

Abstract
Crystal structures of the extracellular ligand-binding region of the metabotropic glutamate receptor, complexed with an antagonist, (S)-(α)-methyl-4-carboxyphenylglycine, and with both glutamate and Gd3+ ion, have been determined by x-ray crystallographic analyses. The structure of the complex with the antagonist is similar to that of the unliganded resting dimer. The antagonist wedges the protomer to maintain an inactive open form. The glutamate/Gd3+ complex is an exact 2-fold symmetric dimer, where each bi-lobed protomer adopts the closed conformation. The surface of the C-terminal domain contains an acidic patch, whose negative charges are alleviated by the metal cation to stabilize the active dimeric structure. The structural comparison between the active and resting dimers suggests that glutamate binding tends to induce domain closing and a small shift of a helix in the dimer interface. Furthermore, an interprotomer contact including the acidic patch inhibited dimer formation by the two open protomers in the active state. These findings provide a structural basis to describe the link between ligand binding and the dimer interface.
Metabotropic glutamate receptors (mGluRs) are a class of G-protein coupled receptors that possess seven transmembrane regions and couple with a variety of second messenger systems, including the activation of phosphoinositide hydrolysis and the regulation of adenylyl cyclase. To date, eight subtypes are known, which are categorized into three groups according to sequence similarity, location in the nervous system, and responses to agonists/antagonists (1–3). mGluRs exert a number of effects on neural excitability and synaptic transmission at most glutamatergic synapses in the central nervous system, performing crucial roles in changing synaptic efficacy or plasticity.
We have recently reported the crystal structures of the extracellular ligand-binding region of the homodimeric mGluR subtype 1 (m1-LBR) in the complex with glutamate and in the two unliganded forms (4). These structures suggested that the “Active” and “Resting” conformations of m1-LBR are modulated through the dimer interface. The bi-lobed protomer is composed of two domains, LB1 and LB2, and flexibly changes the domain rearrangement to form the “open” or “closed” conformation. Glutamate binding increases the population of the “Active” conformer, designated as “closed–open/A,” which adopts the “Active” type of the dimeric interface with each of the “closed” and “open” protomers. The ligand-free state exhibits two different structures, “closed–open/A” and “open–open/R” (the “Resting” dimer with two open protomers). These results suggested that the m1-LBR is in dynamic equilibrium, and that glutamate binding stabilizes both the “Active” dimer and the “closed” protomer.
To obtain clearer insights into the structural basis of receptor activation, we have determined the crystal structure of m1-LBR complexed with an antagonist, (S)-(α)-methyl-4-carboxyphenylglycine (S-MCPG) (5–7). Furthermore, we have solved the crystal structure in the presence of both the native agonist, l-glutamate, and gadolinium ions, because metal cations, including Gd3+, modulate the signaling of mGluRs (8–11). Here we report the new structural basis of agonism and antagonism, which accounts for the receptor activation mechanism.
Materials and Methods
Structure Determination of the Antagonist-Bound Form.
Overproduction and purification of the m1-LBR (residues 33–522) were done as described (12). Crystals of the complex with S-MCPG were obtained under the conditions used for the ligand-free form (0.2 M Tris⋅HCl, pH 8.5/1.9 M ammonium sulfate) (4) with 10 mM S-MCPG. The crystals belong to the space group P41212, with unit cell dimensions of a = b = 112.14 Å and c = 289.91 Å. Intensity data were collected with the R-AXIS V image plate detector (Rigaku, Tokyo) at the BL24XU beamline (λ = 0.8360 Å) of SPring8 (Hyogo, Japan). The data were processed with mosflm (13) and were reduced with scala in CCP4 (14) with an Rmerge of 0.086 within 20- to 3.3-Å resolution. Molecular replacement by using amore (15) was successful when the probe [Protein Data Bank ID code (PDB) 1EWK] was separated into two domains. Under the noncrystallographic symmetry (NCS) restraint between the two subunits, the structure was refined by the use of cns (16) to provide final Rcryst and Rfree values of 0.257 and 0.314, respectively (20- to 3.3-Å resolution). Other crystallographic statistics are shown in Table 1.
Table 1.
Data collection and refinement statistics
| S-MCPG complex | Glutamate/Gd3+ complex | |
|---|---|---|
| Data collection | ||
| Wavelength, Å | 0.8340 | 1.0000 |
| Resolution limit, Å | 3.3 | 4.0 |
| Rmerge | 0.086 (0.493*) | 0.10 (0.230†) |
| Completeness, % | 97.8 (99.3*) | 90.1 (75.6†) |
| I/σ(I) | 8.5 (1.6*) | 6.7 (2.9†) |
| Multiplicity | 4.3 (4.7*) | 5.0 (2.8†) |
| Refinement | ||
| Resolution, Å | 20–3.3 | 12–4.0 |
| Number of reflections | 26032 | 7182 |
| Rwork | 0.257 (0.461‡) | 0.237 (0.277§) |
| Rfree¶ | 0.314 (0.445‡) | 0.259 (0.332§) |
| Deviation from ideality | ||
| Bond length, Å | 0.009 | 0.003 |
| Bond angle, ° | 1.3 | 0.9 |
3.37–3.30 Å.
