

Abstract
Wentworth et al. [Wentworth, P., Jones, L. H., Wentworth, A. D., Zhu, X. Y., Larsen, N. A., Wilson, I. A., Xu, X., Goddard, W. A., Janda, K. D., Eschenmoser, A. & Lerner, R. A. (2001) Science 293, 1806–1811] recently reported the surprising result that antibodies and T cell receptors efficiently catalyze the conversion of molecular singlet oxygen (1O2) plus water to hydrogen peroxide (HOOH). Recently, quantum mechanical calculations were used to delineate a plausible mechanism, involving reaction of 1O2 with two waters to form HOOOH (plus H2O), followed by formation of HOOOH dimer, which rearranges to form HOO—HOOO + H2O, which rearranges to form two HOOH plus 1O2 or 3O2. For a system with 18O H2O, this mechanism leads to a 2.2:1 ratio of 16O:18O in the product HOOH, in good agreement with the ratio 2.2:1 observed in isotope experiments by Wentworth et al. In this paper we use docking and molecular dynamics techniques (HierDock) to search various protein structures for sites that stabilize these products and intermediates predicted from quantum mechanical calculations. We find that the reaction intermediates for production of HOOH from 1O2 are stabilized at the interface of light and heavy chains of antibodies and T cell receptors. This inter Greek key domain interface structure is unique to antibodies and T cell receptors, but is not present in β2-microglobulin, which does not show any stabilization in our docking studies. This result is consistent with the experimentally observed lack of HOOH production in this system. Our results provide a plausible mechanism for the reactions and provide an explanation of the specific structural character of antibodies responsible for this unexpected chemistry.
Recently, Wentworth et al. (1) reported surprising results that antibodies can convert molecular oxygen to hydrogen peroxide and that the antibodies catalyze the oxidation of H2O to H2O2 by singlet oxygen molecules, 1O2 (2). This observation suggests that in addition to the well known antigen recognition function of antibodies, they may also promote destruction of the molecules to which they bind. This finding could have implications in the function (and malfunction) of the immune system and in the evolution of this system.
Investigations of the long-term photo-production of H2O2 by antibodies and non-Ig proteins reveal a remarkable difference (2). Wentworth demonstrated that the sustained high concentrations of H2O2 produced recursively could not have been by the oxidation of the amino acids in the antibodies. Thus, production of H2O2 by antibodies remains linear for a much longer period than for all non-Ig proteins tested (up to >50 mol equivalents of H2O2). Furthermore, if the H2O2 generated during the assay is removed, antibodies are able to resume H2O2 production at the same initial rate as at the start of the experiment, whereas other proteins that produce H2O2 do so by the photo-oxidation of the amino acids (e.g., tyrosine, tryptophan) and are not able to resume the same initial rate of H2O2 production. These experiments strongly suggest that the antibodies play a catalytic role in converting 1O2 plus water to H2O2.
Through isotopic labeling experiments Wentworth et al. (1) concluded that water was oxidized by the 1O2 generated. However, the experiments have not provided a mechanism to understand how the antibodies and T cell receptors (TCR) carry out this remarkable and unexpected chemistry. They observed that only antibodies and TCR catalyze this reaction, which implies that these molecules probably have unique structural features not present in other proteins. One unique feature of these systems is the interfaces created by the Greek key motifs. However, β-microglobulin also has a Greek key motif but does not convert 1O2 to H2O2.
The goal of this paper is to determine which sites in the antibodies (and TCR) play a role in the process by which 1O2 interacts with H2O to produce H2O2. A companion paper (3) presents quantum mechanical (QM) calculations that delineate plausible chemical reaction mechanisms for this chemistry, which are summarized in Results. Briefly, this mechanism involves formation of HOOOH from the reaction of 1O2 with H2O dimer, followed by complexation with another HOOOH to form a dimer that rearranges to form two HOOH plus O2. For a system with 18O H2O, this mechanism leads to a 2.2:1 ratio of 16O:18O in the product HOOH, in good agreement with the ratio 2.2:1 observed in isotope experiments by Wentworth et al. (1).
In this paper we use docking and molecular dynamics (MD) techniques to search various protein structures for sites that stabilize these products and intermediates predicted from QM calculations. That is, we consider here only catalytic processes. We use the HierDock docking and MD protocol (4) to find antibody sites that might stabilize the reaction intermediates. These HierDock studies considered high-resolution (<2.0 Å) crystal structures known to catalyze this chemistry (several Fab fragments of antibodies with varying sequence homology and TCR) and other structures (β2-microglobulin) known not to. We find that all antibodies and TCR have unique sites that stabilize the QM intermediates and products, whereas no such sites are found for the β2-microglobulin. The deduced catalytic sites are at the interface of light and heavy chains of the antibody and TCR.
