

Abstract
We present an approach that allows rapid determination of the topology of Escherichia coli inner-membrane proteins by a combination of topology prediction and limited fusion-protein analysis. We derive new topology models for 12 inner-membrane proteins: MarC, PstA, TatC, YaeL, YcbM, YddQ, YdgE, YedZ, YgjV, YiaB, YigG, and YnfA. We estimate that our approach should make it possible to arrive at highly reliable topology models for roughly 10% of the ≈800 inner-membrane proteins thought to exist in E. coli.
Keywords: bioinformatics‖fusion protein
An important first step in the characterization of an integral membrane protein of the helix bundle class (1) is to determine its membrane topology—i.e., the number of transmembrane α-helices and the overall in/out orientation of the protein relative to the membrane. In Escherichia coli, this step is usually accomplished by using reporter enzymes such as PhoA or LacZ fused to different portions of the membrane protein (2). In general, the number of fusions that need to be made and analyzed for a complete topology determination is equal to or larger than the number of transmembrane helices in the protein, thus requiring a significant experimental effort.
In the absence of experimental information, one can use various topology prediction methods to gain an idea of a protein's topology. The best current methods predict the correct topology with a success rate of 65–70% (3, 4) and thus provide a reasonable guide to minimizing the number of fusion proteins that have to be made for a given membrane protein (5). Recently, we have shown that the reliability of a given topology prediction can be estimated by comparing the predictions from a number of different prediction programs (6): when all methods agree, the topology is virtually certain to be correct, whereas the fraction of correct predictions drops with increasing levels of disagreement between the different methods.
Here, we suggest that the amount of experimental work needed to establish a topology should be inversely related to the reliability of the theoretical topology prediction, and we provide data that allows the topology for 12 E. coli inner-membrane proteins to be deduced from a combination of topology predictions and single C-terminal reporter-protein fusions. Given that there are only ≈60 experimentally determined topologies for E. coli inner-membrane proteins available in the literature (6, 7), our 12 additional topologies represent a substantial increase in topology information. From topology predictions for the whole complement of E. coli inner-membrane proteins (≈800 proteins), we estimate that the topology for an additional ≈75 proteins can be rapidly mapped by using our approach.
Materials and Methods
Enzymes and Chemicals.
Unless otherwise stated, all enzymes were obtained from Promega. T7 DNA polymerase and [35S]Met were obtained from Amersham Pharmacia. Taq polymerase, T4 ligase, and oligonucleotides were obtained from GIBCO/BRL. PhoA antiserum was obtained from 5 Prime→3 Prime (Boulder, CO). The alkaline phosphatase chromogenic substrate p-nitrophenyl phosphate (PNPP) (Sigma 104 phosphatase substrate) was obtained from Sigma.
Strains and Plasmids.
Experiments were performed in E. coli strains MC1061 [ΔlacX74, araD139, Δ(ara, leu)7697, galU, galK, hsr, hsm, strA] (8), CC118 [Δ(ara-leu)7697 ΔlacX74 ΔphoA20 galE galK thi rpsE rpoB argE(am) recA1] (9), and BL21(DE3)pLysS [F− ompT hsdSB (rB−mB−) gal dcm (DE3) pLysS]. PhoA fusion constructs were expressed from a modified version of the pHA-1 plasmid (10)—originally derived from the pING1 plasmid (11)—by induction with arabinose. Green fluorescent protein (GFP) fusion constructs were expressed from a modified pET28(a+) vector constructed by Waldo et al. (12) by induction with isopropyl-β-d-thiogalactoside (IPTG).
DNA Techniques.
