

Abstract
Hsp104 from Saccharomyces cerevisiae is a hexameric protein with two AAA ATPase domains (N- and C-terminal nucleotide-binding domains NBD1 and NBD2, respectively) per monomer. Our previous analysis of the Hsp104 ATP hydrolysis cycle revealed that NBD1 and NBD2 have very different catalytic properties, but each shows positive cooperativity in hydrolysis. There is also communication between the two domains, in that ATP hydrolysis at NBD1 depends on the nucleotide that is bound to NBD2. Here, we extend our understanding of the Hsp104 ATP hydrolysis cycle through mutagenesis of the AAA sensor-2 motif in NBD2. To do so, we took advantage of the lack of tryptophan residues in Hsp104 to place a single tryptophan in the C-terminal domain (Y819W). The Y819W substitution has no significant effects on folding stability of the C-terminal domain or on ATP hydrolysis by NBD1 or NBD2. The fluorescence of this tryptophan changes in response to ATP and ADP binding, allowing the Kd and Hill coefficient to be determined for each nucleotide. By using this site-specific probe of binding, we analyze the effect of mutating the conserved arginine residue in the sensor-2 motif in Hsp104 NBD2. An R826M mutation causes nearly equal decreases in affinity of NBD2 for both ATP and ADP, indicating that at this site, the sensor-2 provides binding energy, but does not act to sense the difference between these nucleotides. In addition, the rate of ATP hydrolysis at NBD1 is decreased by the R826M mutation, providing further evidence for interdomain communication in the Hsp104 ATP hydrolysis cycle.
Proteins in the AAA superfamily function to remodel other protein structures and complexes in an extraordinary variety of biological circumstances. Hsp104 and its Escherichia coli homolog ClpB are members of the Clp/Hsp100 subfamily of AAA proteins that act with Hsp70 and Hsp40 cochaperones to return aggregated proteins to their folded, active state (1–3). Hsp104 is required for thermotolerance, resolubilizing protein aggregates generated by severe stress (4, 5), and for the propagation of several yeast prions (6–8), mediating conversions between the soluble and aggregated forms of the prion protein (8–10). Hsp104 has two AAA modules per protomer (N- and C-terminal nucleotide-binding domains NBD1 and NBD2, respectively) and assembles into a hexamer (11). Cooperative ATP hydrolysis at both NBD1 and NBD2 is required for Hsp104 function in thermotolerance and [PSI+] maintenance (12).
Crystal structures of a number of AAA proteins reveal a common two-domain nucleotide-binding fold for this superfamily (see ref. 13 for review), consisting of a domain with a Rossmann fold (central β-sheet flanked by α-helices) and a separate C-terminal domain. ATP or ADP binds at the interface between the two domains, and conserved residues from each domain form the active site for hydrolysis. These proteins have the Walker A and Walker B motifs shared by other P-loop type nucleotide triphosphatases (NTPases) (14). In addition, two other AAA-specific sequence motifs in the Rossmann domain seem to have a role in ATP hydrolysis analogous to those of conserved amino acids in other P-loop NTPases (see refs. 15 and 16 for review). In the N-ethylmaleimide-sensitive fusion protein (NSF) D2 domain, residues in the sensor-1 motif form a hydrogen-bonding network that positions a water molecule 4 Å from the γ-phosphate of ATP (17, 18), and this water seems to act as the nucleophile for ATP hydrolysis in other AAA proteins (12). Also, a highly conserved arginine (Arg-765 in Hsp104 NBD2) in the Box VII motif projects from one subunit into the active site of the neighboring subunit and may act as an “arginine finger” (19, 20) to stabilize the developing negative charge in the transition state.
In contrast to the domain with the Rossmann fold, there is little sequence conservation in the C-terminal domain, with the exception of the AAA sensor-2 motif. This sequence variability may be one factor in the remarkable diversity of function of AAA proteins. For example, in ClpA, a Clp/Hsp100 protein with two ATPase domains that acts in proteolysis, the C-terminal domain of NBD2 forms the interface with the ClpP protease (21, 22). Hsp104 and ClpB do not interact with a protease, and the C-terminal domain must have another function, such as binding to substrates, cofactors, or a regulatory protein. In the few AAA proteins with known structures, residues in the sensor-2 motif project from the C-terminal domain into the active site, and contacts between sensor-2 and nucleotide probably regulate the activity of this domain by means of conformational changes associated with nucleotide binding or hydrolysis. However, the nature of this regulation is an unanswered question.