These results suggest a specific structural characteristic of antibodies that is responsible for this unexpected chemistry. Armed with such specific predictions it should be possible to design experimental tests that would help verify or discard some of the plausible mechanisms. The predictions about specific important sites in the antibody could be used to design mutation studies in the antibodies and TCR to provide detailed tests on the role of the antibody.
Because the proposed mechanism does not require an energy or electron source (other than 1O2) one might be able to use these insights to design nanoscale biomimetics to carry out this remarkable chemistry in very different environments.
Methods presents the methods used in the HierDock protocol, Results summarizes the QM results, Discussion describes the sites in antibody found to stabilize the catalytic intermediates, and Conclusions discusses the results.
Methods
To identify plausible catalytic sites in the antibodies, we used the HierDock (4) protocol to search the entire antibody structure for sites that would bind to the reaction intermediates in Fig. 1 by using the structures obtained from QM (3). HierDock uses a hierarchical strategy of coarse grain docking and fine grain MD methods (including continuum solvation forces) to sample possible binding sites for ligands in the protein to determine binding sites and energies. HierDock has been applied successfully to such membrane-bound proteins as the olfactory receptors (4) and outer membrane protein A of Escherichia coli (D.D., N.V., W. B. Floriano, K. S. Kim, N. V. Prasadarao, and W.A.G., unpublished data) and to phenylalanyl t-RNA synthetase (P. Wang, N.V., D. A. Tirrell, and W.A.G., unpublished data).
Figure 1.

Gas phase structures (optimized using quantum mechanics; see ref. 3 for various clusters and transition states). These structures were used in the docking studies.
In this paper we first used HierDock to search the entire Fab structure for low energy binding sites. Here we partitioned the entire Fab antibody structure into four docking regions that could be searched in parallel. First, we carried out a coarse grain search in each region to generate a set of conformations for ligand binding. This procedure used DOCK 4.0 (7) to generate 20,000 configurations, of which 100 were ranked using the DOCK scoring function. Docking of the intermediates and products of this reaction was done using rigid ligand option in DOCK4.0.
We then selected the 20 best conformations from DOCK in each region and subjected each ligand to annealing MD to further optimize the conformation in the local binding pocket while allowing both the ligand and binding cavity (residues with an atom within 5 Å of the binding ligand) to move. In this step the ligand and the binding cavity in the protein were heated and cooled from 50 K to 600 K in steps of 10 K (0.05 ps at each temperature) for one cycle. This annealing step allows the protein cavity to readjust for the interaction with the ligand. This fine grain optimization was performed using MPSim (8) and a full atom force field (FF) (DREIDING) (9).
In addition, we used the surface generalized Born (SGB) continuum solvent method (10) to obtain forces and energies resulting from the polarization of the solvent by the charges of the ligand and protein. The SGB method allows us to calculate the change in the overall binding conformation resulting from differential solvation to obtain accurate binding energies. The charges on the various ligands were obtained from quantum mechanics (Mulliken population densities at the atom centers), whereas the charges for the protein were from CHARMM22 FF (11). A dielectric constant of 80.37 was used for the solvent field in the SGB calculation and 2.0 for the inside of the protein.
From the 20 trajectories of annealing calculations in each docking region, we selected the 20 best conformations. The relative binding energies of the 20 best structures in each region were compared (DREIDING FF with solvation) to decide which of the four docking regions leads to good binding energies for the ligands. In addition to the binding energies we also examined the population density of good binding structures in each region. The most populated regions of good structures (structures with good binding energies) were chosen for analysis.
Results
Summary of Results from QM Calculations and Plausible Mechanisms.
The QM studies (3) lead to plausible mechanisms for formation and decomposition of HOOOH and related compounds. The most plausible mechanism involves several steps: (i) Reaction of 1O2 with two waters to form HOOOH plus H2O (reaction 1 in Fig. 1); (ii) formation of HOOOH dimer; (iii) unimolecular rearrangement of HOOOH dimer to form [HOO—HOOO + H2O] (reaction 2 in Fig. 1); (iv) unimolecular rearrangement of this complex to form HOOH—OOO + H2O; (v) unimolecular rearrangement of this complex to form HOOH product + HOOOOH (reaction 3 in Fig. 1); (vi) fission of the HOOOOH to 2 HOO and association to form cyclic HOO dimer (singlet or triplet); (vii) rearrangement of cyclic HOO dimer to form HOOH product plus 1O2 or 3O2.
For a system with 18O H2O, this mechanism leads to a 2.2:1 ratio of 16O:18O in the product HOOH, in good agreement with the ratio 2.2:1 observed in isotope experiments by Wentworth et al. (1).