All plasmid constructs were confirmed by DNA sequencing using T7 DNA polymerase. The genes encoding the E. coli MarC, PstA, TatC, YaeL, YcbM, YddQ, YdgE, YedZ, YgjV, YiaB, YigG, and YnfA proteins were amplified from E. coli JM109 by using Taq polymerase. The genes were cloned by using primer-introduced sites 5′ XhoI and 3′ KpnI into the previously constructed pHA-1 plasmid (13) which carries a phoA gene lacking both the 5′ segment coding for the signal sequence and the first five residues of the mature protein; it is immediately preceded by a KpnI site. A second XhoI site present downstream of the phoA gene in the original pHA-1 plasmid was changed to PstI by PCR mutagenesis to facilitate cloning. In all PhoA constructs, an 18 amino acid linker (VPDSYTQVASWTEPFPFC) was present between the membrane protein part and the PhoA moiety. By using a new set of PCR primers, the same membrane-protein-encoding genes, a truncated version of YaeL, YigG, YedZ (missing the last predicted transmembrane segment), leader peptidase (Lep), inverted leader peptidase (Lep-inv), ExbB, SecY, and SecF were amplified from either in-house plasmids or genomic DNA and, by using primer-introduced sites 5′ NdeI and 3′ BamHI or EcoRI, were cloned into the C-terminal GFP fusion-expression vector constructed by Waldo et al. (12).
Expression of PhoA Fusion Proteins.
E. coli strain CC118 transformed with the modified pHA-1 vector carrying the relevant PhoA-fusion constructs under control of the arabinose promoter was grown at 37°C in M9 minimal medium supplemented with 100 μg/ml ampicillin/0.5% fructose/100 μg/ml thiamin and all amino acids (50 μg/ml each) except Met. An overnight culture was diluted 1:25 in fresh medium, shaken for 3.5 h at 37°C, induced with arabinose (0.2%) for 5 min, and labeled with [35S]Met (75 μCi/ml; 1 Ci = 37 GBq). After 2 min, samples were acid-precipitated with trichloroacetic acid, 10% final concentration, resuspended in 10 mM Tris/2% SDS, immunoprecipitated with antisera to PhoA, washed, and analyzed by SDS/PAGE. Gels were scanned in a FUJIX Bas 1000 phosphorimager and analyzed by using the MACBAS v.2.31 software.
Expression of GFP-Fusion Proteins.
E. coli strain Bl21(DE3)pLysS transformed with a C-terminal GFP(S65T, F64L)-fusion vector (12) carrying the appropriate GFP-fusion constructs under control of the T7 promoter was grown overnight at 37°C in LB medium containing 50 μg/ml kanamycin and 30 μg/ml chloramphenicol. An overnight culture was diluted 1:50 in 15 ml of fresh medium with antibiotics and growth was continued at 37°C. When OD600 reached 0.3–0.4, cells were grown for another 4 h in the presence of 0.4 mM IPTG. Cells were subsequently harvested and resuspended in 1 ml of buffer containing 50 mM Tris⋅HCl at pH 8.0, 200 mM NaCl, and 15 mM EDTA. One-hundred microliters was removed, recentrifuged, and resuspended in 40 μl of SDS/PAGE solubilization (SB) buffer. The remaining suspension was sonicated, and the high-speed spin fraction was isolated and resuspended in 100 μl of sonication buffer, as described (14). For GFP fusions with high fluorescence and the low-fluorescent protein MarC/GFP, 15 μl and 25 μl of whole-cell suspension, respectively, was used for separation in standard SDS/12% polyacrylamide gel. For the remaining proteins with low fluorescence, 15 μl of high-speed spin fraction was incubated with equal volume of SB buffer and used for separation in a standard SDS/12% polyacrylamide gel. Proteins were subsequently transferred from the gel to a poly(vinylidene difluoride) membrane by means of Western-blotting. Blots were decorated with Lep antibody, or with a GFP-specific antibody (Novagen) and were developed by using the alkaline phosphatase system according to the instructions of the manufacturer (Sigma).
PhoA Activity Assay.