Current models for the function of AAA proteins suggest that they use ATP hydrolysis to drive the unfolding of a substrate protein or to otherwise disrupt protein–protein interactions (13). However, the mechanisms linking ATP binding and hydrolysis to “protein remodeling” remain largely undefined. This is a particularly difficult problem for proteins with more than one AAA active site, as experiments to describe the ATP hydrolysis cycle must be able to separate the contribution of each domain to the activity (catalysis or binding) being measured. We have described a method to measure the steady-state kinetics of ATP hydrolysis in the Hsp104 hexamer that allowed the catalytic activity of both NBD1 and NBD2 to be resolved (12). Both sites exhibit positive cooperativity, but otherwise have very different properties: NBD1 has a high catalytic rate but a low apparent affinity (kcat = 76 min−1, Km = 170 μM, Hill coefficient = 2.3); and NBD2 has a 300-fold slower rate of hydrolysis but binds with higher apparent affinity (kcat = 0.3 min−1, Km = 5 μM, Hill coefficient = 1.6). Moreover, there is communication between the two sites: a mutation in the sensor-1 motif of NBD2 (N728A) that decreases its rate of ATP hydrolysis also causes significant decreases in the kcat and Km for hydrolysis at NBD1 (12).
A necessary complement to measuring enzymatic activity is to measure nucleotide binding in a site-specific manner. Hsp104 has no tryptophan residues, making it possible to introduce single tryptophan substitutions to act as intrinsic fluorescent probes of binding to either NBD1 or NBD2. Here, we develop a single tryptophan substitution (Y819W) in the C-terminal domain of NBD2 of Hsp104 for use as a probe for nucleotide binding to this site. With this probe, we examine the role of the AAA sensor-2 motif in NBD2. A mutation in sensor-2 (R826M) directly affects nucleotide binding to NBD2, but the site is still functional in ATP hydrolysis. This is a fundamentally different effect than the loss of catalytic activity that results from the N728A mutation (12), yet both mutants affect catalysis at NBD1. Generally, our results provide insight into the catalytic mechanism of the AAA proteins. Moreover, they shed new light onto the means by which NBD1 and NBD2 communicate in the Hsp104 hexamer.
Materials and Methods
Limited Proteolysis.
Limited V8 proteolysis of Hsp104 (a 1 μM solution) purified from Saccharomyces cerevisiae (23) was performed at 37°C in 20 mM Hepes, pH 7.5/10 mM MgCl2/150 mM NaCl. A 1:75 (wt/wt) Hsp104 to V8 protease was used. Reactions were stopped with 5 mM 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) and SDS/PAGE sample buffer or by precipitation with 20% trichloroacetic acid (TCA). Fragments in sample buffer were resolved by SDS/PAGE using a 10–17% acrylamide gradient gel and visualized by Coomassie blue staining, or after transfer to a poly(vinylidene difluoride) (PVDF) membrane, by immunoblotting with Abs 1–2 or 8–1. TCA precipitates were resuspended in 0.1% trifluoroacetic acid and 6 M guanidine HCl then loaded onto a Delta-Pak C4 column (Waters). Fragments eluting in a 10–60% acetonitrile gradient were analyzed by electrospray-ionization mass spectrometry.
Plasmid and Strain Construction.
For bacterial expression of Hsp104, we created the pNOTAG vector by modifying pJC25 (24). The SmaI site in the polylinker was replaced with SacI and the lacI gene was inserted into the AatII site. The Hsp104R gene (25) was subcloned into the NdeI-SacI sites of pNOTAG, yielding pNOTAG Hsp104. To create pNOTAG Hsp104Y819W, the SpeI-SacI cassette from pGALHsp104Y819W (A. Cashikar, J. Glover, and S.L.L., unpublished data) was used to replace the wild-type cassette. To create pNOTAG Hsp104R826M, a mutant cassette was created by 2-step PCR, using pNOTAG Hsp104 as a template, then used to replace the SpeI-SacI fragment in pNOTAG Hsp104. pNOTAG Hsp104Y819W R826M was made by the same procedure, using pNOTAG Hsp104Y819W as a template.
For production of the Hsp104549–908 fragment, DNA encoding amino acids 549–908 was amplified by PCR from pGALHsp104R then subcloned into pPROEXHTb (GIBCO) as a BamHI-SacI fragment. Y819W and R826M variants were constructed by inserting the appropriate mutagenic SpeI-SacI cassette from the pNOTAG plasmids into pPROEX Hsp104549–908. The fidelity of all mutagenesis was verified by DNA sequencing.
For gene replacement in yeast, the R826M SpeI-SacI cassette was inserted into pRS306Hsp104 (12). The resulting integrating plasmid, pRS306Hsp104R826M, was linearized with Eco81I and transformed into S. cerevisiae strain 74D-694a [PSI+] (Mata, ade1–14, trp1–289, his3Δ-200, ura3–52, leu2–3, lys2). An hsp104R826M strain was isolated and identified by PCR as described (12) followed by BsrDI digestion.
Protein Purification.
