

Abstract
The fact that natural β-sheet proteins are usually soluble but that fragments or designs of β structure usually aggregate suggests that natural β proteins must somehow be designed to avoid this problem. Regular β-sheet edges are dangerous, because they are already in the right conformation to interact with any other β strand they encounter. We surveyed edge strands in a large sample of all-β proteins to tabulate features that could protect against further β-sheet interactions. β-barrels, of course, avoid edges altogether by continuous H-bonding around the barrel cylinder. Parallel β-helix proteins protect their β-sheet ends by covering them with loops of other structure. β-propeller and single-sheet proteins use a combination of β-bulges, prolines, strategically placed charges, very short edge strands, and loop coverage. β-sandwich proteins favor placing an inward-pointing charged side chain on one of the edge strands where it would be buried by dimerization; they also use bulges, prolines, and other mechanisms. One recent β-hairpin design has a constrained twist too great for accommodation into a larger β-sheet, whereas some β-sheet edges are protected by the bend and reverse twist produced by an Lβ glycine. All free edge strands were seen to be protected, usually by several redundant mechanisms. In contrast, edge strands that natively form β H-bonded dimers or rings have long, regular stretches without such protection. These results are relevant to understanding how proteins may assemble into β-sheet amyloid fibers, and they are especially applicable to the de novo design of β structure. Many edge-protection strategies used by natural proteins are beyond our current abilities to constrain by design, but one possibility stands out as especially useful: a single charged side chain near the middle of what would ordinarily be the hydrophobic side of the edge β strand. This minimal negative-design strategy changes only one residue, requires no backbone distortion, and is easy to design. The accompanying paper [Wang, W. & Hecht, M. H. (2002) Proc. Natl. Acad. Sci. USA 99, 2760–2765] makes use of the inward-pointing charge strategy with great success, turning highly aggregated β-sandwich designs into soluble monomers.
All-β folds are a common, major category of protein structure, and as produced in the cell they fold up successfully with only a few isolated cases of aggregation or insolubility. In contrast, fragments or peptides from β-sheet proteins have long had a reputation for insolubility (1), and it is now known that the amyloid fibrils produced in some disease states or by harsh treatment in vitro have a β-sheet structure (2). Also, de novo designs of β proteins almost all have severe problems of insolubility and aggregation (3–6); many designs originally intended as globular β-sheet proteins have turned out instead to be valuable as models for amyloid-fiber formation. It seems that natural β-sheet proteins know something we do not: those natural proteins only get in trouble after long periods of time or under hostile conditions, but de novo β designs are seldom soluble and monomeric even as first produced.
In hindsight, it seems obvious that the edges of completely regular β-sheets or β-sandwiches are inherently aggregation-prone, because they are already set up to form further β H-bonding with any other β strands they encounter. This is a problem in “negative design” (7), because the protein sequence incorporates factors dictated not by the desired final structure (the monomeric β fold) but by the need to avoid a different final structure (the edge-to-edge β aggregate). On first recognizing this problem some time ago, we sampled the edges of β-sandwich proteins [reported in a retrospective review (4)], and suggested addition of an inward-pointing charge to the edges of our de novo betabellin design. That step was never tried because of the untimely death of our collaborator Bruce Erickson, but now those ideas have prompted the protein design work reported by Wang and Hecht (8). Increased recent interest in the design of β structure adds to the importance of this issue; therefore, to learn the details of how natural β-sheet edge strands are designed to avoid aggregation, the present study systematically surveys their properties in all classes of all-β structure.
