

Abstract
The flavoprotein nitroalkane oxidase (NAO) from Fusarium oxysporum catalyzes the oxidation of nitroalkanes to the respective aldehydes with production of nitrite and hydrogen peroxide. The sequences of several peptides from the fungal enzyme were used to design oligonucleotides for the isolation of a portion of the NAO gene from an F. oxysporum genomic DNA preparation. This sequence was used to clone the cDNA for NAO from an F. oxysporum cDNA library. The sequence of the cloned cDNA showed that NOA is a member of the acyl-CoA dehydrogenase (ACAD) superfamily. The members of this family share with NAO a mechanism that is initiated by proton removal from carbon, suggesting a common chemical reaction for this superfamily. NAO was expressed in Escherichia coli and the recombinant enzyme was characterized. Recombinant NAO has identical kinetic parameters to enzyme isolated from F. oxysporum but is isolated with oxidized FAD rather than the nitrobutyl-FAD found in the fungal enzyme. NAO purified from E. coli or from F. oxysporum has no detectable ACAD activity on short- or medium-chain acyl CoAs, and medium-chain acyl-CoA dehydrogenase and short-chain acyl-CoA dehydrogenase are unable to catalyze oxidation of nitroalkanes.
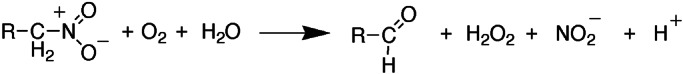
The flavoprotein nitroalkane oxidase (NAO; ref. 1) from the fungus Fusarium oxysporum (ATCC 695) catalyzes the oxidation of nitroalkanes to the corresponding aldehydes or ketones, hydrogen peroxide, and nitrite (Scheme S1). The likely physiological role of the enzyme, which is induced when the fungus is grown in the presence of a nitroalkane (2), is to protect the fungus from nitroalkanes produced by plants as defensive toxins (3). When isolated from fungal cells, the FAD cofactor in NAO is in the form of a 5-nitrobutyl-1,5-dihydro-FAD (4). This form of the enzyme is inactive, but it is converted to an active form upon the decay of the adduct to yield FAD. The ability of NAO to catalyze the oxidation of neutral nitroalkanes is unique among flavoproteins studied to date (5). Studies of the mechanism of formation of the flavin adduct and steady-state kinetic isotope effects with the active enzyme are consistent with substrate oxidation involving cleavage of the CH bond of the substrate to form a carbanion. Although carbanion intermediates have been proposed for other flavoprotein oxidases, in virtually all cases, the data can be accommodated equally well or better by a hydride transfer mechanism (6). The much lower pKa value of a nitroalkane α-proton vs. that in an amino or a hydroxy acid makes formation of a carbanion by NAO far more energetically favorable. Thus, this enzyme seems to present a unique opportunity for study of carbanion formation during catalysis by a flavoprotein. Moreover, inasmuch as nonenzymatic carbanion formation from nitroalkanes has been heavily studied as a model for ionization of carbon acids (7), study of the mechanism of the enzyme-catalyzed reaction can provide insights into the basis for the increased rates of the latter.
Scheme 1.
Until now, the only structural information regarding NAO has been its N-terminal sequence and the sequence of a putative active-site peptide (8, 9); this has been sufficient to rule out the existence of homologous proteins in the available databases, which do not include F. oxysporum. We describe here the cloning of the fungal enzyme, its expression in Escherichia coli, and the mechanistic and evolutionary insights provided by knowledge of its amino acid sequence.
Materials and Methods
Materials.