Alkaline phosphatase activity was measured by growing strain CC118 transformed with the modified pHA-1 plasmid carrying the appropriate PhoA-fusion constructs in liquid culture for 2 h in the absence of arabinose and then for 1 h in the presence of 0.2% arabinose (2). Mean activity values were obtained from two independent measurements and were normalized by the rate of synthesis (mean of three experiments) of the fusion protein determined by pulse-labeling of arabinose-induced CC118 cells as described above. Normalized activities were calculated as:
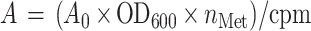 |
where A0 is the measured activity, OD600 is the cell density at the time of pulse-labeling, nMet is the number of Met residues in the fusion protein, and cpm is the intensity of the relevant band measured on the phosphorimager. To correct for between-gel variations, a YnfA/PhoA fusion-protein sample was run as a standard on all gels, and the cpm values were normalized to the YnfA/PhoA band.
Analysis of GFP Fusions.
GFP-fusion expressing cells (200 μl) previously suspended in buffer containing 50 mM Tris⋅HCl at pH 8.0, 200 mM NaCl, and 15 mM EDTA was transferred into a 96-well Nunc plate (Nunclone, Denmark), and triplicate GFP emissions were measured and averaged for each sample using a FLOUstar microtiter plate reader, excitation filter 485 nm, and emission filter 508 nm (BMG LabTechnologies, Offenburg, Germany).
Identification of E. coli Inner-Membrane Proteins.
The full set of E. coli ORFs was downloaded from the EcoGene database (15) at http://genolist.pasteur.fr/Colibri/and putative membrane proteins with a minimum of two predicted transmembrane helices were identified by TMHMM v.1.0 (16).
Prediction Methods.
Five topology prediction methods—TMHMM 1.0 (16), HMMTOP 1.0 (17), MEMSAT 1.8 (18), TOPPRED 2.0 (19, 20), and PHD 2.1 (21)—were used in their single-sequence mode (i.e., information from homologous proteins was not included) because local installations that would run in multiple-sequence alignment mode could not be obtained. All user-adjustable parameters were left at their default values. We also used the most current web-versions of TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), HMMTOP (http://www.enzim.hu/hmmtop/), MEMSAT (http://bioinf.cs.ucl.ac.uk/psipred/), and PHD (http://dodo.cpmc.columbia.edu/predictprotein/). For PHD and MEMSAT, the multiple-sequence alignment modes were used.
Results
Target Protein Selection.
The basic observation behind the topology mapping approach proposed here is that the reliability of a given predicted topology is very high when many different prediction methods agree. In particular, in a previous analysis of 60 E. coli inner-membrane proteins with experimentally determined topologies (6), we found 20 for which five different prediction methods all gave the same prediction (see Materials and Methods); the prediction was correct in all cases. Four methods of five agreed on a further 12 proteins; the prediction was correct for 10 of these.
In E. coli, the method of choice for topology mapping of inner-membrane proteins is fusion-protein analysis using suitable reporter proteins such as PhoA (2). For proteins where reliable topology predictions can be made, very few fusion-protein constructs should be needed to confirm the predicted topology. Hence, we selected all proteins of 764 predicted E. coli inner-membrane proteins (16) where all five prediction methods referred to above agreed on the periplasmic or cytoplasmic location of the N terminus, but where one or two of the methods disagreed with the majority for one and only one transmembrane helix. Assuming that the correct topology is one of the two predicted topologies (this is always the case when this analysis is applied to the 60 E. coli inner-membrane proteins in our previous study; data not shown), the correct topology then can be chosen once the location of the C terminus of the protein is known. By using the fusion-protein approach, experimental data on a single C-terminal fusion should thus suffice to derive the full topology. As a first test of this idea, we chose 12 proteins with between 3 and 7 predicted transmembrane helices from the initial set of 85 predictions, and, for each protein, fused two different topology reporter proteins to its C terminus (Table 1). After an early update of the prediction methods, all five methods gave the same predicted topology for YiaB, but we nevertheless retained this protein in the study.
Table 1.