Depending on the products from steps vi and vii, this QM based mechanism leads to a net reaction of
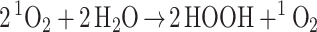 |
1 |
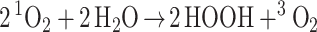 |
2 |
The net reaction in Eq. 1 has a molecularity of 2 HOOH formed from each 1O2 is in agreement with the experimental results from Wentworth et al. This excellent agreement with the experiments gives some credence to the QM-based mechanism.
To determine sites in antibodies and TCR that might play a role in enhancing these catalytic processes, we searched for sites in the antibody that bind the HOOOH product of reaction 1 (part of P1 and of R2 in Fig. 1); the HOOOH dimer (R2) of reaction 2 (Fig. 1); and the HOOH product of steps v and vii (part of P3 in reaction 3).
Given the clusters of binding sites favorable for these stable intermediates or products, we also examined whether they would stabilize the following reaction intermediates: TS1, the H2O—H2O—1O2 transition state of reaction 1 (step i); TS2, the transition state for reaction 2 (step iii); and TS3, the transition state for reaction 3 (step v).
The gas-phase structures for important intermediates and complexes are summarized in Fig. 1. Some additional comments are: (i) Xu et al. (3) finds that the barrier for the direct reaction of 1O2 with H2O to form HOOOH is over 60 kcal/mol, whereas the reaction of 1O2 with H2O dimer (R1 of Fig. 1) has a barrier (TS1) of ≈30 kcal/mol. (ii) There are two stable structures for the monomer: trans (P1 of Fig. 1) and cis (shown in R2 of Fig. 1). The cis structure is 2.4 kcal/mol higher in energy than the trans structures. We docked both conformations. (iii) Xu et al. (3) find 12 stable but distinct structures for the dimer (HOOOH)2. The most relevant for the formation of HOOH is R2 in Fig. 1. This structure is 4.9 kcal/mol more stable than the cis-monomer.
The Catalytic Site in Antibodies for Catalytic Transformation of 1O2 and H2O to HOOH.
Binding sites in the Fab antibody fragment [crystal structure (4c6.pdb)].
To seek plausible reaction sites for various steps in the QM mechanism we used the 1.2-Å Fab crystal structure 4c6.pdb (X. Y. Zhu, N. A. Larsen, and I. A. Wilson, private communication), which is the highest resolution Fab crystal structure available. The crystal structure was supplemented by adding hydrogens at standard geometries (as given by DREIDING) and hydrated counterions Na+ and Cl− were also added to charged side chain residues to maintain neutrality (12).
The crystal structure was optimized using the force field (FF), charges, and continuum solvation methods described in Methods. This minimized structure has a coordinate RMS error of 0.71 Å to all atoms of the crystal structure. (The experimental resolution of the crystal structure is 1.2 Å.) This finding indicates that the FF, charges, and solvation methods are sufficient to describe the system. We used this optimized 4c6 Fab structure in the HierDock protocols to search for sites in the 4c6 Fab structure that strongly bind HOOOH, HOOOH dimer, and H2O2 (see Eq. 1). In addition, we examined the stabilization of the transition states in Eq. 2 at the predicted binding sites for Eq. 1.
Binding sites for HOOOH monomer and dimer.
We find three sites (denoted I1, I2, I3) that strongly bind HOOOH monomer and dimer. Two of these sites (I1, I2) are at the interface of VH and VL and one site (I3) is between CH1 and CL, as shown in Fig. 2a. To help a reader to locate sites I1, I2, I3 in the three-dimensional structure, Table 1 lists the residues at each site within 5 Å of the bound HOOOH dimer. It is interesting that near I1 is Trp-109 on the heavy chain that is conserved across all antibodies and could be a potential sensitizing residue for the singlet oxygen.
Figure 2.