All proteins were expressed and purified by using a BL21-RIL codon plus strain (Stratagene). For purification of full-length Hsp104 mutants, cells were grown at 37°C in Circle Grow (Qbiogene, Carlsbad, CA) to an OD600 of 0.8, then 1 mM isopropyl-β-d-thiogalactoside (IPTG) was added and growth was continued at 21°C for 4 h. Cells were lysed by sonication in 20 mM Tris (pH 7.7), 1 mM EDTA, and 25 mM NaCl, with protease inhibitors (Roche Molecular Biochemicals). Protein was purified by sequential chromatography on DEAE Sepharose (Amersham Biosciences), Fast Flow Blue (Amersham Biosciences), and hydroxyapatite (Bio-Rad) columns, essentially as described for Hsp104 purified from S. cerevisiae (23). After the hydroxyapatite column, protein was concentrated to 4 mg/ml then passed over a Sephacryl S-400 (Amersham Biosciences) column.
For purification of Hsp104549–908 and variants, bacteria were grown at 37°C in Circle Grow to an OD600 of 0.8, 1 mM isopropyl-β-d-thiogalactoside was added, and growth was continued at 25°C for 4 h. Cells were lysed by sonication in 50 mM sodium phosphate (pH 8.0), 500 mM NaCl, and 1.4 mM β-mercaptoethanol (buffer A) with EDTA-free protease inhibitors (Roche Molecular Biochemicals). Cleared lysates were applied to an Ni-nitrilotriacetic acid (NTA) column (Qiagen, Chatsworth, CA). Proteins were eluted with buffer A and 200 mM imidazole. The polyhistidine tag was removed by using TEV protease (GIBCO), leaving the nonnative amino acids GAMGSG at the N terminus. Cleaved Hsp104549–908 was purified further by using a DEAE Sepharose column.
For all proteins, glycerol was added after the final column to 10% concentration, and proteins were flash-frozen and stored at −80°C. Protein concentration was determined by absorbance at 276 nm. Concentrations mentioned in the text refer to the concentration of monomer.
Molecular Mass Determination by Static Light Scattering.
Molecular mass determinations were performed by light scattering, using unfractionated solutions of 200 nM protein as described (12).
Urea Denaturation.
Urea titrations were performed in 20 mM Hepes pH 7.5/200 mM NaCl at 25°C. CD was measured in a Jasco (Easton, MD) J-715 at 225 nm; fluorescence was measured in a Spex (Jobin-Yvon, Edison, NJ) Fluoromax-3 with λex at 295 nm and λem at 365 nm. Experiments were begun with a 1 μM protein solution without urea, followed by sequential additions of a 1 μM protein solution in 8 M urea. After each addition, the sample was stirred for 1 min. CD or fluorescence was averaged over 30 sec.
CD data were fit to a three-state model describing an unfolding transition that occurs through a stable intermediate:
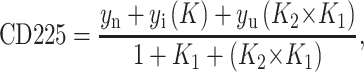 |
1 |
where yn, yi, and yu are the signals from the native, intermediate, and denatured states, respectively. K1 and K2 are equal to exp((ΔGD-NH20 + m[D]/RT) for transition 1 and transition 2, where D is denaturant. yn, yi, and yu were assumed to have a linear dependence on D.
Because Y819W is a domain-specific probe, fluorescence was fit to a two-state unfolding model:
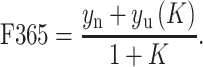 |
2 |
ATP Hydrolysis.
ATP hydrolysis was measured as described (12). Data were fit to a model describing cooperative hydrolysis at two independent sites:
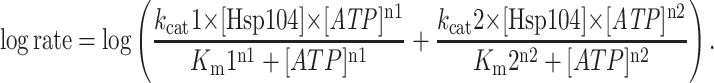 |
3 |
Nucleotide Binding.
Nucleotide binding was measured in 20 mM Hepes, pH 7.5/20 mM NaCl/10 mM MgCl2 at 4°C. Tryptophan fluorescence was measured in a Spex Fluoromax-3 with λex at 295 nm and λem at 346 nm. Nucleotide was added in sequential increments from a stock solution (20 mM for ATP, 2 mM for ADP) to a 400 nM protein solution. After each addition, the sample was stirred for 3 min. Fluorescence was averaged over 10 sec. Buffer-subtracted fluorescence was corrected for dilution from nucleotide addition. Data were fit to the Hill equation:
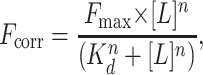 |
4 |
where L is nucleotide and n is the Hill coefficient.
In Vivo Assays of Hsp104 Function.
Thermotolerance and mating tests for analysis of [PSI] propagation were done as described (12).
Results
Homology Model of the C-terminal Proteolytic Fragment.