Predicted and experimentally determined topologies for the 12 proteins analyzed in this study
| Protein | N-tail | TOPPRED | TMHMM | HMMTOP | MEMSAT | PHD | C-tail PhoA | C-tail GFP |
|---|---|---|---|---|---|---|---|---|
| MarC | P | 6P | 5C | 6P | 6P | 6P | P | P |
| PstA | C | 6C | 6C | 6C | 6C | 7P (6C) | C | ? |
| TatC | C | 6C | 6C | 5P | 6C | 6C | C | C |
| YaeL | P | 4P | 5C (4C) | 5C | 4P | 5C | ? | P* |
| YcbM | C | 6C | 7P | 6C | 7P | 7P (6C) | C | C |
| YddQ | C | 5P | 5P | 6C | 6C | 6C | C | C |
| YdgE | P | 4P | 4P | 4P | 3C | 4P | P | P |
| YedZ | C | 6C | 6C | 6C | 6C | 5P (6C) | C | C |
| YgjV | P | 5C | 5C | 5C | 5C | 6P (5C) | n.d. | C |
| YiaB | C | 4C | 4C | 4C | 4C | 4C | C | C |
| YigG | C | 4C | 4C | 4C | 4C | 3P | ? | P* |
| YnfA | P | 4P | 4P (4C) | 4P | 3C | 4P | P | P |
Column 1 gives the name of the protein in the Colibri database (15), column 2 the location of the N-tail as predicted by all five methods (C, cytoplasmic; P, periplasmic), columns 3–7 the number of transmembrane helices and the C-terminal location predicted by the different methods, and columns 8–9 the location of the C-terminus as determined experimentally by PhoA and GFP fusions. Predictions that agree with the experimentally determined C-terminal location are indicated in bold-italics. Differences between the original predictions and the latest web versions of the prediction programs are shown in parenthesis. The detailed topologies derived for each of the proteins are published as supporting information on the PNAS web site, www.pnas.org. ?, ambiguous result;
, confirmed by truncated fusion protein; n.d., not done.
Fusion-Protein Topology Reporters.
We chose E. coli alkaline phosphatase (PhoA) as a reporter of periplasmic location and GFP as a cytoplasmic reporter. PhoA has been widely used in topology studies as it folds into an enzymatically active conformation only in the periplasm (22, 23). In contrast, GFP folds efficiently in the cytoplasm but does not form active enzyme when targeted to the periplasm by a Sec-type signal peptide (24). GFP does fold properly, however, when attached to cytoplasmic domains of inner-membrane proteins (14). As shown below, GFP indeed can be used to discriminate between cytoplasmic and periplasmic domains in inner-membrane proteins and, thus, nicely complements the PhoA fusions.
Topology Mapping of 12 E. coli Inner-Membrane Proteins.
We obtained C-terminal PhoA fusions to 11 of the 12 proteins listed in Table 1, all of which could be expressed in the phoA− strain CC118 (9), could be immunoprecipitated by a polyclonal PhoA antiserum, and were of the expected sizes (data not shown). Alkaline phosphatase activities (corrected for different expression levels as measured by the incorporation of [35S]Met during a 2-min pulse and for the number of Met residues in each protein) were measured according to ref. 2 and are shown in Fig. 1 Upper. In the initial analysis, we chose conservative cutoff values; we considered all fusion proteins with an activity <1,000 (on the arbitrary scale used in the panel) as having a cytoplasmic C terminus and those with activities >3,000 as having a periplasmic C terminus (these cutoffs are somewhat ad hoc, and may be more tightly constrained when more data are available). This left two fusions—YaeL and YigG—for which the location of the C terminus could not be unambiguously deduced from the PhoA-fusion data, plus YgjV for which we did not obtain a PhoA fusion.
Figure 1.

PhoA acticities (Upper) and GFP fluorescence-emission intensities at 508 nm (Lower) for the 12 E. coli inner-membrane protein GFP/PhoA fusions studied here. Note that the PhoA activity for YdgE (13.100 units) and the GFP fluorescence intensity for YgjV (65.500 units) are off scale, and that the YgjV/PhoA fusion (indicated by *) was not made. Horizontal lines indicate the thresholds used to decide on the periplasmic or cytoplasmic location of the different fusion proteins.
