

Abstract
The completion of the human genome project and the development of high-throughput approaches herald a dramatic acceleration in the pace of biological research. One of the most compelling next steps will be learning the functional roles of all proteins. Achievement of this goal depends in part on the rapid expression and isolation of proteins at large scale. We exploited recombinational cloning to facilitate the development of methods for the high-throughput purification of human proteins. cDNAs were introduced into a master vector from which they could be rapidly transferred into a variety of protein expression vectors for further analysis. A test set of 32 sequence-verified human cDNAs of various sizes and activities was moved into four different expression vectors encoding different affinity-purification tags. By means of an automatable 2-hr protein purification procedure, all 128 proteins were purified and subsequently characterized for yield, purity, and steps at which losses occurred. Under denaturing conditions when the His6 tag was used, 84% of samples were purified. Under nondenaturing conditions, both the glutathione S-transferase and maltose-binding protein tags were successful in 81% of samples. The developed methods were applied to a larger set of 336 randomly selected cDNAs. Sixty percent of these proteins were successfully purified under denaturing conditions and 82% of these under nondenaturing conditions. A relational database, FLEXProt, was built to compare properties of proteins that were successfully purified and proteins that were not. We observed that some domains in the Pfam database were found almost exclusively in proteins that were successfully purified and thus may have predictive character.
With the application of large-scale and high-throughput (HT) approaches to biological and medical questions, biology has embraced a new era of technology development and information collection. The great task lying ahead is to elucidate the functions of all proteins encoded in the genomes of sequenced model organisms. This process involves collection of information about the temporal, spatial, and physiological regulation of proteins, their interaction partners, biochemical activities, posttranslational modifications, and the mutual influence of all these parameters on the physiology of the organism. Over the past several decades, biologists and biochemists have amassed a large collection of powerful tools for the study of individual proteins. However, compared with the study of nucleic acids, the HT study of proteins is still in its infancy. The next great challenge in biology will be to adapt these tools and develop new ones that enable the simultaneous and parallel study of thousands of proteins.
The elucidation of biochemical activity and protein–protein interactions are central aspects of understanding protein function. Protein microarrays provide one platform for biochemical experiments to be carried out at extraordinary pace (1–3). However, this exciting technology calls attention to the question of how thousands of proteins can be rapidly expressed and isolated for use on this and other HT platforms. Early efforts in model organisms show significant promise. Zhu et al. (3) cloned all open reading frames (ORFs) of the yeast Saccharomyces cerevisiae by gap-repair and expressed them as glutathione S-transferase (GST) fusion proteins in the same organism. The expressed proteins were purified and used to produce a high-density protein array (2). However, the fact that the coding sequences (CDSs) are locked into the vector in which they have been assembled, and cannot be transferred into alternative expression constructs, constitutes a major disadvantage if tags other than GST or other expression systems are required. In addition, the ability to purify proteins from their natural cell types does not easily extrapolate to proteins of other model organisms or human proteins. Thus, there is a significant need for flexible methods that enable the rapid expression and purification of proteins in heterologous systems in a HT format.
The increased availability of comprehensive cDNA collections will facilitate the expression and study of all proteins encoded in a given genome (4). Many of these collections are being assembled in recombinational cloning systems, which exploit site-specific or homologous recombination to capture the cDNAs into a master vector in which they are maintained. Because untranslated regions are of variable length and may contain stop codons, which interfere with the expression of fusion proteins, there is a need for repositories in which untranslated regions have been removed and only the CDSs have been captured (5, 6). To express proteins, the CDSs can be transferred into any desired protein expression vector by using a universal, simple, single-step procedure that is well suited to HT operations (7).
Escherichia coli cells offer a robust, convenient, and inexpensive expression system for the production and purification of human proteins. Bacteria are easily grown in a HT format and are widely used to express human proteins for use in research and as pharmaceuticals. The existing literature regarding protein expression in E. coli has focused on the optimization of conditions for individual proteins. However, because proteins frequently differ significantly in their physical and chemical properties, it is difficult to apply conditions that work well from one protein to another. Thus, there is a need to define expression and purification conditions that are amenable to hundreds and thousands of proteins in parallel.
Affinity tags are widely used to produce proteins of high purity in a single-step procedure (8). Because polypeptide-purification tags can be genetically attached to any protein, they are suited to HT operations. In addition to determining the chemistry to be used in protein purification, these tags can influence the behavior of the fusion protein during various steps of protein expression, purification, and utilization. A number of different purification tags have been described, each with different features that influence the stability, solubility, and expression level of recombinant proteins in bacteria (9). We wished to examine these properties as they relate to the success of HT protein purification and employ them in the context of an inexpensive and easy method for the rapid purification of a diverse set of proteins in parallel.