To aid in design of a fluorescent probe for nucleotide binding to Hsp104 NBD2, we generated a homology model of this binding site, using the NSF D2 domain bound to ATP (18) as a template. Only the 11 conserved regions of the AAA superfamily (26) were included in the model. The two sequences share 21.2% identity and 43.6% similarity in these regions, sufficient to model the fold of Hsp104 NBD2 (residues 578–853) and the location of highly conserved active site residues. The model consists of a domain with a Rossmann fold (residues 578–772) and a smaller α-helical domain (residues 773–853) (Fig. 1). Nonconserved regions, such as the highly charged 56 amino acids at the C terminus of Hsp104 and several loops, were not included in the model. Because of the high conservation of active site residues, the coordinates for ATP from the NSF structure were included in the model, placing ATP at the interface between the two domains. Tyr-819 is located near the sensor-2 motif at this interface, and its proximity to the active site suggests that the fluorescence of a tryptophan residue introduced at this position might be sensitive to nucleotide binding.
Figure 1.

Ribbon diagram of a homology model of residues 578–853 of Hsp104 created with SWISS-MODEL (30). The predicted position of ATP from the 1NSF Protein Data Bank coordinates (18) is shown in orange, and that of R826 is shown in red. The location of the Y819W substitution is shown in purple.
Limited Proteolysis Defines Two Stable 40-kDa Fragments.
To ensure that the Y819W substitution did not cause a structural defect, we sought a system that could be used for equilibrium unfolding measurements to define the stabilities of each domain in the NBD2 AAA module. We used limited proteolysis to define the physical boundaries of a suitable protein fragment. During V8 proteolysis of Hsp104, two closely migrating fragments of 40 kDa were resistant to cleavage for up to 256 min (Fig. 2A). Select time points from the digest were immunoblotted with Ab 1–2, which binds the extreme N terminus (residues 1–15), or Ab 8–1, which binds the extreme C terminus (residues 894–908). One of the two 40-kDa fragments was recognized by Ab 1–2, whereas the other was recognized by Ab 8–1 (Fig. 2B). The exact molecular masses of the 40-kDa N- and C-terminal fragments were 40,409 and 40,270 Da, respectively, (determined by mass spectrometry, data not shown). Thus, the two fragments result from V8 cleavage after Glu-360 and after Glu-548 (Fig. 2C). The Hsp104549–908 fragment represents a single site model of the NBD2 AAA module.
Figure 2.

Limited proteolysis. (A) Time course of V8 proteolysis monitored by SDS/PAGE with Coomassie blue staining. (B) Immunoblots with Ab 1–2 or Ab 8–1 of select time points. (C) Location of V8 protease cleavage sites (arrows) relative to the Ab 1–2 and Ab 8–1 epitopes and to the Walker A and B, sensor-1, Box VII, and sensor-2 motifs. Predicted boundaries of the Rossmann domains (26) are shown as gray boxes.
Secondary Structure and Equilibrium Denaturation.
The CD spectrum in Fig. 3A shows that the Hsp104549–908 recombinant fragment is well-folded, with mixed α,β secondary structure. In AAA proteins of known structure, the two conserved domains are contiguous in sequence and each has a separate hydrophobic core, suggesting that in Hsp104549–908, the two predicted domains should unfold independently. Because we observed dimerization of Hsp104549–908 at high protein concentrations (by size-exclusion chromatography and light scattering, data not shown), our equilibrium denaturation experiments were done at a protein concentration of 1 μM to maintain the protein as a monomer, so that domain unfolding can be distinguished from a disruption of the interaction between subunits.
Figure 3.

Equilibrium unfolding of Hsp104549–908. (A) CD spectrum of a 40 μM solution of Hsp104549–908. Urea denaturation of Hsp104549–908 (B) and Hsp104549–908 Y819W (C) monitored by CD at 225 nm. (D) Urea denaturation of Hsp104549–908 Y819W monitored by fluorescence at 365 nm.
After titration of Hsp104549–908 with urea, two well-separated unfolding transitions were visible by CD at 225 nm (Fig. 3B). One occurs with a transition midpoint at a urea concentration of 2.5 M (transition 1), and the second occurs with a midpoint at 4.2 M urea (transition 2). The data were fit to a three-state model (N ↔ I ↔ U, where I presumably represents an intermediate with one domain folded and the second domain unfolded) to determine the free energy of the folded state (ΔGD-NH20), and the m value, which is proportional to the surface area buried after folding, for each transition. For transition 1, ΔGD-NH20 and m were −4.1 kcal mol−1 and 1.7 kcal mol−1 M−1, respectively, whereas for transition 2, the values were −10.4 kcal mol−1 and 2.5 kcal mol−1 M−1 (Table 1).
Table 1.