Methods
Coordinate files are from the Protein Data Bank (PDB) web site (9) at http://www.rcsb.org/pdb. The SCOP Structural Classification of Proteins web site (10) at http://scop.mrc-lmb.ac.uk/scop, Version 1.55, was used to choose a wide-ranging and representative set of β-sheet structures. For β-helices, each of the 15 families was sampled: PDB IDs 1KAP, 2PEC, 1QCX, 1QJV, 1TYU, 1CZF, 1DAB, 1DBG, 1EZG, 1LXA, 3TDT, 2XAT, 1QRE, 1HV9, 1EWW. For β-propellers, each of the six folds was sampled: 1HXN, 1TL2, 1CRU, 1GOTc, 1AOQ, 1G61. For open single sheets, each of the four high-resolution folds in the SCOP all-β category was included (1PIN, 1HCB, 4BCL, 1OSP), supplemented by 14 files from the α+β folds, chosen to be open single sheets with widely varying shapes and topologies: 1FUS, 7RSA, 3IL8, 1IGD, 1MOL, 1FV1, 1BKF, 1CSEi, 1CTF, 1GADo, 3SSI, 1E7X, 1SPH, 1PNE. For βsandwiches, each fold with an example at 2-Å resolution or higher was represented, plus extra immunoglobulins and lectins for studying conservation of β-edge features: 2RHE, 8FAB, 1TEN, 2MCM, 1XSO, 1AMX, 1TTA, 1ES6, 1HOE, 1PLC, 1RSY, 1CZY, 1STM, 1AMM, 1YGE, 1PSG, 1SLU, 1CZS, 1AOL, 1KNB, 1ALY, 1SFP, 1CB8, 1THW, 1CQ3, 1NLS, 1GBG, 1SLT, 1D2S, 1SAC, 1A8D, 1KIT, 1SLI, 1XNB, 1RIE, 1VJS, 2ARC, 1WAP.
Kinemages were made in prekin (with main chain, side chain, H-bonds, heterogens, and sometimes ribbons) and examined in MAGE (11) to identify and tabulate their β edge-strand features. Figures were made in MAGE, with output rendered in RASTER3D (12). Twist and bend of β-strand pairs were calculated as follows: a line was drawn between the midpoints of each pair of peptides opposite one other on adjacent β strands, and their midpoints were joined into a segmental center line for the strand pair; twist per residue is the dihedral angle of two adjacent peptide lines measured around the midline; bend is the complement of the angle formed by the n − 2, n, n + 2 midline points. For β-sandwiches or other edges with two exposed backbone elements, a charged side chain is defined as “inward-pointing” if its Cα—Cβ bond points toward the other backbone element.
When needed, hydrogens were added and optimized by REDUCE (13). Solvent accessibilities and all-atom contacts were calculated in PROBE (14) and displayed as dot surfaces in MAGE. Amino acid replacements and rotations were made and analyzed with the interactive MAGE/PROBE system (15).
Results
Representative, high-resolution β-sheet structures were chosen from about 75 β-sheet folds in the SCOP database (see Methods for details). Edge strands that form multimer interfaces were considered separately. Within each structure, all edge β strands were examined for features that could serve to block further β-sheet H-bonding. Some of those features we expected in advance, whereas others were surprises.
β-Barrels.
One of the simplest ways to avoid edge-to-edge β-sheet aggregation is to have no edges. Well-formed antiparallel βbarrel structures accomplish this by having continuous β H-bonding all of the way around a cylindrical barrel. Those cylinder cross-sections vary from round to quite flattened, and usually the top and bottom splay apart somewhat in twisted, 2-strand β ribbons or irregular loops. For Greek key topologies, pairs of strands may stay H-bonded as they curve next to each other across an end of the cylinder.
Where the β-barrel H-bonding is regular, there are no peptide groups free to aggregate with other β-sheets. Even where the H-bonding is less complete or regular in a highly curved part of the barrel, often the strands have curved toward one another enough to block external strand access. β ribbons exposed at a barrel end are usually highly twisted and may also use other blocking strategies. Some β-barrels have one side with continuous β H-bonding, whereas the other side is sandwich-like; they may be classified either as sandwiches or barrels, but in either case one side uses the barrel strategy and the other side uses the β-sandwich edge strategies described below.
Parallel (β8α8) barrels are even better protected than antiparallel barrels, because they almost always have continuous H-bonding all of the way around and are further covered by an outer cylinder of helices. Other α/β structures are not discussed here because the β-sheet edge is only a small part of the potential interface, and also because their possible aggregates are not as relevant to the structure of amyloid fibers.
β-Helices.
Parallel β-helix structures can have either two or three connected β-sheets, and their wide helical turns can be either left-handed or right-handed. For all types the parallel β H-bonding is quite continuous through the body of the structure, but a regular β-helix exposes at each end a ring of β strands poised for further β H-bonding.