Materials and Methods
PCR and BP Recombination Reaction.
Specific PCR primers were designed to amplify only the CDSs by using the nearest-neighbor algorithm. Brain or placental first-strand cDNA served as template in the first PCR. All clones were assembled in the Gateway recombinational cloning system manufactured by Invitrogen. Recombination sites were attached in a second PCR. The final PCR products were visualized on an agarose gel, and correctly sized bands were excised. The DNA was isolated by filtration, and 5 μl of the flow-through was used in a capture reaction according to the protocol of the supplier of the kit (7).
Destination Vectors and LR Recombination Reactions.
PDEST-17 was used as a His6-expression vector. For the other tags, pCAL-n-Flag, pGEX-2tk, and pMal-2c were adapted to recombinational cloning by insertion of the appropriate recombination cassette using a blunt site in the multiple cloning sites and subsequent determination of the correct orientation. Transfer reactions were done using the protocol in ref. 7 with the following changes: the final reaction volume was 10 μl, and all other components were used at half the recommended volume except that 1 μl of LR Clonase enzyme (Invitrogen) and 3 units of DNA topoisomerase I were used. The mixture was either frozen or immediately used for transformation of DH5α cells.
Transformations into DH5α Cells and DNA Minipreps.
Transformations were done in 20 μl or 100 μl final volume as described in ref. 10. Up to 384 colonies were plated robotically on 25 cm × 25 cm LB agar plates, which contained 125 μg/ml ampicillin, by using a Tecan (Durham, NC) Genesis robotic sample processor 150. Minipreps were produced robotically by using the Qiagen 96-well Turbo prep.
Protein Expression and Purification in 96-Well Format.
A streak of freshly transformed BL21pLyss was inoculated in 1 ml of TB medium (Teriffic Broth) containing 125 μg/ml ampicillin, 34 μg/ml chloramphenicol, and 2% glucose and grown for 14–16 hr. The OD600 was measured and the cultures were diluted to a final OD600 of 0.1 in 1.5 ml of fresh TB containing the same antibiotics. The cultures were then grown for ≈3.5 hr at 25°C and simultaneously induced with 1 mM isopropyl β-d-thiogalactoside (IPTG) when the average OD600 of all cultures was 0.7–0.9. After 1.5 hr of growth at 25°C, a 75-μl aliquot for Western blot analysis was removed, the OD600 was measured, and the remainder of the liquid culture was harvested. The pellets were frozen to −20°C.
Frozen cell pellets were thawed for 5 min at room temperature and resuspended at 4°C in 100 μl of lysis buffer. Resuspension was achieved by agitating the 96-deep well block on a Beckman shaker for 5 min at 600 rpm while mixing the cells and the buffer with an inverted 96-pin device. Then 10 μl of 200 μg/ml lysozyme and 0.1% Triton X-100 for His6 or GST and 0.05% for CBP and MBP purifications was added, and mixing was continued for 30 min at 300 rpm. Subsequently, 10 μl of DNase mix [900 mM MgSO4/100 mM MnCl2/0.5 μg/ml DNase (Sigma D-4527)] was added and mixing was continued at 300 rpm for another 15 min. During these incubations, two Whatman GF/C plates were prepared the following way: The filter was wetted with lysis buffer. The purification matrix was pipetted into the second plate and equilibrated by addition of 200 μl of lysis buffer followed by a centrifugation. The purification plate was sealed at the bottom with aluminum foil and placed on top of a rubber cushion. The filtration plate was placed on top of the purification plate and the lysates were transferred into this filtration plate by centrifugation for 2 min at 2,000 × g. Then the purification plate was sealed on top and rotated for 45 min at 4°C. After binding, the seals were removed and the lysates were separated from the beads by a centrifugation at 16 × g for 1 min 30 sec at 4°C. The matrix was washed by repeated addition of 260 μl of urea wash buffer and centrifugation. Finally, the proteins were eluted by addition of elution buffer (urea wash buffer containing 0.7 M imidazole, pH 8.0) followed by a 5-min incubation and a centrifugation. After the last elution, the plates were centrifuged once more at 2,000 × g for 5 min to remove the remainder of liquid.