Folding stabilities of the two domains in the Hsp104 NBD2 AAA module
| WT
|
Y819W
|
R826M
|
||
|---|---|---|---|---|
| CD225 | CD225 | F365 | CD225 | |
ΔG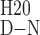 1, kcal
mol−1 1, kcal
mol−1
|
−4.1 ± 0.2 | −4.5 ± 0.4 | −4.1 ± 0.2 | −3.8 ± 0.4 |
| m1, kcal mol−1 M−1 | 1.7 ± 0.1 | 1.8 ± 0.2 | 1.7 ± 0.1 | 1.5 ± 0.2 |
ΔG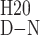 2, kcal
mol−1 2, kcal
mol−1
|
−10.4 ± 0.4 | −10.3 ± 1.1 | NA | −10.6 ± 1.3 |
| m2, kcal mol−1 M−1 | 2.5 ± 0.1 | 2.4 ± 0.2 | NA | 2.4 ± 0.3 |
NA, not applicable.
Y819W Fluorescence As a Conformational Probe of the Hsp104 C-terminal Domain.
We incorporated the Y819W substitution into Hsp104549–908 and performed urea denaturation on the purified fragment. As measured by CD at 225 nm, the denaturation of this variant also occurs via two transitions (Fig. 3C). The values of ΔGD-NH20 and m for the two domains were the same as for the wild-type fragment (Table 1), indicating that the Y819W mutation did not significantly perturb the structure of the protein.
In contrast to CD, which is a probe of the unfolding of both domains, fluorescence of the Y819W probe should only be sensitive to conformational transitions involving the C-terminal domain. Complete denaturation of Hsp104549–908 Y819W with 6 M urea caused an increase in the intensity of tryptophan fluorescence and a shift in the λmax from 345 nm to 352 nm (data not shown). By measuring tryptophan fluorescence at 365 nm (where the largest difference between the folded and unfolded states was observed) as a function of urea concentration, only the unfolding transition between 1 and 4 M urea was observed (Fig. 3D). The fluorescence data were fit to a two-state model, and values for ΔGD-NH20 and m were −4.0 kcal mol−1 and 1.6 kcal mol−1 M−1, identical to those determined for transition 1 by CD at 225 nm (Table 1). These data indicate that the C-terminal domain is the low stability domain, whereas the domain with the Rossmann fold has greater stability.
Hexamer Formation and ATP Hydrolysis by Hsp104Y819W.
We next incorporated the Y819W substitution into full-length Hsp104. The substitution had no deleterious effect on hexamer stability, as the salt dependence of assembly was similar to that of wild-type Hsp104 (ref. 12; Fig. 4A). Furthermore, under low salt conditions (used to stabilize the hexamer), steady-state ATP hydrolysis by Hsp104Y819W showed the two kinetic transitions characteristic of the wild-type hexamer (Fig. 4B). Fitting the data to an equation describing hydrolysis at two independent allosteric sites yielded kinetic parameters that were very similar to those of the wild-type protein. The two-fold decrease in kcat and Km for hydrolysis at NBD2 (kcat2 and Km2; Table 2, bottom rows) represents a very minor perturbation of Hsp104 properties. In addition, this substitution does not affect Hsp104 function in thermotolerance in vivo (A. Cashikar, J. Glover, and S.L.L., unpublished data).
Figure 4.

Nucleotide binding and hydrolysis by Hsp104Y819W. (A) Molecular mass of Hsp104Y819W as a function of NaCl concentration. (B) ATP hydrolysis by Hsp104Y819W (●), fit to a model describing two independent sites (solid line). The dashed line represents hydrolysis by wild-type Hsp104. ATP (C) and ADP (D) binding to Hsp104Y819W fit to a single-site Hill equation.
Table 2.
Steady-state kinetics of ATP hydrolysis by Hsp104 and mutants
| Wild-type | Y819W | R826M | |
|---|---|---|---|
| kcat1, min−1 | 76 ± 4.5 | 97 ± 6.5 | 34 ± 1.8 |
| Km1, μM | 170 ± 12 | 172 ± 10 | 206 ± 9 |
| n1 | 2.3 ± 0.1 | 2.1 ± 0.1 | 1.7 ± 0.1 |
| kcat2, min−1 | 0.27 ± 0.05 | 0.11 ± 0.05 | 1.2 ± 1.1 |
| Km2, μM | 4.7 ± 1.1 | 2.5 ± 1.7 | 19 ± 13 |
| n2 | 1.6 ± 0.2 | 1.5 ± 0.4 | 1.4 ± 0.1 |
Cooperative Nucleotide Binding to NBD2 in the Hsp104 Hexamer.