4.20–4.00 Å.
3.33–3.30 Å.
4.13–4.00 Å.
7% of the diffraction data.
Structure Determination of the Glutamate/Gd3+-Bound Form.
The m1-LBR in complex with the agonist and the gadolinium ion was crystallized under the same conditions used for the glutamate-bound form of m1-LBR (0.2 M Hepes, pH 7.5/20% polyethylene glycol 4000/1 mM l-glutamate) (4), except for the presence of 0.5 mM GdCl3. The crystal data [space group P3221; a = b = 145.3 (Å), and c = 76.75 (Å)] are different from the previous data, suggesting that the gadolinium ions affect the crystal packing. The absorption edges (1.563 and 1.711 Å), measured at beamline BL40B2 at SPring8, correspond to the LII and LIII edges of gadolinium [1.5612 and 1.7094 Å (17)], proving the presence of the element in the crystal. X-ray diffraction images were recorded on a Quantum 4R charge-coupled device detector (Area Detector Systems, Poway, CA) at the same beamline (λ = 1.000 Å). Because the crystal was too sensitive to find adequate conditions for cryoprotection, the diffraction data had to be collected at 298 K. Consequently, radiation damage was so serious that only partial data could be obtained with one crystal. The partial data of four crystals, indexed with mosflm (13), were combined and scaled [by using scala (14)] to obtain the complete data set, with an Rmerge of 0.101 within a 20- to 4.0-Å resolution range. The application of amore (15) provided a clear solution of the molecular replacement by using the closed conformation of m1-LBR (chain A of PDB 1EWK) as a search model. Refinement by use of cns (16) provided the converged structure with an Rcryst value of 0.237 and an Rfree value of 0.259 for the 12- to 4.0-Å resolution data. Other crystallographic statistics are shown in Table 1.
Calculation of Interprotomer Nonbonded Energy.
Models of the m1-LBR dimer with variable interdomain angles were constructed by using the coordinates of PDB 1EWK. The initial models, consisting of 14,180 atoms, including the ligand (glutamate) and the Mg2+ ion, were energy-minimized with presto Ver. 3 (18) to alleviate unreasonable steric collision. The AMBER PARAM96 force field (19) was applied for the potential energy calculation, except for the ligand, for which the partial charge was calculated with MOPAC2000 (Fujitsu, Kawasaki, Japan) by using the AM1 Hamiltonian. The electrostatic interactions (dielectric constant 4.0) were exactly evaluated with no cutoff procedure.
Results and Discussion
Mechanism of Antagonism by S-MCPG.
The crystal structure of m1-LBR in complex with S-MCPG contains two protomers in the asymmetric unit. The two NCS-related protomers, which form a dimeric structure, equivalently bind the antagonist (Fig. 1B) and, as a result, are well superimposed, with a rms deviation (rmsd) of 0.38 Å for 453 Cα atoms. The conformation of the protomer is the “open” form, in which the bound S-MCPG is accessible to the bulk solvent region. The two open protomers symmetrically dimerize with the “Resting” interface. Thus, this symmetric dimer is defined as “open–open/R,” which was observed in the ligand-free state (4). This antagonist-bound complex strongly supports the previous proposal that the open–open/R structure indeed corresponds to the “Resting” state of the ligand-binding region.
Figure 1.

(A) Dose–response curves of S-MCPG (filled diamonds) and R-MCPG (filled squares) in inhibiting [3H]quisqualate binding to m1-LBR. Each reaction contained 0.75 μg of purified m1-LBR. The binding assay was done as described (12, 20). (B) Stereo-pair diagram of m1-LBR complexed with S-MCPG, viewed from the perpendicular direction to the dimer interface. The LB1 and LB2 domains are colored blue and red, respectively, except for the B (cyan), C (green), and F (yellow) helices. The two protomers in the dimeric m1-LBR are distinguished by saturated and light colors. The magenta Corey–Pauling–Koltun model represents the bound S-MCPG. These protomers are related by an NCS 2-fold axis (black arrow). (C) Schematic diagrams of the recognition of S-MCPG (Left) and glutamate, observed in both the closed (Center) and open (Right) protomers of the closed-open/A dimer (4). Yellow and green boxes represent residues forming direct and water-mediated interactions, respectively, with the corresponding ligand. The bound water molecule is depicted by the circled “W.” The recognition of each ligand is established by polar interactions (broken lines) and van der Waals interactions (with W110). (D) Structures of the glutamate-bound form [closed–open/A (4); red for closed, blue for open] superimposed onto the S-MCPG complex (green) by their LB1 domains. The side chain of Y236 blocks the S-MCPG complex from closing. This figure was generated as if the front protomer (saturated color) in B were viewed from the left.