Buffers and Matrices for the Purifications.
His6 denaturing conditions.
Lysis buffer: 100 mM NaH2PO4/10 mM Tris⋅HCl/6 M guanidinium hydrochloride/10 mM 2-mercaptoethanol, pH 8.0; wash buffer: 100 mM NaH2PO4/10 mM Tris⋅HCl/8 M urea, pH 8.0; elution buffer: wash buffer containing 0.5 M imidazole, pH 8.0. Ni-NTA matrix from Qiagen was used for purifications under denaturing conditions.
His6 nondenaturing conditions.
Lysis and wash buffer: 50 mM NaH2PO4/500 mM NaCl/10% glycerol, pH 8.0; elution buffer: wash buffer containing 150–500 mM imidazole, pH 8.0 Ni-NTA manufactured by Qiagen and Talon matrix manufactured by CLONTECH were used.
CBP.
For CBP purifications the buffers A, B, and D were used as described in ref. 15. CBP-agarose was purchased from Stratagene and Pharmacia and washed thoroughly.
GST.
Lysis and wash buffer I: 140 mM NaCl/10 mM Na2HPO4/2.7 mM KCl/1.8 mM KH2PO4/2 mM EDTA/10% glycerol, pH 7.3; wash buffer II: wash buffer I, but 500 mM NaCl and 0.1% Triton X-100; elution buffer: wash buffer II containing 20 mM reduced glutathione. Glutathione-agarose was purchased from Pharmacia and equilibrated in wash buffer I.
MBP.
Lysis and wash buffer: 20 mM Tris⋅Cl/500 mM NaCl/10% glycerol/2 mM EDTA, pH 7.4; elution buffer: wash buffer containing 20 mM maltose. Amylose resin was purchased from NEB and washed thoroughly with wash buffer before use.
SDS/PAGE and Western Blot Analysis.
For SDS/PAGE analysis, 4–20% Criterion precast gradient gels with 26 wells were used. GelCode Coomassie blue reagent from Pierce was used to visualize protein bands on the gels. Western blotting was done as described (11). Antibodies and dilutions were as follows: anti-His4 antibody from Qiagen at 0.1 μl/ml in 3% (wt/vol) BSA; M2 monoclonal anti-FLAG from Pierce at 1:1000 in Blotto (50 mM Tris⋅HCl, pH 7.4/100 mM NaCl/5% nonfat dry milk); Z-5 polyclonal anti-GST from Santa Cruz at 1:1000 in Blotto; and anti-maltose-binding protein (MBP) antibody from NEB at 1:1000 in Blotto. The signal was visualized by using Pierce Femto- or Pico-West luminol reagent and was detected on a Chemidoc from Bio-Rad or film.
Purification of Kinases from Insect Cells and Kinase Reactions.
The baculoviruses for cyclin E/GST-cdk2 and cyclin D1/GST-cdk4 were kind gifts of H. S. Chou (Smith–Kline Beecham, Philadelphia) and J. Zhao (Univ. of Rochester), respectively. Cells were lysed in cyclin D buffer as described (8) and purified according to the recommendations of the manufacturer. Kinase assays were performed as described (12).
Database and Informatics.
The FLEXProt database was created by using Microsoft Access. External data for the FLEXProt database were parsed from the SwissProt, LocusLink, and Gene Ontology databases. Query results were visualized by using spotfire software. The Pfam database IDs of the mentioned domains are as follows: 00001, 7-TM; 000010, HLH; 00017, SH2; 00018, SH3; 00022, actin; 00069, protein kinase; 00071, ras; 00229, TNF; 00812, Ephrin; 00917, MATH; 02176, TRAF; and 02421, ferrous transport (13).
Results
Test Construct Assembly.
Protein affinity purification tags can profoundly influence stability, solubility, and expression levels of human proteins expressed in bacteria (9). We wished to identify polypeptide purification tags with robust chemistry and that have favorable effects on yield and purity of many different proteins when used in parallel protein purification. After reviewing the literature, we selected four purification tags for experimental evaluation: the His6 tag (14), the 4-kDa calmodulin-binding peptide (CBP) (15), the 26-kDa GST (16), and the 42-kDa MBP (17).