Next, we asked whether the tryptophan fluorescence of Hsp104Y819W would change after addition of nucleotide, thus providing a site-specific probe of binding to NBD2. Addition of ATP to a solution of hexameric Hsp104Y819W caused a saturable increase in its fluorescence intensity, with the maximum difference between the nucleotide-bound and unliganded states observed at 346 nm (Fig. 4C). ADP caused a similar increase (Fig. 4D), although the magnitude of the change was greater (compare values of Fmax in Table 3). For both nucleotides, the fluorescence increase showed a sigmoidal dependence on nucleotide concentration, indicative of positive cooperativity in binding. Fitting the ATP binding data to a single-site Hill equation yielded a dissociation constant (Kd) of 69 μM and a Hill coefficient of 1.8. ADP bound more tightly to NBD2 than ATP, with Kd = 9 μM and a Hill coefficient of 1.7 (Table 3). Notably, similar binding affinities for ATP and ADP were measured for Thermus thermophilus ClpB by using a fluorescent nucleotide analog; however, the protein was monomeric under the conditions used and cooperativity was not detected (27).
Table 3.
Cooperative nucleotide binding by Hsp104 and mutants
| Y819W | Y819W R826M | ||
|---|---|---|---|
| ATP | Fmax × 106 | 1.39 ± 0.01 | 1.43 ± 0.04 |
| Kd, μM | 69 ± 1 | 460 ± 20 | |
| n | 1.8 ± 0.1 | 2.1 ± 0.1 | |
| ADP | Fmax × 106 | 2.05 ± 0.06 | 2.14 ± 0.06 |
| Kd, μM | 9.1 ± 0.3 | 49 ± 2 | |
| n | 1.7 ± 0.1 | 1.9 ± 0.1 |
Mutagenesis of the Sensor-2 Motif.
The conserved arginine in the sensor-2 motif of AAA proteins (Arg-826 in Hsp104 NBD2) projects from the C-terminal domain into the nucleotide binding site and seems to interact with the phosphate groups (Fig. 1). We created a R826M mutation to probe the nature of this interaction. Three mutant proteins were purified: Hsp104R826M, for analysis of hexamer assembly and ATP hydrolysis activity; Hsp104 Y819W R826M, for measurement of ATP and ADP binding; and the fragment Hsp104549–908 R826M, for analysis of domain folding stability.
Hsp104R826M was fully competent to form hexamers, as seen in the NaCl dependence of assembly (Fig. 5A), and the folding stabilities of both domains in the Hsp104549–908 R826M fragment were not significantly different from wild type (Fig. 5B and Table 1). The primary effect of this mutation was on ATP and ADP binding. For each nucleotide, R826M caused a nearly 6-fold decrease in affinity, with no significant effect on the Hill coefficients (Fig. 5 C and D, and Table 3). Because stability of the C-terminal domain is unaltered, the binding defect is likely caused by removal of a specific contact between the nucleotide and Hsp104, as expected, rather than to a structural perturbation. As observed with wild-type Hsp104 (12), ATP hydrolysis by Hsp104R826M showed two kinetic transitions (Fig. 5E), but there were three differences between the two proteins. First, the reduced affinity for ATP and ADP was reflected by an increase in the Km for ATP hydrolysis at NBD2 (Km2, Table 2). This mutation also caused an approximate 4-fold increase in kcat at this site (kcat2, Table 2). Last, the kcat of NBD1 was decreased (kcat1, Table 2), reflecting allosteric communication between NBD1 and NBD2.
Figure 5.

Biochemical properties of Hsp104R826M. (A) Molecular mass of Hsp104R826M as a function of NaCl concentration. (B) Urea denaturation of Hsp104549–908 R826M monitored by CD at 225 nm. ATP (C) and ADP (D) binding to Hsp104Y819W R826M fit to a single-site Hill equation. (E) ATP hydrolysis by Hsp104R826M (□), fit to a model describing two independent sites (solid line). The dashed line represents hydrolysis by wild-type Hsp104.
Effect of the R826M Sensor-2 Mutation in Vivo.
We introduced the R826M mutation into the Hsp104 locus by gene replacement, creating a hsp104R826M [psi−] strain, to determine its effect on Hsp104 function in vivo. The effects of this mutation on [PSI+] propagation and thermotolerance were compared with those of the hsp104N728A mutant, which abolishes ATP hydrolysis at NBD2 and is a dominant severe loss of function allele in vivo. Analysis of [PSI+] propagation was performed by using suppression of the nonsense codon in the ade1–14 allele, with the presence (and strength) of the [PSI+] element detected by yeast colony color on yeast extract/peptone/dextrose (YPD). White color is indicative of the strong [PSI+] phenotype, whereas colonies that are red have less Sup35 protein in the prion state (e.g., weak [PSI+], cryptic [PSI+], or [psi−]). Thermotolerance was measured as the ability of the mutant strains to survive exposure to 52°C for increasing lengths of time after a pretreatment at 39°C to induce the heat shock response.