Nitroethane, nitropropane, and nitromethane were obtained from Aldrich. Phenylmethylsulfonyl fluoride and FAD were obtained from Sigma. DTT was obtained from Research Organics. Isopropyl β-d-thiogalactopyranoside (IPTG) was from Inalco Pharmaceuticals, San Luis Obispo, CA; l-1-tosylamido-2-phenylethyl chloromethyl ketone-treated trypsin was purchased from Worthington; butyryl and octanoyl CoA were obtained from Sigma. Oligonucleotides were custom synthesized on an Applied Biosystems model 380B DNA synthesizer by the Gene Technology Laboratory of the Biology Department of Texas A&M University. Restriction endonucleases NcoI, SphI, PstI, HindIII, BamHI, and T4 ligase were obtained from New England Biolabs; Pfu DNA polymerase was obtained from Stratagene; Taq polymerase was from Promega or Qiagen, Chatsworth, CA. Plasmids were purified by using mini- and midi-kits from Qiagen. Nytran membranes were purchased from Schleicher & Schuell. α-32P-dATP was obtained from Amersham Pharmacia; leupeptin, pepstatin, and lysozyme were obtained from Roche Molecular Biochemicals; DEAE-Sepharose Fast Flow was purchased from Amersham Pharmacia. E. coli strain BL21(DE3) from Novagen was used for protein expression; strains NovaBlue (Novagen) and XL1-Blue (Stratagene) were used during DNA cloning protocols. DNA sequencing was carried out with the Applied Biosystems BigDye kit and analyzed by the Gene Technology Laboratory of the Biology Department of Texas A&M University. NAO was purified from F. oxysporum (ATCC 695) as described (10). The activated FAD-containing form of the enzyme was prepared according to Gadda et al. (4). Human kidney medium-chain acyl-CoA dehydrogenase (MCAD) and Bacillus subtilis short-chain acyl-CoA dehydrogenase (SCAD) were generous gifts from Colin Thorpe (Univ. of Delaware, Newark) and Marian Stankovich (Univ. of Minnesota, Minneapolis), respectively.
Isolation of Partial NAO Gene.
F. oxysporum was grown in 500 ml of yeast extract-peptone-dextrose medium (YEPD) medium with 0.5 ml nitroethane at 28°C for 24 h. The DNeasy plant maxi kit (Qiagen) was used to isolate genomic DNA from 0.33 g cells. This genomic DNA was used as the starting template for two sequential PCRs using Taq polymerase. The primers for the first reaction were GTIGAYTTYAARYTIWSNCC and GGICCISWIGTNGTDATRTG. The second reaction was carried out with the product of the first reaction as template and the primers AGTCTAAAGCTTCARGCITTYGCIAAYACNGT and CTAAGTGGATCCGGYTCICCIARDATYTGRTA. The final DNA product from these reactions (about 700 bp) was digested with BamHI and HindIII and ligated into pUC18. Twelve plasmids (pUCNAO1 through pUCNAO12) that contained the sequences of the oligonucleotides from the second PCR were sequenced from the BamHI site to the HindIII site.
Isolation of NAO cDNA.
F. oxysporum was grown in 1 liter YEPD medium with 0.1% nitroethane at 28°C; aliquots were removed after 38, 47, 62, 71, 86, and 110 h. The cells (≈4 g for each time point) were harvested, and RNA was isolated from about 200 mg cells for each time point with the RNeasy plant kit from Qiagen; mRNA then was isolated with the Oligotex kit from Qiagen. To ascertain the optimal incubation time to obtain an mRNA preparation enriched in the mRNA for NAO, a 5 μg sample of mRNA for each time point was analyzed by probing a Northern blot. The probe used was the radioactively labeled 700 bp BamHI–HindIII fragment from pUCNAO9.
Three grams of cell pellet from the 110-h time point was used for a scaled-up RNA preparation followed by mRNA preparation using the same protocols. The resulting mRNA was sent to Stratagene to construct a custom cDNA library in the Uni-ZAP XR λ vector. A λ suspension (1.5 × 1010 plaque-forming units/ml) that resulted from one amplification of this original library was used as the template for all further cloning.
Two PCRs were performed on the cDNA library with Qiagen HotStarTaq. Oligonucleotide primers based on internal sequences obtained from the genomic DNA, GACAACGCATGCAAGATCAGCCCC' and CCCGTGATTCTTTGCGATTCACC, and flanking sequences from the Uni-ZAP vector were used. The products were purified with the QIAquick kit and sequenced in both directions.