To complement the PhoA results, we tested whether GFP could be used as a marker for cytoplasmic localization. We have found previously that GFP fused to cytoplasmic parts of E. coli inner-membrane proteins folds into an active conformation with high fluorescence at 508 nm (14). In contrast, GFP is incorrectly folded and does not fluoresce when targeted to the periplasm of E. coli by a Sec-type signal peptide (24), suggesting that it would likewise be inactive if fused to periplasmic segments in inner-membrane proteins.
To test this idea, we made C-terminal GFP fusions to the inner-membrane proteins Lep (which has a periplasmic C terminus; ref. 25), Lep-inv (with a cytoplasmic C terminus; ref. 26), ExbB (cytoplasmic C terminus; ref. 27), SecY (cytoplasmic C terminus; ref. 28), and SecF (cytoplasmic C terminus; ref. 29). Typical GFP fluorescence emission spectra from liquid cultures expressing Lep/GFP and Lep-inv/GFP are shown in Fig. 2A. When grown at 37°C, only cells expressing the Lep/GFP fusion (the only protein with a periplasmic C terminus) lack the typical emission at 508 nm (Fig. 2B), confirming that GFP fusions may be used for topology studies. Conservative first-approximation cutoff levels for deciding on the cytoplasmic/periplasmic location of the GFP reporter were chosen as indicated (i.e., 15.000 and 5.000 intensity units; presumably, with more data, these cutoffs can be refined).
Figure 2.

Test of GFP as a marker for C-terminal membrane protein topology. (A) GFP fluorescence emission spectra of cells expressing Lep-inv/GFP and Lep/GFP grown at 37°C. (B) GFP fluorescence-emission intensities at 508 nm for GFP fusions to test proteins with known topology. Only Lep/GFP has a periplasmically located C terminus. (C) Immunoblots using Lep (Upper) and GFP (Lower) antisera of whole-cell lysates prepared from cells overexpressing Lep-inv/GFP and Lep/GFP. Lane 1, Lep/GFP, uninduced; lane 2, Lep/GFP, induced at 25°C; lane 3, Lep/GFP, induced at 37°C; lane 4, Lep-inv/GFP, uninduced; lane 5, Lep-inv/GFP, induced at 25°C; lane 6, Lep-inv/GFP, induced at 37°C.
Immunoblotting of Lep/GFP and Lep-inv/GFP with antibodies to Lep and GFP also was performed after growth both at 25°C and 37°C (Fig. 2C). Lep-inv/GFP is stably expressed at both temperatures, whereas Lep/GFP is largely degraded to smaller fragments that react with Lep but not GFP antibodies during growth at 37°C. Thus, it seems that GFP is unstable at 37°C when expressed as a fusion to a periplasmic domain of an inner-membrane protein. A low but measurable level of fluorescence was seen when cells expressing Lep/GFP were grown at 25°C (data not shown), caused by either inefficient targeting/translocation (and hence some cytoplasmically localized GFP) or a limited degree of productive folding in the periplasm at this temperature. In any case, we conclude that growth at 37°C allows a better discrimination between cytoplasmic and periplasmic GFP fusions than growth at lower temperatures. We also note that the fluorescence measurements should be carried out on whole cells rather than isolated membranes, because for some proteins with a cytoplasmic C terminus, a significant fraction of the molecules undergo proteolysis, releasing much of the active GFP into the cytoplasm (data not shown).