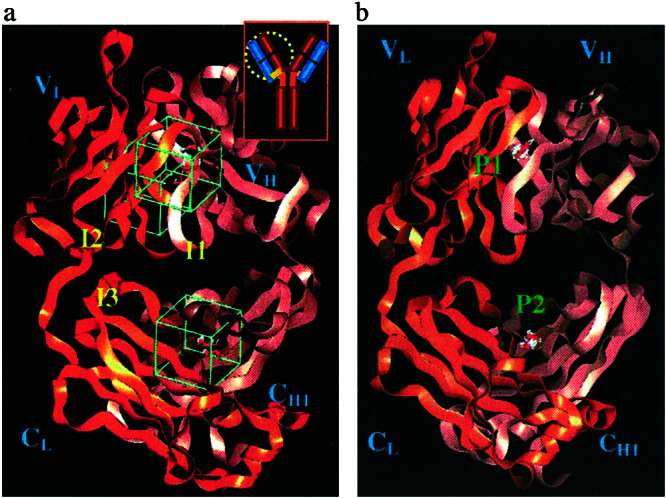
(a) Clustering sites for docking of HOOOH dimers. All sites are located between the VL and VH interface. This shows regions I1 and I3 in front. Region I2 is opposite region I1 in the back. Inset shows where this region is relative to the overall Ig. Regions I1–I3 are in the IGKD unique to antibodies and TCR. (b) Clustering sites for docking of H2O2. Region P1 is situated within the β-barrel created by the VH and VL interface. P2 is located between the CH and CL interface. Regions P1–P2 are in the IGKD unique to antibodies and TCR.
Table 1.
List of residues in the 4c6 Fab structure in the three predicted binding sites I1, I2, and I3 of the HOOOH dimers
| I1
|
I2
|
I3
|
|||
|---|---|---|---|---|---|
| VL | VH | VL | VH | CL | CH1 |
| Ser-48 | Gln-3 | Asp-1 | Lys-45 | Val-164 | Leu-147 |
| Lys-50 | Leu-4 | Pro-100 | Glu-47 | Leu-165 | Lys-149 |
| Arg-51 | Gly-107 | Tyr-101 | Trp-48 | Asn-166 | Phe-172 |
| Ser-108 | Thr-102 | Asn-61 | Ser-167 | Ala-174 | |
| Trp-109 | Pro-62 | Ser-181 | Pro-173 | ||
| Gly-110 | Ser-63 | Ser-182 | Val-175 | ||
| Thr-183 | Tyr-181 | ||||
| Thr-182 | |||||
| Leu-183 | |||||
| Ser-184 | |||||
The boldface residues are strictly conserved across 37 aligned sequences of Fab. The residues in italics are conservative replacements.
All three sites are at the interface of two Greek key domains and hence we call this interface region inter Greek key domain interface, or IGKD. The two xenon-binding sites reported by Wentworth et al. (1) in the 4c6 structure close to the sites I1 and I2. Thus Xe1 is 18.4 Å from Site I1, whereas Xe2 is 11.8 Å from site I1 and 13.0 Å from site I2.
Because the QM predicted mechanisms require two H2O for the reaction with 1O2, we would expect that the reaction site should have ordered water clusters at this site. Indeed Fig. 4 a and b shows that the crystal has higher ordered water clusters at sites I1, I2, and I3, with several water dimers and trimers
Figure 4.
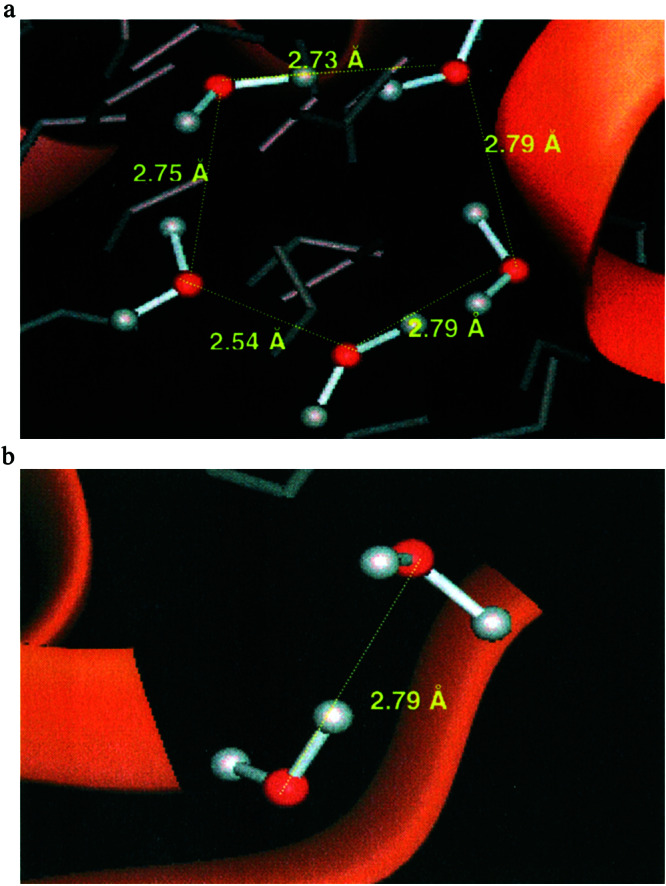
Ordered water molecules found in the x-ray structure at the IGKD interface of the Fab antibody fragment. (a) A pentamer ring of H2O molecules with each hydrogen bonded to two others. (b) An example water dimer where hydrogen from one water molecule is pointing toward the oxygen of the other molecule.