S-MCPG is bound into the interdomain cleft, where glutamate is found in the closed–open/A structure (4). On the side of the LB1 domain, the same residues involved in glutamate recognition (Y74, W110, S165, T188, and K409), except for S186, are used for S-MCPG recognition (Fig. 1C). The LB2 domain also participates in S-MCPG recognition with D208, Y236, and D318 (Fig. 1C), which similarly interact with glutamate (4), even in the different protomer conformations: open for the antagonist and closed for the agonist. Notably, these three residues cannot reach glutamate in the open conformation because of the large spacing between the two domains. Similarly, the open conformation separates R323 not only from glutamate but also from S-MCPG, whereas this side chain makes a salt bridge with the agonist in the closed protomer. This recognition scheme implies that the role of the antagonist is to wedge the open protomer conformation. In fact, the binding of S-MCPG restricts the open angle within a narrow range (24–25°), in contrast to the considerable polymorphism of the other open protomers, where open angles range from 21 to 32° (Fig. 2, Table 2). S-MCPG is longer by two carbon atoms than glutamate, and thus the antagonist collides with the LB2 domain, in particular at Y236, when the two domains approach each other (Fig. 1D). This structural feature of the open protomer wedged by this ligand may provide hints for the design of new antagonists.
Figure 2.
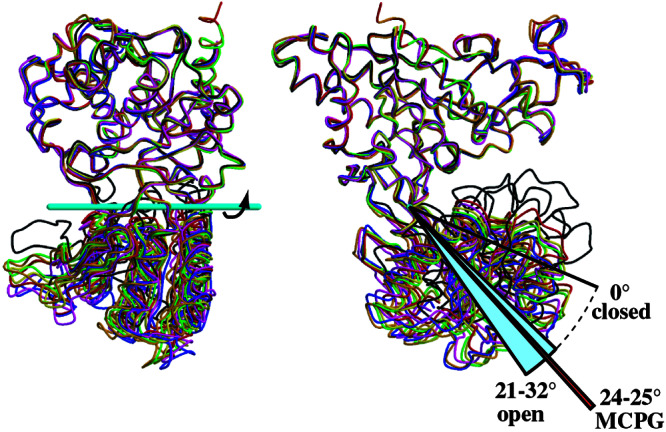
Two orthogonal views of the six open protomers superimposed on the closed protomer (complexed with glutamate; black) by using the LB1 domains [blue, glutamate-bound form (closed-open/A); magenta, ligand-free form (closed–open/A); red and orange, ligand-free form (open-open/R); yellow and green, S-MCPG complex (open–open/R)]. Although the LB1 domains are well superimposed (rmsd 0.44–0.67 Å), the deviations including the LB2 domain increase to 2.9–4.3 Å. However, the application of a simple rotation around the cyan bar significantly reduced the deviation to ≤1.0 Å (Table 2).
Table 2.
Superimposition of the m1-LBR protomers
| Protomer | rmsd, Å* | Open angle, °† | rmsd after rotation, Ň |
|---|---|---|---|
| Closed | |||
| Closed–open/A (+glutamate)§ | — | 0 | — |
| Closed–open/A (ligand-free)§ | 0.58 | 0.0 | 0.58 |
| Closed–closed/A (+glutamate, + Gd3+)¶ | 0.38 | 0.0 | 0.38 |
| Open | |||
| Closed–open/A (+glutamate)‖ | 4.3 | 32 | 0.67 |
| Closed–open/A (ligand-free)‖ | 3.7 | 26 | 1.0 |
| Open–open/R (ligand-free)** | 2.9 | 21 | 0.78 |
| 4.0 | 29 | 0.91 | |
| Open–open/R (+S-MCPG)** | 3.2 | 24 | 0.57 |
| 3.4 | 25 | 0.73 |
Cα atoms are superimposed on the closed protomer of the glutamate-bound closed–open/A structure.
Rotation angle of the LB2 domain around the axis in Fig. 2.
Calculated with the structure after application of the “open angle” rotation.
The closed protomer is used for the calculation.
The two closed protomers are related by crystallographic symmetry.
The open protomer is used for the calculation.
These crystal structures contain a dimer (two protomers) in their asymmetric units.
[3H]Quisqualate binding to m1-LBR is displaced by S-MCPG (20), whereas its stereoisomer, (R)-(α)-methyl-4-carboxyphenylglycine (R-MCPG), does not inhibit binding (Fig. 1A). R-MCPG can be placed on the binding site for S-MCPG without any steric collision. However, R-MCPG is unable to form the same polar interactions as S-MCPG (Fig. 1C) because of the different steric configuration of the α amino and α carboxyl groups around the chiral center, the Cα atom. Therefore, R-MCPG is unlikely to form a stable complex with the receptor.