C-terminal GFP fusions were made to the inner-membrane proteins in Table 1, and the fluorescence intensity at 508 nm was recorded for each construct after growth at 37° C (Fig. 1 Lower). Immunoblots with GFP antibodies also were made, Fig. 3. A clear pattern is seen in Fig. 1: proteins with high PhoA activities have low GFP fluorescence, whereas the opposite is true for proteins with low PhoA activities. The PhoA and GFP results are completely consistent for 8 of the 11 proteins for which both kinds of fusions were made. PstA has a reasonably high GFP fluorescence, and the low PhoA activity clearly favors a cytoplasmic localization. For the two proteins for which the PhoA results may be considered somewhat uncertain, YaeL and YigG, the low GFP fluorescence strongly suggests a periplasmic C terminus for both. To confirm this interpretation, C-terminal GFP fusions also were made to the last predicted cytoplasmic domains of YaeL (at Gly-416) and YigG (at Ala-104) and, as a control, to the last predicted periplasmic domain in YedZ (at Val-169). For YedZ, GFP fluorescence decreased as expected from 31.300 to 1.700 units for the full-length vs. truncated protein, whereas for YigG, the GFP fluorescence increased from 2.500 to 15.200 units, and for YaeL, fluorescence increased from 1.000 to 10.000 units for the full-length vs. truncated proteins (data not shown), thus reconfirming the use of GFP as a topology marker and the topology deduced for these proteins.
Figure 3.

Western blots of GFP fusions decorated with GFP antibody. Lane 1, YgjV/GFP (theoretical Mw = 44 kDa); lane 2, YcbM/GFP (51 kDa); lane 3, YiaB/GFP (36 kDa); lane 4, TatC/GFP (54 kDa); lane 5, YedZ/GFP (48 kDa); lane 6, YddQ/GFP (58 kDa); lane 7, PstA/GFP (58 kDa); lane 8, YdgE/GFP (37 kDa); lane 9, YnfA/GFP (37 kDa); lane 10, YigG/GFP (40 kDa); lane 11, MarC/GFP (50 kDa); lane 12, YaeL/GFP (75 kDa). GFP itself has a molecular mass of 25 kDa and is detected as a proteolytic fragment for YgjV, YddQ, PstA, and MarC. Lanes 1–7 and 11 show whole-cell fractions (the fusions in lanes 1–7 all have high GFP fluorescence), and lanes 8–10 and 12 show purified membrane fractions of fusions with low GFP fluorescence (see Materials and Methods). For the latter, blots of whole-cell lysates do not show detectable signals (data not shown).
We did not obtain a PhoA fusion for YgjV, but the very high GFP fluorescence clearly indicates a cytoplasmic C terminus.
The C-terminal orientations deduced from the PhoA and GFP fusions are compared with the predictions from the different programs in Table 1 (the detailed topologies derived for each of the proteins are published as supporting information on the PNAS web site, www.pnas.org). For this limited set of proteins, TOPPRED performs best with 10 correct predictions. However, given the small number of proteins and the way in which these particular proteins have been selected, this observation cannot be taken as a general measure of relative predictive performance. We also note that the majority prediction is correct for 9 of the 12 proteins. Consistent with our previous finding (6) that predictions where four of the five methods agree (“4/1 majority”) are more reliable than when only three methods agree (“3/2 majority”), seven of the eight predictions with a 4/1 majority are correct, whereas only one of the three predictions with a 3/2 majority is correct. Because updated versions of PHD, TMHMM, HMMTOP and MEMSAT are available as web servers, we reanalyzed all proteins with these new versions with their default settings. Differences compared with the original predictions are indicated in Table 1. PHD in particular performs better in this test.
Discussion
In this paper, we report membrane topologies for 12 E. coli inner-membrane proteins, to be added to the ≈60 previously known E. coli inner-membrane protein topologies (6, 7). Our topology mapping approach takes advantage of the good predictive abilities of current topology prediction methods (3), and our recent observation that the reliability of a given prediction is very high when multiple prediction methods agree on the result (6).