The biochemical and biophysical properties of proteins may vary significantly from one protein to another. To define conditions and identify a purification chemistry that works well when fused to a variety of proteins, a test set of 32 full-length human genes was chosen (see Fig. 2). As it has been observed that protein expression in bacteria may depend on the size of the recombinant protein (16), the test set included proteins with a broad range of molecular weights (see Fig. 2). In addition, the set contained proteins that localize to different subcellular compartments and that have different biochemical activities. Integral membrane and secreted proteins were excluded because these classes of proteins require separate optimization and purification methods (18). The test set was assembled in the Gateway recombinational cloning system to enable the rapid creation of the required expression constructs (7). The sequence-confirmed cDNAs were then transferred overnight into each of the expression vectors to create 128 expression constructs. All transfer reactions were confirmed by analytical PCR.
Figure 2.
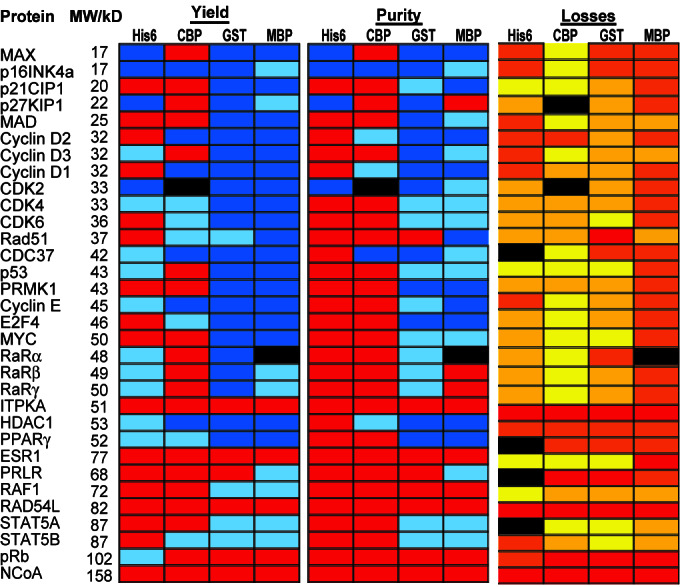
Summary of all test set protein purifications: All 128 proteins were purified by using the respective affinity tag, and total yield and purity of the purified proteins were analyzed by GelCode staining and image analysis. Losses were characterized by Western blot analysis of five key fractions. Yield: red, <300 ng; light blue, 300 ng to 1 μg; dark blue, >1 μg. Purity: red, <10% purity or no detectable band; light blue, 10–30% purity; dark blue, >30% purity. Losses: red, protein degraded in vivo; orange, >60% of lost protein in the flow-through; dark yellow, losses evenly distributed between flow-through and matrix; bright yellow, >60% of lost protein was found on the matrix.
Protein Expression in Vivo.
The 128 fusion proteins were expressed under conditions that were shown in preliminary experiments to decrease degradation and give satisfactory yield for most fusion proteins as opposed to maximum yield for some proteins. The chosen conditions have been shown previously to favor the production of intact soluble protein for individual proteins (19). Under these conditions, nearly all of the fusion proteins were expressed, as determined by Western blot analysis on whole cells using the respective antibodies (Fig. 1). However, within each set a decrease in expression levels was observed with increasing protein size. It has been observed previously that larger human proteins tend to be expressed at lower levels in bacteria (16).
Figure 1.

Expression of 32 test set proteins fused to four different purification tags. The 32 genes in the test set were transferred into each of the four expression vectors, transformed into BL21 cells, and cultured and induced as described. 10 μl (≈1%) of each culture was lysed directly in Laemmli buffer and analyzed by Western blotting using antibodies against the peptide tags as indicated. Bands of the correct size are indicated by a dot on the right side of the band.
Of the four tags, the GST fusion proteins were most prone to proteolysis in vivo. In some cases, such as p21Cip1 and Rad51, only the full-length fusion protein and the GST band were detectable, suggesting that the GST moiety was cleaved off in some molecules. For other proteins, like E2F4, multiple proteolytic fragments were visible, indicating that the fusion partner has been degraded in a step-wise fashion, with GST as a stable end-product.
For some proteins—ITPKA, RAD54L, and NCoA—only degradation products were detectable regardless of the fusion tag. Since neither of the corresponding cDNAs has any mutation, this behavior suggests an inherent instability of these proteins when expressed in bacteria. Even though the retinoblastoma protein (pRb, 110 kDa) was expressed as a His6-fusion protein, it could not be observed as a fusion with any of the larger tags.
HT Protein Purification Under Denaturing Conditions.