4-Carboxyphenylglycine has been used as a lead compound for mGluR antagonist development (21, 22). Its derivative, S-MCPG, is a nonselective antagonist for group I and II receptors (5–7), whereas it has little or no activity with group III receptors (6, 22). All of the residues that directly recognize the antagonist are completely conserved among all of the groups, except for Y74. This tyrosine residue is variable among mGluRs (Tyr in group I, Arg in group II, and Lys/Asn/Gln in group III). Therefore, these direct contacts are not sufficient to discriminate the group III receptors from the others. Instead, the discrimination may be accomplished by the other residues, which would not make direct interaction with S-MCPG but interact with it through water molecules. Otherwise, the residues specific to the group III receptors may cause steric inhibition of the S-MCPG binding.
Role of Gd3+ Ion in Receptor Activation.
The structure of the m1-LBR protomer complexed with glutamate and the Gd3+ ion (Fig. 3A) is essentially the same as that of the closed conformation with glutamate (4). The rmsd value was calculated to be 0.38 Å by using the 449 Cα positions. The dimeric structure consists of the two protomers related by a crystallographic 2-fold axis. The dimer interface formed by the LB1 domains (LB1 interface; Fig. 3B) is also the same as that observed in the complex with glutamate. According to the conformational notation of the LB1 interface, which determines the R and A conformations (4), this dimer is defined as the A state. The conformation of the complex related by the exact 2-fold symmetry is different from the closed–open/A of the Gd3+-free complex, where the two LB1 domains are related by the pseudo-2-fold symmetry, but the LB2 domains are asymmetric. Thus, the present structure can be defined as “closed–closed/A,” which was not observed in the previous study.
Figure 3.

(A) Structure of m1-LBR complexed with glutamate and Gd3+ ion, viewed from perpendicular (Left) and parallel (Right) directions to the dimer interface, with the anomalous difference Fourier map (green cages; contoured at 5σ). The coloring is the same as in Fig. 1B. The yellow Corey–Pauling–Koltun model represents the bound glutamate. As the asymmetric unit contains one protomer, its symmetry mate, related by a crystallographic 2-fold axis (black arrows), is drawn by light colors to represent the dimer structure. (B) grasp (28) electrostatic surface representation (negative, red; neutral, white; positive, blue) of the two dimer interfaces, encircled by broken lines.
An anomalous difference Fourier map clearly revealed three gadolinium sites bound to the protomer (Fig. 3A). Site 1 is located on the crystallographic 2-fold axis. Sites 2 and 3 are too distant from any protein atoms to affect interprotomer or interdomain interactions, and therefore neither appears to modulate receptor activation. This notion is in agreement with the fact that the residues interacting with Gd3+ at sites 2 or 3 are not conserved among mGluRs. In the vicinity of site 1, a new interprotomer interaction is found between the LB2 domains. This contact site is designated as the LB2 interface (Fig. 3B). The LB2 interface is markedly negative because of the cluster of four acidic residues, D191, E233, E238, and D242 (Fig. 4). Among them, E238 and D242 lie close enough to interact with the Gd3+ ion, which alleviates the electrostatic repulsion between the two protomers (Fig. 4A). Similar interactions of Gd3+ with acidic residues were observed in the other crystal structures (23–25). On the other hand, the closed–open/A dimer (4) also involves the LB2–LB2 contact, in the absence of the Gd3+ ion (Fig. 4B). In addition to the four acidic residues forming the acidic patch, K260 participates in the interprotomer LB2–LB2 contact in the closed–open/A structure. The amino group of K260 in the closed protomer is located at the position similar to the site 1 Gd3+ ion to form interprotomer salt bridges with E238 and D242 in the open protomer. The same basic side chain of the open protomer is also salt-bridged to the acidic side chain of D242 of the closed one but not to E238, because of the asymmetry.
Figure 4.
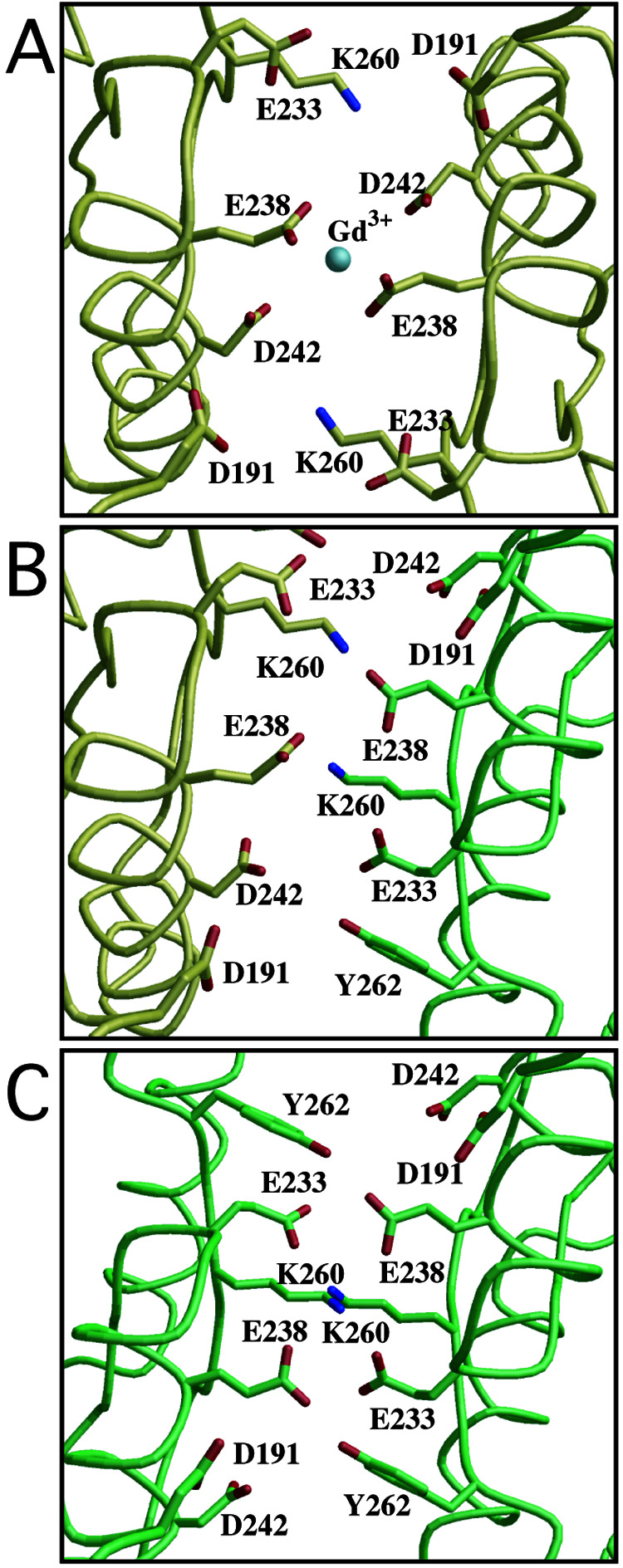