Although the QM calculations use a second H2O to catalyze the reaction of 1O2 with H2O, it is possible that these IGKD sites that stabilize the water clusters might also be able to replace the catalytic role of the H2O (that is, protons from the amino acids surrounding these sties might play similar roles).
Binding sites for product H2O2.
The same HierDock procedure was used to search for sites in the 4c6 antibody structure that would stabilize the product H2O2. Here we find the two clusters (P1 and P2 shown in Fig. 2b) containing most of the highest binding structures. P1 is at the base of the antigen-binding site, completely overlapping the Xe2 site reported by Wentworth et al. (1). P2 is between the CL and CH1 domains and overlaps region I3. Both P1 and P2 are in the hydrophobic region between the barrel-like interface of the variable and constant domains. In contrast to I1, I2, and I3, sites P1 and P2 do not exhibit bound water in the crystal structure, indicating that they are buried hydrophobic pockets.
The results derived from our docking studies of the intermediates and the product suggest that this catalytic reaction takes place in the interface regions of the variable and constant domains. This is supported by experimental evidence that shows strongly bound water dimers and trimers in these regions and the Xe binding studies suggesting that these regions are hydrophobic. Both predicted regions seem to be ideal for the reactions because of their ability to stabilize the key intermediates of the reaction cascade.
We also verified that the sites I1, I2, and I3 also stabilize the transition states for the reaction by performing a HierDock calculation for TS1, TS2, and TS3 (defined in Fig. 1 and Eq. 2) in the I1, I2, and I3 regions of the 4c6 structure. The transition state structures were kept rigid in all these docking studies. We found that the transition states cluster favorably in these regions.
Binding sites for HOOOH dimers, monomers, and H2O2 in other Ig Fab fragments.
The formation of H2O2 has been observed for a large number of antibodies (over 200), all of which have been observed (1, 2) to catalyze the conversion of 1O2 to HOOH. This conserved catalytic activity suggests that the reaction center is highly conserved across all antibodies. This may seem surprising because these antibodies include a reasonable diversity in sequences. However, the sites I1–I3 and P1–P2 we find to be important are associated with a unique structural motif of the fold in antibodies (and TCR), which might be rather insensitive to sequence. To test whether these sites would stabilize the intermediates for a range of antibodies, we selected three high-resolution (<2 Å) Fab structures (PDB ID codes 2fb4, 1c5c, and 1e60) that have maximally diverse sequences. This selection of structures was accomplished using the CLUSTALW sequence alignment program (5). The three Fab structures selected have sequence identities of 47–68% with each other and with 4c6.pdb.
HierDock was performed across the entire antibodies to prevent a bias toward any particular sequence in docking protocol. In each case we find three clusters corresponding to I1–I3 and two corresponding to P1–P2 at the same positions as for 4c6. Thus the bound HOOOH dimer and monomer cluster along the VH and the VL interface of the IGKD for all three additional structures. This study confirms that IGKD fold is important in the catalysis of this reaction, and the commonality of the binding sites for different sequences supports the IGKD region as the catalytic site.
Predicted binding sites of intermediates in TCR.
Experimentally it is known that TCR produces HOOH from 1O2, just as for antibodies. To determine whether our procedure would explain this observation, we examined TCR (PDB ID code 1tcr), which has the Greek key motif and the IGKD just as in antibodies. Again we used the HierDock protocol to perform an unbiased search for binding site across all regions of the TCR.
We found that the HOOOH monomers and dimers cluster at the heavy and light chain interface (sites I1–I3) of the TCR, consistent with the experimental observation that TCR does produce H2O2. Because the sequence similarity between 4c6 and TCR is only 25%, this suggests that the essential feature is structural not sequence-specific. These results support the conclusion that it is the IGKD interface created by the arrangement of Ig domains that is required for the stabilization of the intermediates.
Predicted Binding Sites of Intermediates in β2-microglobulin.
β2-microglobulin has the characteristic Greek key motif present in antibodies, but it is monomeric and hence does not have the barrel-like interfacial structure of the TCR and the Fab region of antibodies. Consequently, we use HierDock to perform an unbiased search for binding sites across all regions of β2-microglobulin (PDB ID code 1duz) to find favorable binding regions for HOOOH monomer, its dimer, and the transition states. However, we found no common consensus-binding region for the monomer and dimer in β2-microglobulin. The bound structures did not have a high population of docked conformations in any one region.