The cDNA for NAO was subcloned directly from the λ library into the expression vector pET21d by generating a PCR product that contained the entire coding region flanked by EcoRI and BamHI sites. The PCR used the primers TAGTTACACTTTCCAGTACTTGGATCCAATGGTTGACTTCAAACTTTC and AGATTCTTATGTATGATGAATTCGACACCATCACAGGCGAGATTT. The PCR product was purified, digested with BamHI and EcoRI, purified again, sequenced in its entirety in both directions, and ligated into pET21d. The correct construct, pETNAO1, was sequenced in both directions from the BamHI to the EcoRI site.
The internal NcoI site of the NAO cDNA was removed from pETNAO1 by translation-silent mutagenesis with the Stratagene QuikChange method. Next, a new NcoI site at the start codon was introduced with the same method. The resulting plasmid was digested with NcoI and religated. This step aligned the start codon of the NAO cDNA optimally with the T7 RNA polymerase promoter. This plasmid, pETNAO4, was sequenced from the T7 promoter to the T7 terminator to confirm the cDNA sequence. Competent E. coli strain BL21(DE3) cells were transformed with plasmid pETNAO4.
Expression of NAO in E. coli.
A single colony of E. coli strain BL21 (DE3) transformed with pETNAO4 was used to inoculate 50 ml of LB containing 100 μg/ml carbenicillin at 37°C. The starter culture was grown for 10–12 h; 5 ml were used to inoculate 1 liter of LB containing 100 μg/ml ampicillin at 37°C. When the A600 value of the culture reached 0.7, IPTG was added to a final concentration of 0.25 mM and the temperature of the culture was lowered to 25°C. After 7 h, cells were harvested by centrifugation at 5,000 × g and 4°C for 25 min and stored at −80°C. Typically, 6 liters of culture yielded approximately 45 g of cells. The cells were suspended in four volumes of 50 mM sodium phosphate, 1 mM EDTA, 0.1 mM FAD, 0.1 mM DTT, 0.1 mg/ml phenylmethylsulfonyl fluoride, 25 μg/ml lysozyme, 5% glycerol at pH 7.0. The suspension was sonicated for 10 min on ice and centrifuged at 10,000 × g for 1 h. The cell-free extract was incubated with 2% streptomycin sulfate for 20 min and centrifuged at 10,000 × g for 20 min. Proteins precipitating between 40% and 65% ammonium sulfate saturation were collected. The ammonium sulfate pellet was resuspended in 50 ml of buffer A (20 mM Hepes/1 mM EDTA/5% glycerol, pH 8.0) containing 0.1 mM FAD and were dialyzed against buffer A. The sample was loaded onto a DEAE-Sepharose Fast Flow column (2.7 × 16 cm) that had been preequilibrated with 1 liter of buffer A. The column was washed with 300 ml of buffer A plus FAD and eluted with a linear gradient from 0–200 mM NaCl (750 ml total) in the same buffer. Fractions with greater than 10% of the peak activity were pooled and then were concentrated by precipitation at 70% ammonium sulfate saturation. The pellet was resuspended in 5 ml of buffer A and dialyzed against 1 liter of the same buffer for 2 h. The purified enzyme was stored at −80°C.
Assays.
The concentration of NAO was determined by the method of Bradford (11) with BSA as standard. The concentration of active NAO with bound FAD was determined spectrophotometrically at 446 nm using an ɛ446 value of 11,700 M−1 cm−1 (4). NAO activity was measured with 25 mM nitroethane as substrate in air-saturated 0.5 mM FAD/50 mM potassium phosphate, pH 7.0 or in 50 mM Hepes/0.5 mM FAD, pH 8.0 by monitoring the rate of oxygen consumption with a computer-interfaced Hansatech Clark oxygen electrode (Hansatech Instruments, Pentney King's Lynn, U.K.) at 30°C, as described (12). Concentrated solutions of substrate were prepared in ethanol, and assays were initiated by the addition of substrate to minimize the amount of nitroalkane anion present. Acyl-CoA dehydrogenase (ACAD) activity was measured in 50 mM Hepes at pH 8.0 by monitoring the reduction of ferrocenium hexafluorophosphate at 300 nm, according to DuPlessis et al. (13). Acyl-CoA oxidase activity was measured in 50 mM Hepes/0.5 mM FAD, pH 8.0 by monitoring the rate of oxygen consumption as described above.