The 15 known families of β-helix proteins quite consistently (27 of 30 ends) use the same straightforward strategy to protect those ends: covering them with a loop of helical or nonrepetitive structure. This allows the β structure to remain regular all of the way to its end, which is then covered up by a separate part of the chain. Fig. 1 shows an example, from the left-handed β-helix of the 1QRE archaeal carbonic anhydrase. If calculated with a 1.4-Å radius probe, the small helical loop buries from solvent four of the nine β-strand H-bond donors and acceptors of the final β-helix turn (two others were already removed by Pro side chains), and it buries all but one of the nine if probed with a 5-Å radius more like a potentially interacting β-sheet protein.
Figure 1.

End view of the left-handed, three-sheet β-helix of the 1QRE archaeal carbonic anhydrase (27). β strands are peach-colored arrows, with the three edge strands emphasized in darker orange. As is true for almost all β-helix ends, those edge strands are covered by a substantial loop, in this case containing an α-helix (in gold).
For five of the 30 β-helix ends one of the sheets rolls inward, typically covering the end of one other sheet and butting perpendicularly into the side of the third one, thus leaving only one edge unprotected. That remaining exposed edge utilizes either prolines or inward-pointing charges. The rolled-edge arrangement only works when the final few strands are antiparallel so the sheets can turn independently. Bulges do not occur in the β-helix ends, mirroring their general absence in parallel β-sheet (16).
The least-covered β-helix end in our sample is in Lpx-A (1LXA), where further β H-bonding is blocked only by a Pro, the charged N terminus, and the three corners between strands, which each make a significant upward protrusion. All nine edge β-strand peptide groups are accessible to solvent at a 1.4-Å radius, but only two separated ones are accessible at a 5-Å radius. Six of the 30 total β-helix ends have strongly upturned corners, 6 have their corners cut across shorter, 12 have outward-pointing prolines, 11 have inward-pointing charges, and 5 are distorted by SS bonds (e.g., 1EWW and 1EZG insect antifreeze); coverage by loops, however, is by far the dominant strategy. Many β-helix proteins form side-by-side dimers or trimers, but none form subunit contacts by using the β-helix ends.
β-Propellers.
β-propeller proteins are known with four, five, six, seven, or eight radial “blades” of up-and-down β-sheet, and there is a five-bladed example with mixed sheet and a βαβ motif at the outer edge (1G61). Fig. 2 shows the β-propeller five of tachylectin (1TL2). The inner blade edges, of course, are completely protected at the propeller center, which constitutes a viable but otherwise rare β-edge strategy. Many of the βpropellers are quite symmetric (as in Fig. 2), with clearly related sequences and conformations in each blade, but for other cases each blade is different (e.g., 1GOTc).
Figure 2.
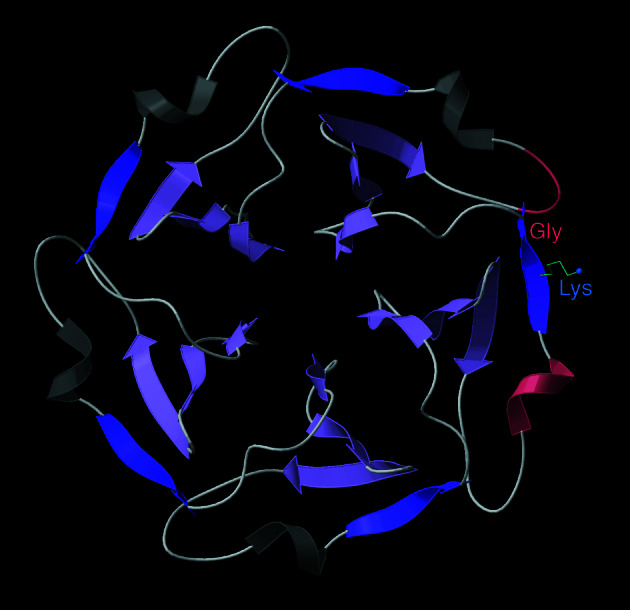
View down the approximate 5-fold axis of the β-propeller five structure of the 1TL2 tachylectin (28). β strands are lilac arrows, with edge strands of the five propeller “blades” in darker purple. For the edge strand on the right, the features preventing further β-sheet interactions are shown: a charged Lys, a Gly β bulge at the end of the strand, a nearby partially covering loop (pink), and a colinear helix (pink) that blocks the first β residue.