Local structures of the LB2 interface of the complexes with glutamate and a Gd3+ ion in the closed–closed/A conformer (A), and with the glutamate in the closed–open/A (B). The interface of the hypothetical open–open/A model, of which both open angles are 30°, is shown in C. Yellow and green represent the closed and open protomers, respectively. The 2-fold axis of the dimer is directed perpendicularly to the paper through the center of each figure. The silver sphere in A denotes the site 1 Gd3+ ion.
These findings suggest a functional role of the Gd3+ ion at the LB2 interface. K260 is close enough to interact with the acidic patch, not only in the closed–open/A structure (Fig. 4B) but also in the closed–closed/A structure (Fig. 4A). Even in the absence of the cation, these two “Active” structures are possible under the environment where water molecules are accessible to the edge of the acidic patch. In fact, the closed–open/A structure without Gd3+ is stable enough to be crystallized (4). Once the Gd3+ binds at the center of the acidic patches, their repulsion is alleviated more efficiently than by K260 alone. As a result, only the closed–closed/A is selectively stabilized. Notably, the core residues in the interface are highly conserved among the mGluR sequences. In particular, two acidic residues, E233 and E238, are completely conserved among all subtypes. K260 is also invariant, except for subtype 7, with Arg at the same position. D242 is replaced with Glu in the other subtypes. The interactions formed by these conserved residues should be common in the mGluRs.
Extracellular calcium ion at several millimolar concentrations was reported to potentiate phosphoinositide signaling by the receptor (9) and to directly activate it in the oocyte system (8). Recently, it was found that the persistent response by glutamate requires extracellular Ca2+ in mGluR1-transfected Chinese hamster ovary cells (10, 11). In this context, the selective stabilization of the closed–closed/A structure is induced not only by Gd3+ but also by Ca2+, because these two metals have similar ionic radii (26) and a similar tendency of binding to carboxyl side chains. In the absence of glutamate, the Gd3+ ion is insufficient to crystallize the P3221 form, suggesting that the metal plays only a subsidiary role in the stabilization of the “Active” dimer. In fact, the receptor is activated by glutamate without extracellular metal cation. Consequently, the cation is unnecessary for the closed–open/A structure (4) and may not be absolutely essential even for the closed–closed/A structure because of the presence of K260. Therefore, it appears reasonable that prolonged receptor activation, but not its ignition, explains the physiological role of the metal ion (11). However, another interpretation is also possible: that the metal itself exhibits an agonistic function (8) by transient formation of the closed–closed/A structure.
Conformational Change of the LB1 Domain Induced by Ligand Binding.
In the previous report, we proposed that receptor activation is modulated by the relocation of the LB1 interface (4). To explore how ligand binding affects the conversion between the A and R conformers, we compared the two crystal structures between the S-MCPG complex (open–open/R) and the glutamate one (closed–open/A). Neither the agonist nor the antagonist induces any global conformational change within the LB1 domain (rmsd 0.49 Å for 279 Cα atoms). Instead, a local conformational change was observed at the B helix (residues 112–123), in which the C terminus is extended by one turn (residues 124–127) in the antagonist-bound complex to expand the LB1 interface slightly (4). In addition, the C helix is apparently pushed away by ≈0.5 Å from the LB2 domain when glutamate is trapped in the closed protomer (Fig. 5), whereas the B helix exhibits a smaller deviation (≈0.2 Å). Even such small alteration in the relative positions of these two helices could modulate the LB1–LB1 contacts in some degree. It is intriguing that the N termini of almost all of the α-helices are directed onto the domain crevice, where glutamate is bound to alleviate the strong dipole–dipole repulsion, particularly between the C and F helices (residues 235–251) (Fig. 5). Although the movement of the C helix is nearly comparable to the average error of structure determination [0.5 Å; estimated from the Luzzati plot (27)] and the rmsd of the superimposition, the agonist may be able to induce the relocation of the LB1 interface through such a subtle perturbation, in coordination with the open–closed movement of the protomers.