This finding indicates that the Ig fold by itself is not sufficient to catalyze the reaction. Rather, we require an interface created by the arrangement of Ig domains, IGKD, to create the environment required for the stabilization of the intermediates. This is consistent with the results of Wentworth et al., who showed experimentally that β2-microglobulin does not produce H2O2 from 1O2. We attribute the lack of H2O2 production in β2-microglobulin to the absence of a hydrophobic interface lined with organized water molecules. This result suggests that the unique feature responsible for the catalysis is the IGKD (only present for antibodies and TCR), not the Greek key fold (which is present in all immunoglobins, including β2-microglobulin and other proteins).
Discussion
Nature of Binding Site for HOOOH Monomer and Dimer.
The two catalytic sites predicted here are at the interface of light and heavy chains of the antibody, a structure unique to antibodies and TCR. This IGKD interface of two Greek key domains is shown in Fig. 3 a and b. The two binding sites are each located on the sides of the barrel-like structural motif (6) at the interface of VH and VL, as shown in the inset of Fig. 3b. This structure has the beta sheets of VH and VL separated by ≈5 Å, favoring the binding of the water sheet observed experimentally. The residues lining these sites shown in bold face in Table 1 are strictly conserved and those in italics are conservative replacements. These results were obtained by performing a CLUSTALW sequence alignment of the 37 Fab sequences having structure resolved to within 2.0 Å. Trp-109 in the I1 binding site is conserved across all antibodies and could be a potential sensitizing residue for the singlet oxygen.
Figure 3.
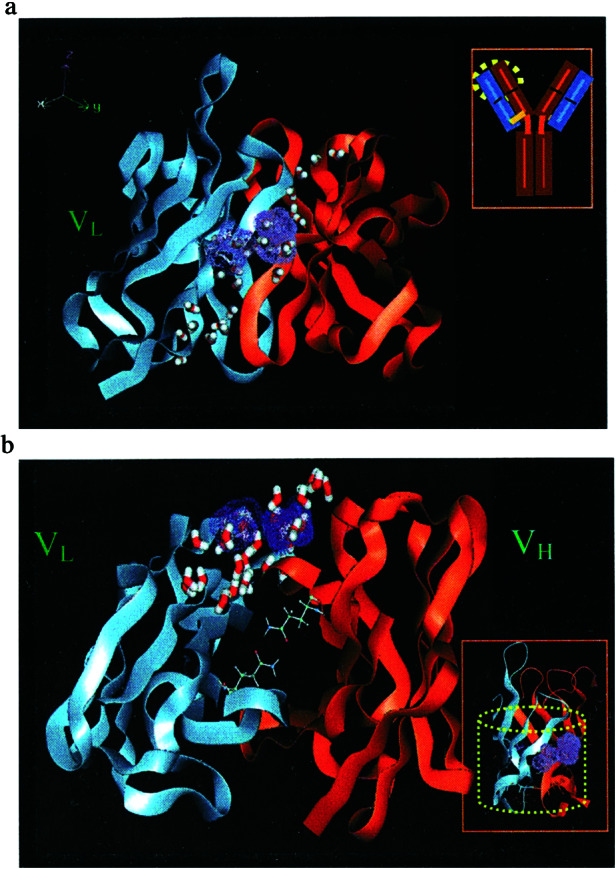
(a) The purple dots indicate two regions of the Fab antibody fragment that bind strongly to the HOOOH dimer and that we conclude are plausible regions for the catalysis of 1O2 plus H2O dimer to form HOOOH. These sites are at the interface of the VH and VL in a region containing well ordered crystallographic waters (shown with half bonds). These regions are in the IGKD unique to antibodies and TCR. Inset shows a schematic antibody structure with a yellow circle to indicate the region magnified. (b) The structure in a is rotated 90° about the horizontal axis to show the hydrophobic channel bounded by Gln-38 from VL and Gln-39 from VH. This forms a hydrogen bond network at the mouth of the barrel. Inset shows the barrel-like structure (containing two Greek keys) unique to antibodies that we suggest is critical to the catalysis.
There are a number of well ordered crystallographic waters on the sides of this interfacial barrel-like structure between the VH and VL, as shown in Fig. 4 a and b. These waters are ordered in dimers (O–O distances ≈2.6 Å), trimers (O–O distances varying from 2.6 to 3.3 Å), and a pentamer cluster (see Fig. 4a) with distances of 2.54–2.79 Å. This pentamer ring of H2O is in region I2 (it is formed by the crystallographic waters: Wat 12, 54, 60, 249, and 339). The water dimers shown in Fig. 4b are Wat 5 and Wat 404. Such well ordered water clusters can be observed only in high-resolution crystal structures, such as 4c6 structure with 1.2 Å resolution.