To determine whether the hsp104R826M allele could support [PSI+], the hsp104R826M [psi−] strain was mated to a HSP104 [PSI+] strain, and the meiotic progeny of the diploid were analyzed for their [PSI+] status. The hsp104R826M/HSP104 heterozygous diploid exhibited a pink color that was similar to that observed in the hsp104N728A/HSP104 diploid, suggesting that the amount of Sup35 protein in the prion conformation was reduced as a result of partial inhibition of wild-type Hsp104 function by Hsp104R826M (Fig. 6A). The level of inhibition was not, however, sufficient to cure this diploid strain of [PSI+], because after sporulation of the hsp104R826M/HSP104 diploid, both HSP104 progeny were [PSI+] (Fig. 6B). Both hsp104R826M progeny grew as red colonies on yeast extract/peptone/dextrose. Mating of the mutant progeny to a wild-type [psi−] strain produced four red colonies after sporulation, demonstrating that the R826M mutant cannot support [PSI+].
Figure 6.

In vivo phenotypes of hsp104R826M. (A) Heterozygous mutant diploids grown on yeast extract/peptone/dextrose (YPD) at 30°C for 5 days. (B) Meiotic progeny from the HSP104/hsp104R826M diploid grown on YPD at 30°C for 5 days. (C) Recovery of wild-type and mutant strains from increasing exposure to 52°C. Five-fold serial dilutions of treated cells, allowed to recover at 30°C for 3 days, are shown.
Relative to the wild-type HSP104 [psi−] strain, the hsp104R826M [psi−] strain showed a loss of thermotolerance, but it was not as severe a defect as that caused by the hsp104N728A allele (Fig. 6C). This phenotype is consistent with the previous observation that mutations that cause only a mild loss of function in thermotolerance (e.g., hsp104T317A, which causes a 10-fold decrease in the rate of ATP hydrolysis at NBD1) are sufficient to cure cells of the [PSI+] prion (12).
Discussion
We have developed a single tryptophan substitution in the C-terminal domain of Hsp104 as a site-specific probe of cooperative nucleotide binding to NBD2. Together with measurement of the steady-state kinetics of ATP hydrolysis of NBD1 and NBD2, this allows a description of how mutations in NBD2 affect its ATP hydrolysis cycle and of how perturbations in this cycle are sensed by the NBD1 ATPase domain. These tools, together with two in vivo assays of Hsp104 function, make Hsp104 a powerful model for understanding the mechanism of a protein with two AAA ATPase domains. Here, we focus on the role of the highly conserved arginine (Arg-826) in the AAA sensor-2 motif of NBD2.
Our results show that in Hsp104 NBD2, the arginine in sensor-2 participates in nucleotide binding but is not required for catalysis. The R826M mutation caused nearly equal decreases in affinity for ATP and ADP, suggesting that it does not act to discriminate between ATP and ADP in the binding site, but instead discriminates between the nucleotide-bound and unliganded states. This finding is consistent with crystal structures showing that this residue in other AAA proteins interacts with the β-phosphate of ADP or ATP (20, 28), although interaction with the γ-phosphate has also been proposed (18, 29). In Hsp104, if ATP and ADP bound to NBD2 promote distinct conformations of the C-terminal domain, residues other than those in sensor-2 must detect the difference between the two nucleotides.
We have shown that a mutation in the AAA sensor-1 motif of Hsp104 NBD2 (N728A) that effectively traps it in the ATP-bound state also affects ATP hydrolysis at NBD1 (12), indicating that the catalytic activity of NBD1 depends on the nucleotide that is bound to NBD2. ATP binding to NBD2 promotes a state at NBD1 with a relatively low kcat and Km; by extension, when ADP is bound to NBD2, kcat and Km at NBD1 must increase. Thus, although fitting the steady-state ATP hydrolysis data by using two independent cooperative kinetic transitions (at NBD1 and NBD2) is a useful approximation, a more accurate fit may require incorporation of two distinct activities at NBD1, weighted by the occupancy time of ATP and ADP at NBD2. Further development of this model will require measurement of the rate constants associated with ATP binding, ATP hydrolysis, and ADP release. The Y819W fluorescent probe developed here will facilitate such measurements.
Our demonstration that the R826M mutation in NBD2 reduces the rate of catalysis by NBD1 provides further insight into the communication between NBD1 and NBD2. The primary effect of R826M was to weaken the affinity of NBD2 for ATP and ADP, which caused a reduction in the kcat for hydrolysis at NBD1. Interestingly, an increase in kcat at NBD2 was observed in this mutant, which may be the result of an elevated rate of product (ADP) release. A quantitative understanding of how this mutation in NBD2 affects NBD1 awaits transient kinetic measurements, as discussed above. However, these observations are consistent with the model that ADP bound to NBD2 promotes a high-activity state of NBD1, such that a decrease in the ADP occupancy time during the hydrolysis cycle at NBD2 should reduce the rate of catalysis at NBD1.