To test the developed HT protein purification platform, proteins were first purified under denaturing conditions by using the His6 tag. Under these conditions, all proteins that were detectable in vivo could be purified successfully, except the 110-kDa pRb (data not shown). (Throughout our experiments, the measure for a “successful purification” was a visible band of the correct size on a Coomassie blue-stained gel. In our process, such a band indicated a total yield of at least 300 ng.) It should be noted that denatured proteins are a useful product in some applications such as antigens to make antibodies or in diagnostic applications. In addition, enzymatic activity can frequently be recovered from proteins purified under denaturing conditions after using a refolding step (20). Given the success of denaturing purification conditions, it may be worthwhile to consider the development of HT renaturation methods.
HT Protein Purification Under Nondenaturing Conditions.
Functional experiments, by definition, require proteins in a native conformation. Thus, HT-compatible conditions for protein purifications under nondenaturing conditions were established, and the 128 fusion proteins purified in parallel were characterized with respect to yield and purity. The data are summarized in Fig. 2. In addition, the protein content of five key steps in each purification process—total lysate, flow-through, wash, matrix-bound, and eluted protein—were qualitatively examined to analyze how each tag performed for each of the 32 test-set proteins. In general, the proteins that did not purify well fell in three basic groups: proteins that degraded in vivo before lysis, proteins primarily lost in the flow-through, and proteins that could not be eluted off the matrix. In some cases, losses were evenly distributed between flow-through and matrix. The fractions in which the majority of protein was lost in each of the 128 protein purifications are indicated in Fig. 2.
His6 tag.
Despite the efficiency with which the His6 tag functioned under denaturing conditions, only four proteins were detected by Coomassie blue staining under nondenaturing conditions, although many more proteins (15/32) could be detected by Western blotting. Of these four, only MAX and p16INK4a were reasonably pure (70%). All His-tagged proteins were lost in the flow-through and/or could not be eluted from the matrix. This was true for both Ni2+ and Co2+ matrices and with a broad range of imidazole concentrations (200–500 mM) or 5 mM EDTA used for elution in the presence of 500 mM NaCl.
CBP tag.
As with the His6 tag, only 5 proteins of 32 proteins that had been purified with the CBP tag could be detected on a Coomassie stained gel and 7 more by Western blotting. Again, p16Ink4a had the highest yield and purity. A loss analysis of the different fractions demonstrated that matrix binding for CBP-fused proteins was more efficient—10/32 proteins were detected in the flow-through. However, 22 of 32 matrix-bound proteins did not elute in 5 mM EDTA even in the presence of 1 M NaCl. Difficulties eluting CBP-tagged proteins have been mentioned in the literature previously (15).
GST tag.
Of the GST-tagged proteins, 26/32 were purified with a yield of at least 300 ng of protein per ml of culture, and 22 of these with a yield of >1 μg/ml of culture based on a comparison with a quantity standard (Fig. 3A). Six of the 32 proteins could not be purified as GST constructs because the full-length protein could not be detected or the GST moiety had been lost in vivo (Fig. 1). The total purity of most proteins was in the range of 30–70%. It is likely that many impurities are degradation products of the recombinant protein, because a similar pattern of bands was observed by an anti-GST antibody on a Western blot. In addition, many of these bands were already detectable in vivo (Fig. 1), indicating that the degradation occurred before cell lysis.
Figure 3.

Test set proteins as purified with the GST tag (A) and the MBP tag (B). Bands of the correct size are indicated by a dot on the right side of the band. Fifteen percent of the total eluate was loaded on a 4–20% gradient SDS-gel and stained with GelCode Coomassie blue reagent.
MBP tag.
The MBP also purified 26/32 proteins to yields of at least 300 ng of protein per ml of original culture, and 18 of these with yields of >1 μg/ml (Fig. 3B). The purity of most MBP-purified proteins ranged from 20% to 70%. Most MBP-tagged proteins were primarily lost in the flow-through. A low binding efficiency of MBP-tagged proteins has been reported previously and is a result of the low affinity of MBP for its matrix (21).
Functional Experiments with GST and MBP Constructs.
To test whether the purification conditions that we used produced biochemically active proteins, proteins tagged with GST and MBP were tested in two different biochemical assays. In the first assay, GST- or MBP-tagged cyclin E, purified from bacteria in 96-well format, was combined with recombinant cdk2 purified from insect cells in standard kinase reactions using histone H1 as a substrate. Fig. 4A shows that both fusion proteins activated histone phosphorylation.
Figure 4.