Here, we have focused on a subset of the E. coli inner-membrane proteins for which five different prediction methods—TOPPRED, TMHMM, HMMTOP, MEMSAT, and PHD—agree on the cytoplasmic or periplasmic location of the N terminus but disagree on one and only one of the predicted transmembrane helices. For such proteins, the topology can be inferred with very high reliability from an experimental determination of the cytoplasmic or periplasmic location of the C terminus, which can easily be accomplished by analysis of one or two suitable C-terminal topology-reporter fusions.
All in all, among the 764 E. coli inner-membrane proteins identified by TMHMM (16), we found 85 proteins that fulfilled our two inclusion criteria. Twelve of these were chosen for this study, and the locations of their C-termini were determined by analyzing C-terminal PhoA and GFP fusions (for one protein, we only obtained a GFP fusion). PhoA is a widely used topological marker for periplasmic loops and tails in inner-membrane proteins (2). To our knowledge, GFP has so far not been used as a topological marker but, by comparing the GFP and PhoA results, we found that high GFP fluorescence consistently reported on the cytoplasmic location of a fusion protein's C terminus. It should be noted that folded, active GFP can be translocated to the periplasm by the so-called Tat pathway (30, 31), but because few if any inner-membrane proteins use this pathway, this is not a significant drawback to our approach. Obviously, other periplasmic and cytoplasmic reporters such as β-lactamase and LacZ (2, 32) also may be used.
For 8 of the 11 proteins for which both PhoA and GFP fusions were analyzed, the PhoA and GFP results were entirely consistent. The topology of the three remaining proteins could be assigned based on clear results for either the GFP or the PhoA fusion (for completeness, GFP fusions to the last predicted cytoplasmic loop also were made for two of these proteins). For one protein, we obtained only the GFP fusion, but this also gave a clear result. None of the five prediction methods correctly predicted all 12 topologies; also, the majority prediction was correct in only 9 of the 12 cases. We further note that the topology determined here for YaeL (a protein that belongs to a newly discovered family of membrane-embedded metalloproteases; ref. 33) is the same as that previously determined for the related Bacillus subtilis protein SpoIVFB as regards the location of the conserved HEXXH and NPDG motifs relative to the inner membrane (34).
In summary, we propose an approach based on combining topology predictions with limited experimental information to rapidly determine the topology of E. coli inner-membrane proteins with very high reliability. Roughly 85 of the ≈800 inner-membrane proteins found in E. coli should be amenable to this approach; for many of the remaining ones, a pair of C-terminal fusions plus a small number of carefully chosen internal fusions should suffice. Our recent observation that partial topologies for which all or most prediction methods agree are also highly reliable (our unpublished data) suggest that full experimental topology determinations should not be necessary in most cases.
Supplementary Material
Acknowledgments
This work was supported by a center grant from the Foundation for Strategic Research to Stockholm Bioinformatics Center and by grants from the Swedish Research Council and the Swedish Cancer Foundation (to G.v.H.).
Abbreviations
- GFP
green fluorescent protein
- Lep
leader peptidase
- Lep-inv
inverted Lep
- PhoA
E. coli alkaline phosphatase
References
- 1.von Heijne G. Q Rev Biophys. 2000;32:285–307. doi: 10.1017/s0033583500003541. [DOI] [PubMed] [Google Scholar]
- 2.Manoil C. Methods Cell Biol. 1991;34:61–75. doi: 10.1016/s0091-679x(08)61676-3. [DOI] [PubMed] [Google Scholar]