Figure 5.
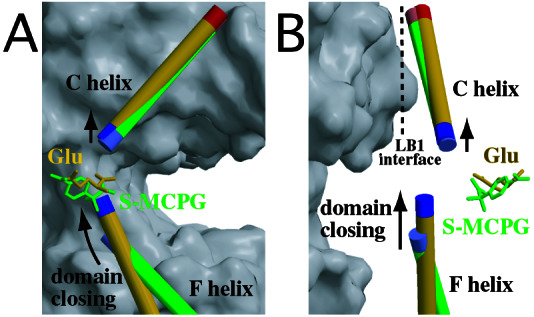
Structures around the C helix at the LB1 interface. The C and F helices of the S-MCPG-bound open protomer (green) and of the glutamate-bound closed protomer (yellow) are drawn with the corresponding ligands (stick models). The blue and red ends of the helices indicate their N and C termini, respectively. The gray surface represents the open protomer forming the dimeric structure of the glutamate-bound closed–open/A. The LB1 interface is approximately parallel to A, and its orthogonal view is on B. Note that the small upward shift of the C helix is presumably caused by dipole–dipole repulsion between the C and F helices, of which the N termini approach each other on domain closing.
Functional Role of the Dimer Interface in Dynamic Equilibrium.
In addition to the conformational link between ligand binding and the relocation of the LB1 interface, other factors may be required to fully account for the activation mechanism, where the agonist simply increases the relative population of the “Active” conformer through dynamic equilibrium (4). The crystal structure of the complex with glutamate and Gd3+ revealed another dimer interface between the LB2 domains. Interprotomer interactions at the LB2 interface significantly differ between the closed–closed (Fig. 4A) and closed–open (Fig. 4B) dimers, when the LB1–LB1 contact is the A state. Therefore, the LB2 interface is capable of sensing the protomer conformation, which seems to be modulated by ligand binding.
To elucidate the functional role of the LB2 interface, we built a hypothetical dimeric model in which the open angles of the two protomers are varied independently. To investigate the effects of the open angles on the dimer formation, the interprotomer nonbonded interaction energy was theoretically computed. Nine protomer models with various open angles (0–40° with 5° intervals) were constructed by using the structure of the closed protomer in the closed–open/A state (PDB 1EWK, corresponding to 0°). Larger open angles caused steric collisions between the two regions, ranging through residues 475–478 and 501–503, respectively. These protomers were combined to build 45 hypothetical A-conformer dimers by using the glutamate-bound closed–open/A structure (PDB 1EWK). The potential energies of the electrostatic (Fig. 6A) and van der Waals (Fig. 6B) interactions formed between the two protomers are shown in Fig. 6. The open–open/A state extensively exhibits higher electrostatic energy, by ≈30 kcal/mol, than the closed–closed/A and closed–open/A states, although solvation energy should be also taken into consideration. The hypothetical open–open/A model (Fig. 4C) suggests that the interprotomer LB2–LB2 contact, involving E233, E238, and K260, generates strong electrostatic repulsion. On the other hand, the interprotomer van der Waals energy is relatively constant. Actually, the invariable LB1–LB1 contact mainly contributes to the interprotomer van der Waals interaction. Therefore, the open–open/A state yields an electrostatically unfavorable dimer interface, which could exclude only the open–open/A state from the components in the dynamic equilibrium of m1-LBR.
Figure 6.