GST- and MBP-tagged proteins are active. Autoradiograms of 32P incorporation. (A) Equal amounts of bacterially purified GST- and MBP-tagged cyclin E were added to GST-cdk2 purified from insect cells in histone H1 kinase reactions. Both constructs activate cdk2 kinase activity. (B) p16Ink4a specifically inhibits kinase activity. Equal volumes of 16 GST-tagged test set proteins were added to kinase reactions using cyclin D1/cdk4 purified from insect cells and C-terminal fragment of pRb as a substrate.
As another functional test, it was examined whether GST-p16INK4a specifically inhibited cyclin D1/cdk4 kinase activity against C-terminal fragment of the retinoblastoma protein. Kinase reaction mixtures were incubated with 16 different GST-tagged proteins purified in HT format. As expected, GST-p16INK4a selectively inhibited the kinase activity (Fig. 4B).
The results from both of these experiments demonstrate that our HT purification method is consistent with obtaining protein that is active in both enzymatic and inhibition assays.
HT Protein Purification.
We wished to estimate what fraction of the human proteome can be purified from bacteria. Therefore, the HT purification methods described above were applied to a large and randomly selected test set of 336 different proteins. As the corresponding cDNAs were not fully sequenced, multiple isolates of each gene (>1,000 isolates total) were expressed as His6 constructs, purified under denaturing conditions, and analyzed for bands of the predicted size. Proteins that were successfully purified were both free of truncation mutations and stable in bacteria. Of the attempted 336 different proteins, 204 full-length proteins were purified successfully (Fig. 5A), which corresponded to a success rate of 60%. Of the 204 proteins, 192 were expressed as GST-fusion proteins and purified under nondenaturing conditions. A band of the correct size was observed for 153/192 proteins, which corresponds to a success rate of ≈80% (Fig. 5B).
Figure 5.

HT protein purifications. Multiple isolates of 336 different proteins were expressed and purified under denaturing conditions. Of these, 204 cDNAs gave rise to a Coomassie blue-stained band of the expected size. The corresponding proteins were expressed as GST fusions and purified under nondenaturing conditions. (A) Seventy-five different proteins successfully purified under denaturing conditions by using the His6 tag. (B) The same 75 proteins fused to GST and purified under nondenaturing conditions. Fifteen percent of the eluate was loaded in all lanes.
FLEXProt Database.
We wondered whether we could identify properties that are common among proteins that either could or could not be purified from bacteria by using our conditions. A relational database, the FLEXProt database (unpublished work), was populated with experimental results, as well as with annotations from public databases. The success of protein expression was related to each of the following factors: isoelectric point (pI), number and frequency of rare codons in the CDS, number and frequency of cysteines and aromatic amino acids in the primary sequence of the protein, Pfam domains, and the subcellular localization in the mammalian cell.
The presence of certain domains and the localization of the proteins in the mammalian cell significantly influenced the purification success. As the chosen expression and purification conditions were biased against some protein classes, it was not surprising that only ≈20% of integral membrane proteins and 30% of secreted proteins were successfully purified (Fig. 6A). To examine whether the presence of specific protein domains could predict the success of expression and purification in one condition, protein purification success was related to protein domains stored in the Pfam database (19). The 336 investigated proteins contained 812 Pfam domains. Fifteen of 16 ras-like proteins, 8/10 kinases, 9/10 SH3-domain and SH2-domain, and 6/8 TRAF- and MATH-domain (Meprin-and-TRAF-homolgy-) containing proteins could be purified (Fig. 6B). In addition to these groups, 3/4 proteins containing a helix–loop–helix domain (HLH), and 3/4 proteins containing an RNA-binding motif, as well as all four proteins involved in ferrous transport, purified well. Unsurprisingly, no member of the tumor necrosis factor family, of the family of seven-transmembrane-domain receptors (7-TM) or of the membrane-integrated Ephrin family purified.
Figure 6.

Correlation of purification success to biological properties of the proteins. The success of protein purification under denaturing conditions was related to the molecular localization of the proteins in the mammalian cell (A) and to Pfam domains of the proteins (B).
Discussion
HT Purification of Human Proteins from Bacteria.