Acknowledgments
We thank John Glover for initial work in the development of the Y819W mutant and Anil Cashikar for assistance with creation of the pNOTAG expression vector. We also thank Annelli Asikainen for technical assistance with protein purification and enzymatic assays, and members of the Lindquist lab for critical reading of the manuscript. This work was supported by the Howard Hughes Medical Institute and National Institutes of Health grants (to S.L.L.). D.A.H. was supported by National Institutes of Health Training Grant 5 T32 GM07183.
Abbreviations
- NBD1
N-terminal nucleotide-binding domain
- NBD2
C-terminal nucleotide-binding domain
References
- 1.Glover J R, Lindquist S. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 2.Zolkiewski M. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 3.Goloubinoff P, Mogk A, Zvi A P, Tomoyasu T, Bukau B. Proc Natl Acad Sci USA. 1999;96:13732–13737. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez Y, Lindquist S L. Science. 1990;248:1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 5.Parsell D A, Kowal A S, Singer M A, Lindquist S. Nature (London) 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 6.Chernoff Y O, Lindquist S L, Ono B, Inge-Vechtomov S G, Liebman S W. Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 7.Moriyama H, Edskes H K, Wickner R B. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sondheimer N, Lindquist S. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 9.Patino M M, Liu J J, Glover J R, Lindquist S. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 10.Paushkin S V, Kushnirov V V, Smirnov V N, Ter-Avanesyan M D. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 11.Parsell D A, Kowal A S, Lindquist S. J Biol Chem. 1994;269:4480–4487. [PubMed] [Google Scholar]
- 12.Hattendorf D A, Lindquist S L. EMBO J. 2002;21:12–21. doi: 10.1093/emboj/21.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogura T, Wilkinson A J. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 14.Saraste M, Sibbald P R, Wittinghofer A. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 15.Maegley K A, Admiraal S J, Herschlag D. Proc Natl Acad Sci USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rees D C, Howard J B. J Mol Biol. 1999;293:343–350. doi: 10.1006/jmbi.1999.3005. [DOI] [PubMed] [Google Scholar]
- 17.Lenzen C U, Steinmann D, Whiteheart S W, Weis W I. Cell. 1998;94:525–536. doi: 10.1016/s0092-8674(00)81593-7. [DOI] [PubMed] [Google Scholar]
- 18.Yu R C, Hanson P I, Jahn R, Brunger A T. Nat Struct Biol. 1998;5:803–181. doi: 10.1038/1843. [DOI] [PubMed] [Google Scholar]
- 19.Karata K, Inagawa T, Wilkinson A J, Tatsuta T, Ogura T. J Biol Chem. 1999;274:26225–26232. doi: 10.1074/jbc.274.37.26225. [DOI] [PubMed] [Google Scholar]
- 20.Putnam C D, Clancy S B, Tsuruta H, Gonzalez S, Wetmur J G, Tainer J A. J Mol Biol. 2001;311:297–310. doi: 10.1006/jmbi.2001.4852. [DOI] [PubMed] [Google Scholar]
- 21.Kim Y I, Levchenko I, Fraczkowska K, Woodruff R V, Sauer R T, Baker T A. Nat Struct Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- 22.Singh S K, Rozycki J, Ortega J, Ishikawa T, Lo J, Steven A C, Maurizi M R. J Biol Chem. 2001;276:29420–29429. doi: 10.1074/jbc.M103489200. [DOI] [PubMed] [Google Scholar]
- 23.Schirmer E C, Lindquist S. Methods Enzymol. 1998;290:430–444. doi: 10.1016/s0076-6879(98)90036-2. [DOI] [PubMed] [Google Scholar]
- 24.Clos J, Brandau S. Protein Expression Purif. 1994;5:133–137. doi: 10.1006/prep.1994.1020. [DOI] [PubMed] [Google Scholar]
- 25.Schirmer E C, Queitsch C, Kowal A S, Parsell D A, Lindquist S. J Biol Chem. 1998;273:15546–15552. doi: 10.1074/jbc.273.25.15546. [DOI] [PubMed] [Google Scholar]
- 26.Neuwald A F, Aravind L, Spouge J L, Koonin E V. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 27.Schlee S, Groemping Y, Herde P, Seidel R, Reinstein J. J Mol Biol. 2001;306:889–899. doi: 10.1006/jmbi.2001.4455. [DOI] [PubMed] [Google Scholar]
- 28.Sousa M C, Trame C B, Tsuruta H, Wilbanks S M, Reddy V S, McKay D B. Cell. 2000;103:633–643. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 29.Bochtler M, Hartmann C, Song H K, Bourenkov G P, Bartunik H D, Huber R. Nature (London) 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 30.Guex N, Diemand A, Peitsch M C. Trends Biochem Sci. 1999;24:364–367. doi: 10.1016/s0968-0004(99)01427-9. [DOI] [PubMed] [Google Scholar]