The β-propeller blades use a wide variety of outer edge-blocking strategies, but two are predominant, each of which occurs on about 30 of the 35 blades in our sample. One strategy is placement of a charged side chain on a low-curvature surface next to the edge strand, where it would be buried by most potential β-sheet associations (e.g., the Lys in Fig. 2). This side chain can be on the edge strand itself, on the next strand in if it is long enough to reach solvent in the isolated β-propeller, or can even be contributed by other neighboring structure. This last arrangement is especially prominent for the αβ-propeller, where the centers of all five edge strands are bracketed by a charged side chain from each of the surrounding helices. Such cases emphasize the fact that these negative design features cannot in general be fully analyzed just by looking at the sequence and conformation of the edge β strand itself.
The second prominent edge strategy in β propellers is to use a β bulge and/or a proline to disfavor further β interactions by providing both a very strong local twist and also a local outward protrusion. A β bulge is an irregular β H-bond pair that has two residues on the bulged strand opposite one on the other strand (16): it twists and protrudes to fit in the extra residue; for a Pro, the twist is needed to bring φ near −70°, and the protrusion is the Pro ring itself. Both Pro and bulges are very rare in the interior of β-sheets, because it is nearly impossible to continue β H-bonding in both directions on the convex side of the β-strand disruption. This same property makes them not only tolerated but actually desirable on edge strands. Each of the five blade edges in tachylectin (Fig. 2) has both a charge and a β bulge. Nearly half of all blade edges are partly protected by loops, but the degree of coverage is generally much less than seen for β-helices. About one in three blade edge strands are short (three or four H-bonds), and about one in three start or end with a colinear helix which protrudes enough to block access to at least one peptide on the edge β strand.
Single β-Sheets.
Single, open β-sheets can be antiparallel or mixed and occur in both
all-β and α+β SCOP categories, usually with helices or loops on
only one side of the sheet but sometimes on both sides. The 34
single-sheet edges in our sample are very similar to the β-propeller
edges, with dominant blocking strategies of bulges and/or prolines (24
of 34) and charged side chains in the central portion (22 of 34). To
effectively block further β-sheet bonding on these single sheets
where there is often no clear direction that counts as “inward,”
16 of those 22 cases have charged side chains on both sides of the
strand, often very close in sequence. About  of the
single-sheet edges are at least partly covered by loops, and about
of the
single-sheet edges are at least partly covered by loops, and about
 are very short (two to four H-bonds). Often a short edge
strand is centered over a much longer adjacent strand, effectively
leaving only very short ends free on that lower strand as well.
are very short (two to four H-bonds). Often a short edge
strand is centered over a much longer adjacent strand, effectively
leaving only very short ends free on that lower strand as well.
β-Sandwiches.
The two-sheet, predominantly antiparallel βsandwich proteins are of special interest here because, with side-by-side pairs of edge β strands exposed, they seem especially susceptible to having main chain dominate an interaction with another β-sandwich. A few β-sandwich proteins do form dimers through edge-strand interactions, providing both direct structural information on such complexes and also control examples of edge strands that lack negative design features adequate to prevent association. β-sandwich folds are very common in natural proteins, they are often a target of protein design, and they form the basis for the generally favored model of β-strand arrangement in amyloid fibers.
For β-sandwich edges, the dominant blocking strategy is an inward-pointing charged side chain, located on the otherwise-hydrophobic strand side that faces the opposite β-sheet (used in 58 of 73 edge-pairs). Fig. 3a shows an example from Con A (1NLS), with a lysine near the center of one edge strand. Lysine, with its flexibility and favorable solvent interaction, occurs most often in this role, followed by Arg, Glu, and Asp in order of decreasing length, followed by His with its more titratable charge; both the order and magnitude of these preferences differ from overall occurrence frequencies. In the isolated β-sandwich such a side chain can form hydrophobic sheet-packing interactions with its aliphatic portion and still expose its charged end. In any edge-to-edge aggregation, however, that charge would necessarily be buried unless it is outside the region that forms intersubunit β H-bonding. This strategy is the minimal change that protects edge strands, involving just one side chain with no distortion of either backbone conformation or regular β-sheet pattern. An inward-pointing charge near the center of one strand protects both paired edge strands from β-sandwich aggregation.