Isolation of Tryptic Peptides of NAO.
NAO (50 μM) in 0.5 ml of water was digested with trypsin [3% (wt/wt) trypsin/NAO]. After 4 h at 37°C, a second aliquot of trypsin [1% (wt/wt) final concentration] was added, and the mixture was allowed to react for a further 15 h at 37°C. The digestion was stopped with trifluoroacetic acid (1% final concentration). Purification of peptides was carried out by HPLC by using a Waters instrument equipped with a model 996 photodiode array detector and a Vydac 218TP54 (Vydac, Hesperia, CA; 4.6 × 250 mm) reverse-phase column at a flow rate of 1 ml/min−1. Eluent A was 0.05% aqueous trifluoroacetic acid, and eluent B was 0.04% trifluoroacetic acid in acetonitrile. The chromatography was carried out with a linear gradient from 5% to 50% B over 90 min. Peptides were collected manually. Automated Edman degradation of purified peptides was carried out on a Hewlett-Packard G1000A protein sequencer at the Protein Chemistry Laboratory of Texas A&M University.
Results
Cloning of NAO.
The N-terminal amino acid sequence of NAO has previously been determined (8); this analysis was repeated to extend the sequence through the first 35 residues. The sequence of a tryptic peptide containing essential tyrosyl and cysteinyl residues has also been described (14). Eleven additional tryptic peptides were isolated and sequenced to aid in the design of PCR primers for isolation of the gene. A nested PCR was then carried out on the genomic DNA of F. oxysporum using primers based on the N-terminal sequence of the protein and on pairs of internal tryptic peptides. One pair of internal peptides allowed isolation of a clean DNA product, 700 bp in length. In one reading frame, this DNA would code for amino acid residues 19–35 of the nonrecombinant protein and a portion of the tryptic peptide used to generate the PCR primer for the second reaction, as well as three other tryptic peptides, confirming its authenticity as a portion of the NAO gene.
Expression of NAO is induced by growth of F. oxysporum in the presence of nitroethane (2). The partial gene was used as a probe in a Northern analysis to measure changes in the level of NAO mRNA in the fungal cells as a function of time after the addition of nitroethane. This procedure showed the mRNA reached a maximum between 86 and 110 h (results not shown). Accordingly, mRNA was isolated from cells grown in the presence of nitroethane for 110 h, and a cDNA library was prepared in λ Uni-ZAP (Stratagene).
Primers based on the sequences of the NAO gene fragment and on the plasmid immediately outside the cDNA insert then were used to isolate the intact cDNA by PCR. Several sources of high-temperature polymerases were tested in isolating the NAO cDNA from this library. The best results were obtained when using Qiagen HotStarTaq, presumably because of the increased priming stringency achieved when using a high-quality enzyme in a hot-start protocol. Two PCR products were obtained that enveloped the entire cDNA for NAO with approximately 250 bp of overlap. The first, 644-bp long, contained the 5′ end of the cDNA and the entire previously determined gene fragment. The second contained the 3′ end and was 1,156-bp long. These PCR products were sequenced in their entirety in both directions. The first PCR product coded for 7 of the 12 peptides sequenced directly. The second PCR product coded for the remaining five peptides, one of which was the previously identified active-site peptide (9, 14). Fig. 1 shows the arrangement of the peptide-coding regions within the entire cDNA. This arrangement would code for a protein of molecular mass 48,028 Da, in agreement with the molecular mass of the nonrecombinant enzyme of 47,955 Da determined by mass spectrometry (8). In addition, the calculated amino acid composition agrees well with the previously determined values (8). In comparing the sequences of the gene fragment and the cDNA, an intron was discovered. A 51-bp sequence from the gene fragment is not in the cDNA and does not code for any of the tryptic peptides.
Figure 1.

Nitroalkane oxidase cDNA sequence and the derived amino acid sequence. Peptide sequences determined from the protein are underlined. The active-site peptide is in bold.