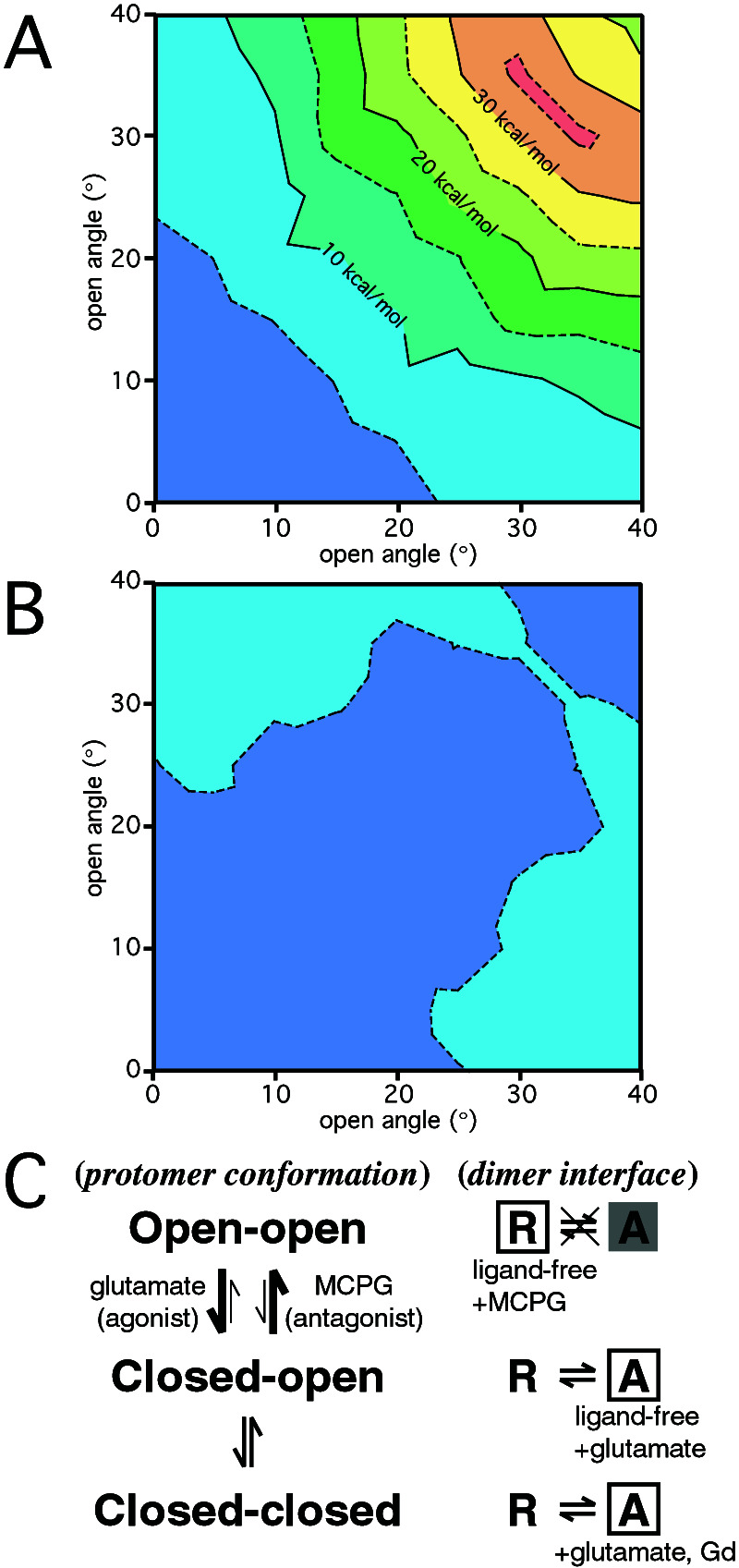
Computed potential energy for the interprotomer nonbonded interactions. Relative electrostatic (A) and van der Waals (B) energies were plotted as a function of the open angles for the two protomers (horizontal and vertical axes). The potential energy difference is contoured at every 5 kcal/mol. The conformations with open angles (5°, 5° and 0°, 0°) exhibit the lowest energies for the electrostatic and van der Waals interactions, respectively. (C) An equilibrium model proposed for m1-LBR. The open boxes indicate the states for which structures have been determined by x-ray crystallographic analyses. The shaded A conformer in the open–open combination forms an energetically unfavorable structure, because of the electrostatic barrier of the LB2 interface.
This exclusion of the open–open/A state suggests another activation mechanism, where the extracellular region of mGluR adopts various conformations in the dynamic equilibrium (Fig. 6C). On agonist binding to mGluR, the population of the open–open dimer decreases, because the agonist induces domain closure. Because of the exclusion of the open–open/A structure, only the R conformer is allowed in the open–open combination. Under these conditions, glutamate binding decreases the open–open/R population and thereby increases the relative population of A conformers in the equilibrium of the five components. On the other hand, S-MCPG, as an antagonist, fixes the open protomers so that the dimeric receptor is forced into the open–open/R state alone. Thus, the LB2 interface appears to play a regulatory role in eliminating the open–open/A state, whereas the LB1 interface contributes to dimerizing the two protomers and determining the receptor activation state. This receptor may be modulated by the equilibrium shift, which is directed by the ligand (agonist or antagonist) and by the structural changes on the LB1 interface, where the structures of the two helices are supposed to change on ligand binding.
Acknowledgments
We are grateful to Dr. S. Nakanishi (Kyoto University) for critical reading of the manuscript and discussions. We also thank Drs. Y. Tsuji and Y. Shimada (Biomolecular Engineering Research Institute) for the m1-LBR preparation and the MCPG-binding experiment, Drs. Y. Katsuya (SPring8 Service), K. Miura, and N. Yagi (Japan Synchrotron Radiation Research Institute) for assistance with synchrotron x-ray experiments, and Dr. D. Sunter (Tocris Cookson, Ltd.) for R-MCPG. We are grateful to Drs. Y. Shimura and H. Toh (Biomolecular Engineering Research Institute) for discussions.