Figure 3.
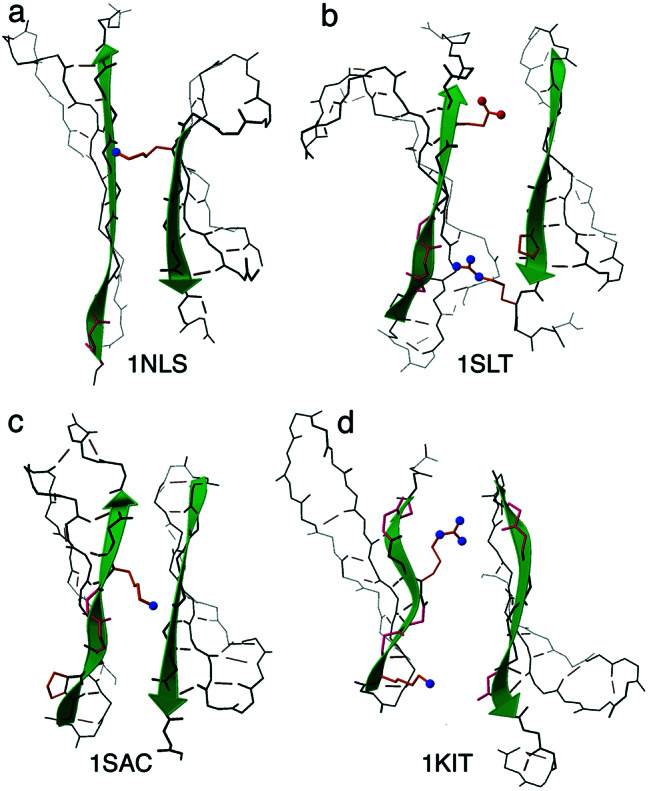
(a–d) A set of equivalent, paired sheet edges from four different β-sandwich protein families having the lectin/glucanase fold. Edge β strands are shown as sea green arrows, β bulges in hot pink, and inward-pointing charged side chains with red or blue balls for their O or N atoms. All examples have at least one bulge and at least one inward-pointing charge, although the positioning and the overall shapes vary. From PDB files 1NLS (29), 1SLT (30), 1SAC (31), and 1KIT (32).
The second major edge strategy in β-sandwiches is the familiar one of a β bulge and/or proline (more bulges, by 46 vs. 30 cases). These protect by forming a protrusion that locally blocks access, by distorting the relative geometry of adjacent peptide groups, and by strongly accentuating the normal right-handed β-strand twist. The resulting twist is near the upper limit of what occurs in β-sheet (usually for isolated two-stranded ribbons); it is completely compatible with regular β H-bonding on the concave side (away from the Pro ring or the extra bulge residue), but it is not compatible with continued β H-bonding across the convex side.
Fig. 3 documents the robustness of edge strategies for four different families in the lectin/glucanase fold. In these, and in the examples from all nine similar families, there is always at least one β bulge and at least one inward-pointing charge. This is not sequence conservation, because the positioning varies widely, but the conservation of strategy speaks to the need for such negative-design features and also to the desirability of their redundancy. Within one homologous family, there is extensive but usually not family-wide conservation of specific protection features; however, when such a feature is lost, its job is taken over by some other protective feature nearby.
Surprisingly, β-sandwich edges can also adopt a reversed twist to discourage β partners. Usually this is accomplished by having a Gly in Lβ conformation at what would normally be an inward-facing position on the edge strand, but still with fully regular β H-bonding. φ,ψ values in the Lβ region (extended, but with +φ) produce an opposite “pleat” to that of normal β, so that instead of the pleats alternating, three residues in a row pleat in the same direction and the strand both curves convexly around the sandwich core and also has the “wrong” left-handed twist. Fig. 4 shows such an example from the 1IGD protein G domain. We found two such cases in single-sheet proteins, one in a β-propeller and ten in β-sandwiches. The 1D2S sandwich is interesting because it manages to produce a convex curve and inverted twist with no Gly, using a string of somewhat unusual φ,ψ's below and left of the φ,ψ plot diagonal and therefore left-twisting (17), but no Lβ conformation.
Figure 4.