The emerging field of functional proteomics requires HT methods to express and purify proteins. The availability of such methods will alleviate a key bottleneck in the application of new proteomic techniques such as protein arrays to human biology. In this paper, we demonstrate the utility of E. coli as an expression system for functional proteomics enterprises. Of more than 200 of 336 different proteins, a product of the expected length could be purified under denaturing conditions, corresponding to a success rate of 60%. As none of the 336 constructs had been fully sequence-verified, it is possible that some cDNAs carry truncation mutations. Further analysis may therefore reveal a slightly higher success rate of proteome-wide expression and purification of human proteins from bacteria. In addition, almost 80% of the 192 proteins out of this subset that were attempted could be purified as full-length proteins by using the GST tag under nondenaturing conditions. Although no special selection methods were used to assemble the initial set of 336 proteins, these numbers represent a reasonable lower estimate for the success rate of HT expression of human proteins in bacteria. The protein yield from each successful purification (>300 ng) is sufficient for the construction of approximately several hundred-protein arrays, which require a few nanograms of proteins per spot (2).
As experience with HT protein purification increases, it is likely that future approaches will use a limited menu of expression and purification conditions that among them could succeed with nearly all proteins (18, 22). As shown here, the use of recombinational cloning facilitates the rapid transfer of the cDNAs to any needed expression vector. This strategy would be particularly effective if proteins could be preassigned to the specific purification method most likely to succeed. For the conditions used here, we found that the presence of certain protein domains correlated with protein expression and purification success. Although this result requires confirmation using more proteins (337 attempted proteins were dispersed over 812 Pfam domains), it suggests that Pfam domains may be a useful marker for the likelihood of successful expression of human proteins in bacteria. Christendat et al. (23) have used biophysical parameters of proteins from Methanobacterium thermoautotrophicum to build a decision tree that results in “final nodes” that are highly enriched in soluble or insoluble proteins. It is not yet known whether their findings apply to human proteins.
As some of the identified domains are quite small in comparison to the whole protein, it is not clear if these domains induce a protein to express well in bacteria or if they merely reflect proteins for which this is true. Examination of larger datasets, especially with large complex proteins that contain both “favorable” and “nonfavorable”' domains will be particularly interesting in this regard.
HT Protein Purification Using Four Affinity Tags.
Under nondenaturing conditions, 82% of test set proteins purified with both the GST- and the MBP-tag. In contrast to the larger tags, relatively few proteins (4 and 5 respectively) could be purified by using the small His6- and CBP-tags under nondenaturing conditions. This low success rate is most likely not an artifact of the HT approach because some proteins purified well in this method, including a positive control, and because the results were the same when the purifications were repeated in individual tubes (data not shown). The fact that proteins fused to the small tags bind inefficiently to the matrix or cannot be eluted specifically suggests either that the tags are inaccessible for binding or that the proteins are not properly folded. Improperly folded proteins in bacteria will often end up in inclusion bodies, which form as a series of intermediates in a process that is time and temperature dependent (24, 25). Our expression conditions may not allow enough time for the formation of inclusion bodies. Thus, even though the proteins in these experiments were in the soluble fraction, they were probably not properly folded, but at an early intermediate state of aggregation.
When expression levels of the 128 fusion proteins were analyzed, increasing molecular weight was accompanied by decreased expression levels. Paradoxically, however, large affinity tags, which increase the net size of the fusion proteins, significantly improved the success rate of purifying large proteins. The solubilizing and stabilizing effect of the large GST- and MBP-affinity tags on human proteins is well documented (25). For GST, the effect is thought to be a result of a soluble fusion partner that increases the solubility of the whole fusion protein simply by its presence (16). In contrast, it has been suggested that MBP acts as a chaperone through a hydrophobic cleft on its surface (26).
The expression of medium-sized human proteins (45–100 kDa) was less consistent than the expression of smaller proteins (<45 kDa). Some medium-sized proteins, such as HDAC1 (51 kDa) or STAT5A (87 kDa), express and purify very well, whereas others, such as ITPKA (52 kDa) or Rad54L (81 kDa), degrade under the same conditions. Our data show that the likelihood of successful protein expression is correlated to the presence of certain protein domains. It is therefore probable that large proteins fail more often because they are more likely than smaller proteins to contain a poisonous domain that is difficult to express. Additionally, the lack of posttranslational modifications in prokaryotes may destabilize some eukaryotic proteins and more apparently larger proteins. As the conditions of protein expression can have a profound effect on the stability of human proteins in bacteria, optimization of expression conditions and strategies will probably further increase the successful expression and purification of human proteins from bacteria.
Acknowledgments
We thank Gerald Marsischky and Leonardo Brizuela as well as the reviewers and editor for helpful suggestions on the manuscript. P.B. was supported by the Boehringer Ingelheim Fond of Germany. The work was partially funded by National Institutes of Health Grant 11-1195-0207.