We consider that these water clusters are the H2O structures that react with 1O2, to form HOOOH, which subsequently reacts with a second HOOOH or 1O2 to form H2O2 and the other reactive intermediates discussed above. Thus the first step of our QM mechanism involves two waters in a dimer-like structure, just as in Fig. 4 a or b, with one water acting as a catalyst in this step.
Thus, the I1 and I2 sites determined using HierDock seem quite appropriate for the reaction to generate HOOOH from 1O2 plus two H2O. This product HOOOH is also favorable in this same site or in I3. Thus it is plausible that a second HOOOH (formed from an additional 1O2 and another H2O dimer) could remain in these regions to combine with the first to form HOOOH dimer at either I1–I2 or I3. This could then form H2O2 as in the QM mechanism. This H2O2 might then migrate to the sites P1–P2 that we find most favorable for H2O2.
A closer look at the interface of light and heavy chains of all antibodies shows that the bottom of the channel or barrel is capped by polar amino acids. For most antibodies these are glutamines forming a hydrogen bond network, as shown in Fig. 3b. We suggest that these residues could serve two functions. (i) They could gate the reactants and various intermediates from entering the hydrophobic channel. Instead, these intermediates would go to the side of the barrel at the interface of light and heavy chain as shown in Fig. 3a. (ii) They could prevent the H2O2 formed from escaping from the bottom of the barrel. This might direct them to be released toward the antigen-binding site. To determine whether these glutamines play a role in capping the products from the 1O2 chemistry, it would be interesting to examine systems where the glutamines are mutated to hydrophobic residues.
For Fab our studies of binding HOOOH and its dimer and of H2O2 suggest the model that the IGKD motif is essential for H2O2 production from singlet oxygen. Because the Fc structure of antibodies have one such IGKD interface compared with two in the Fab structure, this suggests that the efficiency of HOOH production in Fc should be half that of Fab. Indeed Wentworth and Lerner (P. Wentworth and R. A. Lerner, personal communication) have shown that Fc structures have half the efficiency of Fab structures.
Although β2-microglobulin does not have an IGKD, combination of β2-microglobulin with α3 to form class I MHC does lead to an IGKD. Thus we suggest that the β2-microglobulin in the MHC complex would generate H2O2 from 1O2 with the same efficiency as Fc.
Geometric Pathway for the Conversion of 1O2 to HOOH.
A schematic geometric roadmap based on our proposed mechanism is given in Fig. 5 (for the 4c6 Fab structure). (a) We assume that 1O2 may enter the antibody from near the Xe1 (and Xe2) xenon-binding site to migrate through the hydrophobic environment of VH and VL to the IGKD interface region (sites I1 and I2). (b) Here 1O2 can convert the clustered waters at this site to HOOOH. (c) This HOOOH might react with a second 1O2 or it might migrate to the I3 site, where it could react with a second HOOOH. In either case this reaction produces two HOOH. The HOOH products of this reaction might migrate to sites P1 and P2. (d) Subsequently these HOOH might migrate toward the interior of the barrel where H2O2 [or other intermediate such as HOOOH or the (HOO)2 dimer] could react with the antigen. This might mark it for destruction.
Figure 5.

The geometric pathway for the sequence of reactions converting 1O2 water to HOOOH and then to HOOH. Here we assume that 1O2 enters the hydrophobic region near Xe1. At I1 (or I2) it can react with a water dimer (or trimer) to form HOOOH. The HOOOH may stay at I1 (or I2) but it may go to I3, which does not have crystallographic waters. This HOOOH may react directly with a second 1O2 or with the HOOOH from a previous reaction to form the HOOOH dimer—this may occur at I3. The HOOOH dimer can rearrange through a series of steps to form HOOH, which may go to sites P1 or P2 (there are no crystallographic waters at these points). Here the HOOH is positioned close to the region at which antigen may be bound (HOOOH may also go to this region). From here the HOOH (or HOOOH) might react directly with the part of a protein whose antigen is recognized by the antibody.
Such a destructive role of antibodies is consistent with the observation that 1O2 is produced in processes involved with the macrophage engulfing the antigen bound antibody.
Conclusions
Based on the experiments by Wentworth et al. showing that antibodies can catalyze 1O2 to oxidize water to form H2O2, and based on the QM computational studies of Xu et al. showing that the chemical mechanism involves production of HOOOH and subsequent reactions to form a series of products culminating in H2O2, we searched various proteins for special sties compatible with this chemistry.