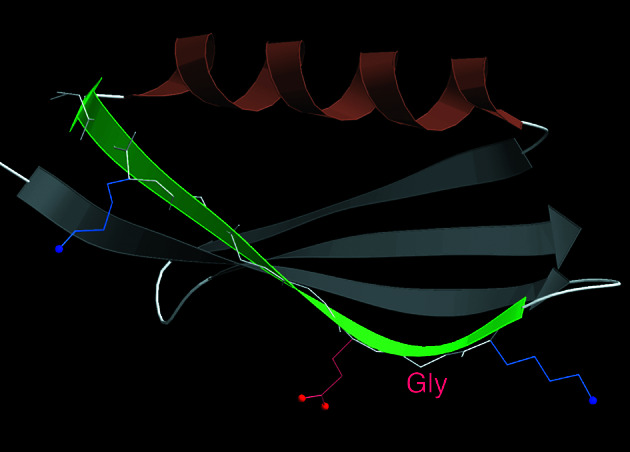
A β-sheet edge strand (green arrow) that uses an Lβ Gly (labeled) to produce a large bend and a negative backbone twist for that end of the strand. The β H-bonding of that strand is completely regular, but the “pleat” is convex outward for three residues in a row (Lys, Gly, Gln). From the 1IGD single-sheet structure of an IGG-binding domain of Streptococcal protein G (33).
β-sandwich proteins make use of all identified edge protection mechanisms, as well as their preferred inward-pointing charge and bulge/Pro strategies. Our 73 β-sandwich edge pairs included 35 covered by loops, 14 rolled-in edges, 12 very short strands, 10 sheet switches (see below), 9 strategically placed charges from neighboring structure, 3 half-barrels, and 2 γ′ conformations that locally tie up two of the H-bonding groups. No nonmultimer edge pair had fewer than two of these strategies. The sheet switch is familiar from Ig variable domains and blue copper proteins, where a strand starts as the edge strand of one sheet, then crosses in an abrupt kink to finish as the edge strand of the opposite sheet. From the perspective of avoiding β aggregation this is a useful arrangement, because it effectively breaks up both exposed edges.
Fold Summary.
Some fold types have unique β-edge protection strategies: β-barrels and the inner edges of β-propellers are protected by the inherent shape of the fold, whereas β-helices cover their ends with loops. Other β-sheet edges use a mixture of local protective features such as charges, β-bulges, prolines, covering loops, short strands, and Lβ glycines. β-sandwich edges particularly emphasize inward-pointing charged side chains.
β-Sheet Multimers.
Dimers, trimers, etc. formed through β H-bond contacts between β-fold subunits are a suitable control set for evaluating the significance of β edge-strand features seen in the isolated folds. Seven such cases were found in our sample: four β-sandwiches (1TTA, 1NLS, 1SLT, 1WAP), two single sheets (1E7X, 1FV1), and one β-barrel (1C4Q). They include five dimers with 2-fold contacts between equivalent strands, and also a 5-mer and an 11-mer that form rings by using contacts between two different strands one on each side of the subunit. The 1SLT dimer uses both strands of the sandwich end equally, but the other dimers have one strand with a dominant, regular contact while the second manages only a few H-bonds if any. Each 2-fold contact has from 8 to 12 total intersubunit β H-bonds, whereas the individual contacts of higher multimers have 4 to 6 β H-bonds.
Fig. 5a is an end view of the two edge β strands that will form the 1TTA transthyretin dimer contact, with a total of 17 available peptide groups and protective features only at the extreme ends. The long strand is dramatically more regular and open than any of the edge strands seen in isolated β-sheet folds. Fig. 5b shows the actual dimer contact of the longer strand, its six β H-bonds well offset from the protective charge. In the shorter strand contact (not shown), a protruding loop at one end allows only one H-bond at each end of the contact, whereas the center is wider apart and H-bonds through water molecules.
Figure 5.