The program PSI-BLAST v.3.2 (15) was used to search the nonredundant databases of protein sequences for related proteins. All of the proteins identified in this search with E scores better than 0.01 were members of the family of ACADs or of unknown function.‖ The pairwise identity of NAO to individual members of the ACAD family was 23–27%, whereas the similarities ranged from 41% to 46%, with equivalent matches to mammalian and bacterial enzymes. (Individual ACAD sequences show pairwise identities with one another as low as 34%.) There was no difference in the alignment to ACADs of different chain length specificity. A representative sequence alignment of NAO with members of the ACAD family is shown in Fig. 2. The similarities and identities are distributed evenly throughout the structures, ruling out an alignment simply because of an FAD-binding domain. Several gaps must be introduced into the ACAD sequences for successful alignment of NAO, as indicated by dashes in the ACAD sequences in Fig. 2. Analysis of the three-dimensional structure of pig kidney medium-chain ACAD showed that the additional sequences in NAO at positions 181, 191, 218, and 327 of pig MCAD correspond to surface loops. The remaining two insertions, at positions 142 and 377, also occurred in loops that are partially accessible to the surface.
Figure 2.

Alignment of F. oxysporum NAO with selected ACAD. The initial alignment was done with the program clustalw and the sequences of human MCAD, LCAD, and SCAD; rat MCAD and SCAD; mouse MCAD; pig MCAD; Drosophila ACAD; C. elegans MCAD; and B. subtilis ACAD. The numbers for human SCAD and pig MCAD are for the mature proteins, whereas those for NAO and B. subtilis ACAD begin with the N-terminal methionine. The active-site base in each is underlined.
Characterization of NAO Expressed in E. coli.
With knowledge of the entire sequence, the cDNA for NAO was subcloned directly from the λ library into the expression vector pET21d by generating a PCR product that contained the entire coding region. After confirmation that the sequence matched that obtained from the library, this plasmid was used to express NAO in E. coli. The recombinant enzyme was produced in large quantities (greater than 4% of the total cell protein). It was possible to simplify the previous purification protocol from two chromatography columns to one; NAO eluted from DEAE-Sepharose in homogenous form as determined by denaturing PAGE (data not shown). Approximately 40 mg of pure NAO can be obtained from 6 liters of culture (Table 1).
Table 1.
Purification of nitroalkane oxidase from E. coli
| Step | Total units, μmol min−1 | Total protein, mg | Specific activity, μmol min−1mg−1 | Yield, % |
|---|---|---|---|---|
| Cell-free extract | 679 | 842 | 0.8 | 100 |
| 40–65% (NH4)2SO4 | 622 | 504 | 1.2 | 92 |
| DEAE-Sepharose | 384 | 36 | 10.6 | 57 |
NAO isolated from F. oxysporum contains 5-nitrobutyl-1,5-dihydro-FAD, formed during induction of the enzyme in the presence of nitroethane (8). In contrast, the FAD in the recombinant enzyme is in the oxidized state, with a spectrum indistinguishable from that of the activated form of the nonrecombinant enzyme (Fig. 3). Incubation of 20 mM nitroethane in 200 mM Hepes at pH 8.0 for 24 h at 25°C generates an approximately equimolar mixture of the neutral and anionic species. Addition of this mixture to recombinant NAO for 1 h at 25°C resulted in complete conversion of the enzyme-bound FAD to the 5-nitrobutyl adduct with a spectrum identical to that of the enzyme isolated from F. oxysporum (Fig. 3). The steady-state kinetic parameters for nitroethane at pH 8.0 and 30°C are comparable for the native and recombinant enzymes, with Vmax and Km values of 14.1 ± 0.7 s−1 and 2.7 ± 0.3 mM for recombinant NAO compared with values of 10.9 s−1 and 2.9 mM for the activated nonrecombinant enzyme (8).
Figure 3.

UV-visible absorbance spectra of recombinant NAO as isolated (solid line) and after reaction with a mixture of neutral and anionic nitroethane (dashed line). Conditions: 50 mM Hepes at pH 8.0 and 25°C.