Abbreviations
- mGluR
metabotropic glutamate receptor
- mGluR1
mGluR subtype 1
- m1-LBR
ligand-binding region of mGluR1
- PDB
Protein Data Bank ID code
- NCS
noncrystallographic symmetry
- rmsd
rms deviation
- S-MCPG
(S)-(α)-methyl-4-carboxyphenylglycine
- R-MCPG
(R)-(α)-methyl-4-carboxyphenylglycine
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1ISS (the S-MCPG-bound form) and 1ISR (the glutamate/Gd3+-bound form].
References
- 1.Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Nature (London) 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- 2.Nakanishi S, Masu M. Annu Rev Biophys Biomol Struct. 1994;23:319–348. doi: 10.1146/annurev.bb.23.060194.001535. [DOI] [PubMed] [Google Scholar]
- 3.Hollmann M, Heinemann S. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 4.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Nature (London) 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 5.Jane D E, Jones P L, Pook P C-K, Salt T E, Sunter D C, Watkins J C. Neurophamacology. 1993;32:725–727. doi: 10.1016/0028-3908(93)90088-k. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi Y, Sekiyama N, Nakanishi S, Jane D E, Sunter D C, Birse E F, Udvarhelyi P M, Watkins J C. J Neurosci. 1994;14:3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sekiyama N, Hayashi Y, Nakanishi S, Jane D E, Tse H-W, Birse E F, Watkins J C. Br J Pharmacol. 1996;117:1493–1503. doi: 10.1111/j.1476-5381.1996.tb15312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubo Y, Miyashita T, Murata Y. Science. 1998;279:1722–1725. doi: 10.1126/science.279.5357.1722. [DOI] [PubMed] [Google Scholar]
- 9.Saunders R, Nahorski S R, Challiss R A. Neuropharmacology. 1998;37:273–276. doi: 10.1016/s0028-3908(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 10.Miyashita T, Kubo Y. Recept Channels. 2000;7:25–40. [PubMed] [Google Scholar]
- 11.Nash M S, Saunders R, Young K W, Challiss R A, Nahorski S R. J Biol Chem. 2001;276:19286–19293. doi: 10.1074/jbc.M007600200. [DOI] [PubMed] [Google Scholar]
- 12.Tsuji Y, Shimada Y, Takeshita T, Kajimura N, Nomura S, Sekiyama N, Otomo J, Usukura J, Nakanishi S, Jingami H. J Biol Chem. 2000;275:28144–28151. doi: 10.1074/jbc.M003226200. [DOI] [PubMed] [Google Scholar]
- 13.Leslie A. In: Crystal Computing V. Moras D, Podjarny A D, Thierry J C, editors. Oxford: Oxford Univ. Press; 1991. pp. 27–38. [Google Scholar]
- 14.Collaborative Computational Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 15.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 16.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 17.Koch B, MacGillavry C H, Milledge H J, Koopmans K, Rieck G D, Bacon G E. In: International Tables for X-Ray Crystallography. MacGillavry C H, Rieck G D, editors. III. Dordrecht, The Netherlands: Reidel; 1985. pp. 157–200. [Google Scholar]
- 18.Morikami K, Nakai T, Kidera A, Saito M, Nakamura H. Comput Chem. 1992;16:243–248. [Google Scholar]
- 19.Kollman P A, Dixon R W, Cornell W D, Chipot C, Pohorille A. In: Computer Simulations of Biological Systems. van Gunsteren W F, editor. Leiden, The Netherlands: ESCOM; 1997. [Google Scholar]
- 20.Okamoto T, Sekiyama N, Otsu M, Shimada Y, Sato A, Nakanishi S, Jingami H. J Biol Chem. 1998;273:13089–13906. doi: 10.1074/jbc.273.21.13089. [DOI] [PubMed] [Google Scholar]
- 21.Watkins J, Collingridge G. Trends Pharmacol Sci. 1994;15:333–342. doi: 10.1016/0165-6147(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 22.Schoepp D D, Jane D E, Monn J A. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 23.Gabelli S B, Bianchet M A, Bessman M J, Amzel L M. Nat Struct Biol. 2001;8:467–472. doi: 10.1038/87647. [DOI] [PubMed] [Google Scholar]
- 24.Bone R, Springer J P, Atack J R. Proc Natl Acad Sci USA. 1992;89:10031–10035. doi: 10.1073/pnas.89.21.10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaudet R, Bohm A, Sigler P B. Cell. 1996;87:577–588. doi: 10.1016/s0092-8674(00)81376-8. [DOI] [PubMed] [Google Scholar]
- 26.Lide D R, editor. CRC Handbook of Chemistry and Physics. Boca Raton, FL: CRC; 1994. [Google Scholar]
- 27.Luzzati P V. Acta Crystallogr. 1952;5:802–810. [Google Scholar]
- 28.Nicholls A, Honig B J. J Comput Chem. 1991;12:435–445. [Google Scholar]