi R; Laha JK Benzylic Methylene Functionalizations of Diarylmethanes. Chem.–Asian J. 2020, 15, 3135–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).McKnight J; Shavnya A; Sach NW; Blakemore DC; Moses IB; Willis MC Reductant-Free Cross-Electrophile Synthesis of Di(Hetero)Arylmethanes by Palladium-Catalyzed Desulfinative C–C Coupling. Angew. Chem., Int. Ed. 2022, 61, No. e202116775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hunter CA Quantifying Intermolecular Interactions: Guidelines for the Molecular Recognition Toolbox. Angew. Chem., Int. Ed. 2004, 43, 5310–5324. [DOI] [PubMed] [Google Scholar]
- (27).Pyridine values were used as an approximation since values for 4-cyanopyridine could not be found. Pike, S. J.; Hutchinson, J. J.; Hunter, C. A. H-Bond Acceptor Parameters for Anions. J. Am. Chem. Soc. 2017, 139, 6700–6706. [DOI] [PubMed] [Google Scholar]
- (28).Pyridine values were used as an approximation since values for 4-cyanopyridine could not be found. Due to the electron-withdrawing nature of the CN group, we hypothesize that 4-cyanopyridine is a weaker base than pyridine. Calculations for BDFEs are in the Supporting Information. Murray PR; Cox JH; Chiappini ND; Roos CB; McLoughlin EA; Hejna BG; Nguyen ST; Ripberger HH; Ganley JM; Tsui E; Shin NY; Koronkiewicz B; Qiu G; Knowles RR Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis. Chem. Rev. 2022, 122, 2017–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.