In light of the sequence similarity of NAO to the ACADs, the ability of the enzyme to carry out the reactions catalyzed by members of the family was determined. In the ACAD assay (13) with 9 μM recombinant NAO, there was no activity with up to 106 μM butyryl-CoA or 59 μM octanoyl-CoA as substrate. Similarly, no oxygen consumption was observed in acyl-CoA oxidase assays with up to 265 μM butyryl-CoA or 150 μM octanoyl-CoA as substrate at concentrations of either fungal or recombinant NAO up to 0.9 μM. Neither acyl-CoA was an inhibitor of NAO from either source with nitroethane as substrate. Conversely, nitroethane was not a substrate for either human MCAD (0.8 μM) or B. subtilis SCAD (0.6 μM) in either the oxidase or dehydrogenase assays. Neither human MCAD nor B. subtilis SCAD showed any activity in the NAO assay with nitroethane, nitropropane, or phenylnitromethane as substrate. Finally, there was no change in the flavin spectrum of human MCAD in the presence of 10 mM nitropropane, and there was no change in the spectrum of NAO upon addition of 626 μM butyryl-CoA.
Discussion
The sequence of NAO described here identifies the enzyme as a member of the ACAD superfamily. Although the identity between NAO and the members of the ACAD family is low, it is well above that expected for random chance. Moreover, the identities are distributed throughout the sequence, so that it is not simply due to an FAD domain. The alignment shown in Fig. 2 shows that NAO contains several short sequences not found in members of the ACAD family. These additional sequences are found in regions corresponding to surface loops in the latter, as is often the case of enzymes with similar folds but slightly different sizes.
The assignment of NAO to the ACAD superfamily is fully consistent with the mechanisms previously proposed for acyl-CoA dehydrogenation and for nitroalkane oxidation. In the catalytic mechanism of the ACAD's glutamate 376 abstracts a proton from the α-carbon of the substrate, as a hydride is transferred from the β-carbon to the flavin (16). We have previously proposed that the NAO reaction is initiated by removal of the α-proton from the nitroalkane substrate by an active-site base to form a carbanion. Examination of the alignment shown in Fig. 2 shows that glutamate 376 of pig MCAD corresponds to aspartate 402 of NAO, suggesting that the latter residue is the active-site base in NAO. An essential tyrosine and an essential cysteine have been identified previously in NAO by modification with tetranitromethane and N-ethylmaleimide, respectively (9, 14). These can now be identified as tyrosine 398 and cysteine 397. Although neither residue is conserved in the members of the ACAD superfamily, the effects of their modification upon the activity of NAO further support the identification of aspartate 402 as the base and the conclusion that NAO shares the three-dimensional structure of the ACADs. Finally, mutagenesis of NAO aspartate 402 to asparagine results in a decrease in activity of three orders of magnitude, consistent with this residue being intimately involve in catalysis (M.P.V. and P.E.F., unpublished observations).
Given that catalysis by both NAO and the ACADs requires proton abstraction from carbon, the question arises as to whether the mechanistic similarities extend further. By analogy to the ACAD mechanism, one can propose a mechanism for NAO in which a hydride is transferred to the flavin from the β carbon of the nitroalkane anion. Displacement of nitrite from this nitroalkene by hydroxide would form a tautomer of the aldehyde product. However, nitromethane is a substrate for NAO, albeit a slow one (12). This observation makes such a mechanism unlikely. Thus, it seems that the overall fold used by NAO and the various ACADs is designed for removal of a proton from a substrate carbon, but the subsequent reactions depend upon the specific enzyme. This possibility resembles the situation in the enolase superfamily described by Gerlt and coworkers (17) in which the common reaction is also proton abstraction from carbon. In that case, a metal ion is used to aid in the enolization of the substrate, thereby lowering the pKa value of the α proton from ≈29 so that it can be removed by an enzymatic base.
The mechanisms by which enzyme specificities evolve are of substantial interest. Several general mechanisms have been proposed (17). In one, the substrate specificity of an enzyme is retained while the catalytic mechanism is modified. In a second, the precursor enzyme already catalyzes the critical initial chemistry, and the substrate specificity is modified. In a third model, the same active-site architecture is adapted to different chemistries and binding specificities. The catalysis by both NAO and ACAD of reactions in which an initial step involves proton abstraction from carbon is consistent with this second model for evolution of enzymes, suggesting that other members of this superfamily also may catalyze carbon-hydrogen bond cleavage as an initial step.