- 3.Möller S, Croning M, Apweiler R. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- 4.Ikeda M, Arai M, Lao D, Shimizu T. In Silico Biol. 2001;2:1–15. [PubMed] [Google Scholar]
- 5.Boyd D, Traxler B, Beckwith J. J Bacteriol. 1993;175:553–556. doi: 10.1128/jb.175.2.553-556.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilsson J, Persson B, von Heijne G. FEBS Lett. 2000;486:267–269. doi: 10.1016/s0014-5793(00)02321-8. [DOI] [PubMed] [Google Scholar]
- 7.Möller S, Kriventseva E, Apweiler R. Bioinformatics. 2000;16:1159–1160. doi: 10.1093/bioinformatics/16.12.1159. [DOI] [PubMed] [Google Scholar]
- 8.Dalbey R E, Wickner W. J Biol Chem. 1986;261:13844–13849. [PubMed] [Google Scholar]
- 9.Lee E, Manoil C. J Biol Chem. 1994;269:28822–28828. [PubMed] [Google Scholar]
- 10.Sääf A, Johansson M, Wallin E, von Heijne G. Proc Natl Acad Sci USA. 1999;96:8540–8544. doi: 10.1073/pnas.96.15.8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston S, Lee J H, Ray D S. Gene. 1985;34:137–145. doi: 10.1016/0378-1119(85)90121-0. [DOI] [PubMed] [Google Scholar]
- 12.Waldo G S, Standish B M, Berendzen J, Terwilliger T C. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 13.Whitley P, Nilsson I, von Heijne G. Nat Struct Biol. 1994;1:858–862. doi: 10.1038/nsb1294-858. [DOI] [PubMed] [Google Scholar]
- 14.Drew D, Nordlund P, von Heijne G, de Gier J. FEBS Lett. 2001;507:220–224. doi: 10.1016/s0014-5793(01)02980-5. [DOI] [PubMed] [Google Scholar]
- 15.Rudd K. Nucleic Acids Res. 2000;28:60–64. doi: 10.1093/nar/28.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krogh A, Larsson B, von Heijne G, Sonnhammer E. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 17.Tusnady G E, Simon I. J Mol Biol. 1998;283:489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 18.Jones D T, Taylor W R, Thornton J M. Biochemistry. 1994;33:3038–3049. doi: 10.1021/bi00176a037. [DOI] [PubMed] [Google Scholar]
- 19.von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 20.Claros M G, von Heijne G. CABIOS. 1994;10:685–686. doi: 10.1093/bioinformatics/10.6.685. [DOI] [PubMed] [Google Scholar]
- 21.Rost B, Fariselli P, Casadio R. Protein Sci. 1996;5:1704–1718. doi: 10.1002/pro.5560050824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akiyama Y, Ito K. J Biol Chem. 1993;268:8146–8150. [PubMed] [Google Scholar]
- 23.Derman A I, Beckwith J. J Bacteriol. 1991;173:7719–7722. doi: 10.1128/jb.173.23.7719-7722.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feilmeier B J, Iseminger G, Schroeder D, Webber H, Phillips G J. J Bacteriol. 2000;182:4068–4076. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfe P B, Wickner W, Goodman J M. J Biol Chem. 1983;258:12073–12080. [PubMed] [Google Scholar]
- 26.von Heijne G. Nature (London) 1989;341:456–458. doi: 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- 27.Kampfenkel K, Braun V. J Biol Chem. 1993;268:6050–6057. [PubMed] [Google Scholar]
- 28.Akiyama Y, Ito K. EMBO J. 1987;6:3465–3470. doi: 10.1002/j.1460-2075.1987.tb02670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pogliano K J, Beckwith J. J Bacteriol. 1994;176:804–814. doi: 10.1128/jb.176.3.804-814.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas J D, Daniel R A, Errington J, Robinson C. Mol Microbiol. 2001;39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- 31.Santini C L, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu L F. J Biol Chem. 2001;276:8159–8164. doi: 10.1074/jbc.C000833200. [DOI] [PubMed] [Google Scholar]
- 32.Broome-Smith J K, Tadayyon M, Zhang Y. Mol Microbiol. 1990;4:1637–1644. doi: 10.1111/j.1365-2958.1990.tb00540.x. [DOI] [PubMed] [Google Scholar]
- 33.Rudner D Z, Fawcett P, Losick R. Proc Natl Acad Sci USA. 1999;96:14765–14770. doi: 10.1073/pnas.96.26.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green D H, Cutting S M. J Bacteriol. 2000;182:278–285. doi: 10.1128/jb.182.2.278-285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.