Our HierDock studies lead to the conclusion that the interfacial motif IGKD, between two Greek keys (present only in antibodies and TCR and not present in β2-microglobulin) is critical to catalysis of 1O2 to oxidize water to form HOOOH and H2O2. For both antibodies and TCR, we found sites (I1–I3) in the region favorable for binding the HOOOH reaction intermediates and sites (P1–P2) favorable for the H2O2 product. Based on these docking results and on the QM calculations, we propose a sequence of steps by which antibodies can produce HOOOH and H2O2 from 1O2. These results suggest that such reactive intermediates as HOOOH and (HOO)2 and the product HOOH are favorably formed in the IGKD paired Greek key barrel region close to the antigen. We speculate that the conversion of 1O2 to HOOOH and/or HOOH might provide for a protective function against singlet oxygen (which can attack dienes and other molecules in cells).
Alternatively these reactive intermediates might react with the antigen to help make the protein recognized by the antibody more susceptible to attack by other enzymes in the macrophage. This might provide a defense mechanism against the proteins having antigens to these antibodies. Here the HOOOH and/or HOOH might react selectively against just the antigen recognized. Based on the detailed prediction of binding sites involved in various steps, one can imagine a variety of biological experiments that might test our QM and HierDock results. Thus selective mutations could be made to enhance or inhibit various steps.
These results suggest a number of experimental tests and provide a guideline for how to build biomimetic nanoscale systems producing HOOH (or HOOOH).
These computational studies provide mechanistic insight to the experimental observations by Wentworth et al. that antibodies and TCR can catalyze the conversion of 1O2 plus water to H2O2. The results gives very close agreement with observed isotope ratio of 2.2:1. In particular the results explain the observed molecularity of 2.0 for the number of HOOH produced per 1O2—this provides strong support for the QM mechanism.
Acknowledgments
We thank Richard Lerner for suggesting this problem and Albert Eschenmoser, Paul Wentworth, Anita Wentworth, Lyn Jones, and Kim Janda for helpful discussions. We also thank Xueyong Zhu, Nicholas Larsen, and Ian Wilson for access to their 1.2-Å structure for the chimeric Fab antibody before publication. This research was funded by National Institutes of Health Grant HD 36385-02. The facilities of the Materials and Process Simulation Center used in these studies were funded by National Science Foundation–Major Research Instrumentation, Defense University Research Instrumentation Program (Army Research Office and Office of Naval Research), and the Beckman Institute. In addition, the Materials and Process Simulation Center is funded by grants from Department of Energy–Accelerated Strategic Computing Initiative–Academic Strategic Alliances Program, Army Research Office–Multidisciplinary University Research Initiative, National Institutes of Health, National Science Foundation, Avery–Dennison, Asahi Chemical, Chevron, 3M, Dow Chemical, Nippon Steel, Seiko-Epson, and Kellogg's. These calculations were made under a SUR Grant from IBM.
Abbreviations
- IGKD
inter Greek key domain interface
- TCR
T cell receptor
- QM
quantum mechanical
References
- 1.Wentworth P, Jones L H, Wentworth A D, Zhu X Y, Larsen N A, Wilson I A, Xu X, Goddard W A, Janda K D, Eschenmoser A, Lerner R A. Science. 2001;293:1806–1811. doi: 10.1126/science.1062722. [DOI] [PubMed] [Google Scholar]
- 2.Wentworth A D, Jones L H, Wentworth P, Janda K D, Lerner R A. Proc Natl Acad Sci USA. 2000;97:10930–10935. doi: 10.1073/pnas.97.20.10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu, X., Muller, R. P. & Goddard, W. A., III (2002) Proc. Natl. Acad. Sci. USA98, in press.
- 4.Floriano W B, Vaidehi N, Goddard W A, III, Singer M S, Shepherd G M. Proc Natl Acad Sci USA. 2000;97:10712–10716. doi: 10.1073/pnas.97.20.10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eddy S R. Proc Int Conf Intell Syst Mol Biol. 1995;3:114–120. [PubMed] [Google Scholar]
- 6.Branden C, Tooze J. Introduction to Protein Structure. New York: Garland; 1998. pp. 306–309. [Google Scholar]
- 7.Ewing J A, Kuntz I D. J Comp Chem. 1997;18:1175–1189. [Google Scholar]
- 8.Lim K-T, Brunett S, Iotov M, McClurg R B, Vaidehi N, Dasgupta S, Taylor S, Goddard W A., III J Comp Chem. 1997;18:501–521. [Google Scholar]
- 9.Mayo S L, Olafson B D, Goddard W A., III J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 10.Ghosh A, Rapp C S, Friesner R A. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 11.MacKerell A D, Bashford D, Bellott M, Dunbrack R L, Evanseck J D, Field M J, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 12.Vaidehi N, Goddard W A., III Proc Natl Acad Sci USA. 1997;94:2466–2471. doi: 10.1073/pnas.94.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]