There are clearly significant differences between the structure of NAO and those of the ACADs, in that each is unable to catalyze the reaction catalyzed by the other. Elucidation of the structural bases for the differences in substrate specificity should provide insight into the evolution of the members of this superfamily. In addition, comparison of the structures of NAO and the ACAD's will provide insight into the fundamental requirements for catalysis of proton abstraction from carbon by the members of this superfamily.
Acknowledgments
We thank Drs. Colin Thorpe and Marian Stankovich for their generous gifts of enzymes, Dr. Larry Dangott for assistance with peptide sequencing, and Pablo Sobrado for carrying out NAO assays with the ACAD.
Abbreviations
- NAO
nitroalkane oxidase
- MCAD
medium-chain acyl-CoA dehydrogenase
- SCAD
short-chain acyl-CoA dehydrogenase
- ACAD
acyl-CoA dehydrogenase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF425595).
The soxC protein has previously been identified as a member of the ACAD superfamily (1). This enzyme catalyzes the oxidation of dibenzothiophene to the corresponding sulfone; its mechanism is unknown. This enzyme was not among those identified in either our blast or psi-blast searches using the nitroalkane oxidase sequence, and its level of identity with NAO is much lower than that of NAO to any individual ACAD. The soxC protein has a phenylalanine in place of the active-site glutamate found in members of the ACAD family, suggesting that its mechanism does not involve proton abstraction.
References
- 1.Denome S A, Oldfield C, Nash L J, Young K D. J Bacteriol. 1994;176:6707–6716. doi: 10.1128/jb.176.21.6707-6716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kido T, Hashizume K, Soda K. J Bacteriol. 1978;133:53–58. doi: 10.1128/jb.133.1.53-58.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanawa F, Tahara S, Towers G H N. Phytochemistry. 2000;53:55–58. doi: 10.1016/s0031-9422(99)00443-4. [DOI] [PubMed] [Google Scholar]
- 4.Gadda G, Edmondson R D, Russel D H, Fitzpatrick P F. J Biol Chem. 1997;272:5563–5570. doi: 10.1074/jbc.272.9.5563. [DOI] [PubMed] [Google Scholar]
- 5.Heasley C J, Fitzpatrick P F. Biochem Biophys Res Commun. 1996;225:6–10. doi: 10.1006/bbrc.1996.1122. [DOI] [PubMed] [Google Scholar]
- 6.Fitzpatrick P F. Acc Chem Res. 2001;34:299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- 7.Bernasconi C F. Adv Phys Org Chem. 1992;27:119–238. [Google Scholar]
- 8.Gadda G, Fitzpatrick P F. Biochemistry. 1998;37:6154–6164. doi: 10.1021/bi973085y. [DOI] [PubMed] [Google Scholar]
- 9.Gadda G, Banerjee A, Fitzpatrick P F. Biochemistry. 2000;39:1162–1168. doi: 10.1021/bi9921743. [DOI] [PubMed] [Google Scholar]
- 10.Gadda G, Fitzpatrick P F. Arch Biochem Biophys. 1999;363:309–313. doi: 10.1006/abbi.1998.1081. [DOI] [PubMed] [Google Scholar]
- 11.Bradford M M. Anal Biochem. 1967;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 12.Gadda G, Choe D Y, Fitzpatrick P F. Arch Biochem Biophys. 2000;382:138–144. doi: 10.1006/abbi.2000.2009. [DOI] [PubMed] [Google Scholar]
- 13.DuPlessis E R, Pellett J, Stankovich M T, Thorpe C. Biochemistry. 1998;37:10469–10477. doi: 10.1021/bi980767s. [DOI] [PubMed] [Google Scholar]
- 14.Gadda G, Banerjee A, Dangott L J, Fitzpatrick P F. J Biol Chem. 2000;275:31891–31895. doi: 10.1074/jbc.M003679200. [DOI] [PubMed] [Google Scholar]
- 15.Altschul S F, Madden T L, Schäffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nuclei Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J-J P, Wang M, Paschke R. Proc Natl Acad Sci USA. 1993;90:7523–7527. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerlt J A, Babbitt P C. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]