Abbreviations
- HT
high-throughput
- GST
glutathione S-transferase
- CDS
coding sequence
- MBP
maltose-binding protein
- CBP
calmodulin-binding peptide.
References
- 1.Walter G, Bussow K, Cahill D, Lueking A, Lehrach H. Curr Opin Microbiol. 2000;3:298–302. doi: 10.1016/s1369-5274(00)00093-x. [DOI] [PubMed] [Google Scholar]
- 2.MacBeath G, Schreiber S L. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 3.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, et al. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 4.Fields S, Kohara Y, Lockhart D J. Proc Natl Acad Sci USA. 1999;96:8825–8826. doi: 10.1073/pnas.96.16.8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walhout A J, Temple G F, Brasch M A, Hartley J L, Lorson M A, van den Heuvel S, Vidal M. Methods Enzymol. 2000;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- 6.Brizuela L, Braun P, LaBaer J. Mol Biochem Parasitol. 2001;118:155–165. doi: 10.1016/s0166-6851(01)00366-8. [DOI] [PubMed] [Google Scholar]
- 7.Hartley J L, Temple G F, Brasch M A. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilsson J, Stahl S, Lundeberg J, Uhlen M, Nygren P A. Protein Expr Purif. 1997;11:1–16. doi: 10.1006/prep.1997.0767. [DOI] [PubMed] [Google Scholar]
- 9.Stevens R C, Yokoyama S, Wilson I A. Protein Sci. 2001;294:89–92. doi: 10.1126/science.1066011. [DOI] [PubMed] [Google Scholar]
- 10.LaBaer J, Garrett M D, Stevenson L F, Slingerland J M, Sandhu C, Chou H S, Fattaey A, Harlow E. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 11.Harlow E, Lane D. Using Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. [Google Scholar]
- 12.Zhao J, Dynlacht B, Imai T, Hori T, Harlow E. Genes Dev. 1998;12:456–461. doi: 10.1101/gad.12.4.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bateman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bornhorst J A, Falke J J. Methods Enzymol. 2000;326:245–254. doi: 10.1016/s0076-6879(00)26058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaillancourt P, Zheng C F, Hoang D Q, Breister L. Methods Enzymol. 2000;326:340–362. doi: 10.1016/s0076-6879(00)26064-3. [DOI] [PubMed] [Google Scholar]
- 16.Smith D B. Methods Enzymol. 2000;326:254–270. doi: 10.1016/s0076-6879(00)26059-x. [DOI] [PubMed] [Google Scholar]
- 17.Sachdev D, Chirgwin J M. Methods Enzymol. 2000;326:312–321. doi: 10.1016/s0076-6879(00)26062-x. [DOI] [PubMed] [Google Scholar]
- 18.Hockney R C. Trends Biotechnol. 1994;12:456–463. doi: 10.1016/0167-7799(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 19.Klein J, Dhurjati P. Appl Environ Microbiol. 1995;61:1220–1225. doi: 10.1128/aem.61.4.1220-1225.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lilie H, Schwarz E, Rudolph R. Curr Opin Biotechnol. 1998;9:497–501. doi: 10.1016/s0958-1669(98)80035-9. [DOI] [PubMed] [Google Scholar]
- 21.Pryor K D, Leiting B. Protein Expr Purif. 1997;10:309–319. doi: 10.1006/prep.1997.0759. [DOI] [PubMed] [Google Scholar]
- 22.Albala J S, Franke K, McConnell I R, Pak K L, Folta P A, Rubinfeld B, Davies A H, Lennon G G, Clark R. J Cell Biochem. 2000;80:187–191. doi: 10.1002/1097-4644(20010201)80:2<187::aid-jcb40>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 23.Christendat D, Yee A, Dharamsi A, Kluger Y, Savchenko A, Cort J R, Booth V, Mackereth C D, Saridakis V, Ekiel I, et al. Nat Struct Biol. 2000;7:903–909. doi: 10.1038/82823. [DOI] [PubMed] [Google Scholar]
- 24.Speed M A, Wang D I, King J. Protein Sci. 1995;4:900–908. doi: 10.1002/pro.5560040509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murby M, Uhlen M, Stahl S. Protein Expr Purif. 1996;7:129–136. doi: 10.1006/prep.1996.0018. [DOI] [PubMed] [Google Scholar]
- 26.Kapust R B, Waugh D S. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]