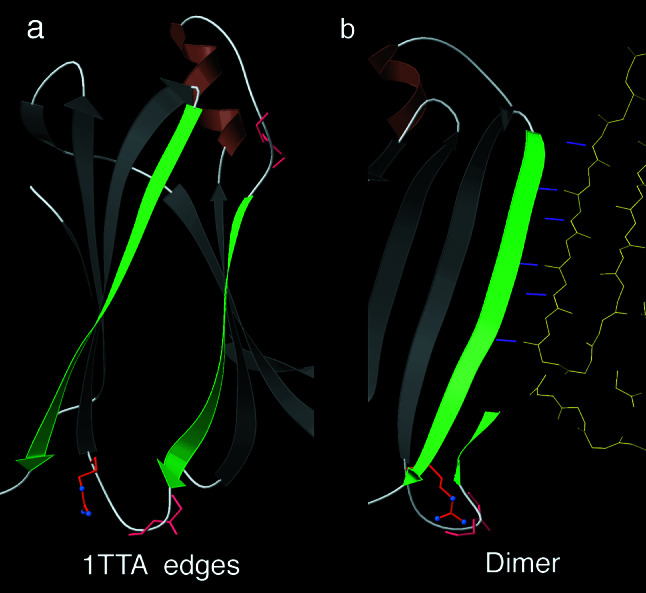
Edge β strands and dimer contacts for the 1TTA transthyretin β-sandwich structure (34). (a) Edge strands (green arrows) in the monomer, with a protective charged Arg and two loops (main chain in pink) only at the extreme ends, and long central stretches of completely regular β strand, especially on the longer strand at left. (b) That longer edge strand in its dimer contact, with six intersubunit β H-bonds (view rotated 90° from a). The shorter edge strand contact (behind; not shown) fits less well, with one H-bond at each end and bridging waters in the center.
Indeed, these multimer β strands are very different from isolated edge strands. They average less than one protective feature per strand (compared with 2.5 per strand for comparable monomers), and those features are (with one exception) always at an extreme end of the regular strand. The one exception is for the MHC contact, which has a charge near its center; that charge turns out to be exposed in the peptide-binding site. Each multimer-contact β strand has between 5 and 14 consecutive peptide H-bonding groups facing outward in regular β conformation and thus available, whereas isolated β-edge strands have only between one and five such consecutive available groups. Three cases of edge strands with between five and ten available regular peptides (1ALY, 1PGS, 1CQ3) turned out to make irregular dimer or trimer contacts by using part of that available strand. In general, it therefore should be considered dangerous for β-sheet aggregation if an edge strand has as many as five or six consecutive unprotected peptide H-bonding groups facing outward in regular geometry.
Discussion
The general conclusion from this work is that to promote solubility over aggregation, the edge strands of β-sheet proteins are covered, made irregular, short, or otherwise unsuitable for further β interactions. This is a case of negative design, because those features do not in general improve the β-sheet structure, but are there to avoid an undesirable alternative structure. Usually several protective features are present at each edge of the β-sheet, presumably providing redundancy against mutations or conformational perturbations. There is redundancy, also, in protecting both ends, because a single available edge would only produce dimers and not larger, more damaging aggregates. Because structures are not rigid, conformational protection may sometimes fail, with a bulge disappearing, a shielding loop moving out of the way, or an Lβ glycine shifting back to normal β conformation. However, this is much less likely in a well folded protein than for isolated strands or hairpins, because the whole protein is optimized to fit the native irregularity.
These same features of β edge strands that help avoid aggregation also help distinguish edge from interior strands in the β-sheet. They should thus be useful in structure prediction, in understanding selectivity during β-sheet folding, and in specifying strand topology in designs of β structure. However, there is the complication that nearly all such strategies significantly lower the β propensity of the edge strand. The presence or absence of such features must therefore always be a compromise, for either natural or designed β-sheet proteins.
The oldest (18) and most enduring observation about β-sheet edge strands is that their sequences are more hydrophilic than those of interior strands. Could edge strands avoid aggregation simply by the hydrophilicity of their “hydrophobic,” inward side (the side toward other structure)? We see only very weak evidence for such an effect, because there are no otherwise-unprotected strands with their inward-pointing residues mainly polar. However, polarity must make a contribution and could well be decisive if the balance were marginal, such as for a small β-hairpin that is only folded part of the time.
Amyloid-fiber formation, in disease states or in the laboratory, involves some sort of edge-to-edge β-sheet aggregation into a long “cross-β” structure with the strands perpendicular to the fiber axis (2). Small β-sheet designs with regular edge strands often form fibers indistinguishable from amyloid (6, 19). Because the structures of amyloid and of the normally avoided β-protein aggregates are so similar, many of the same considerations apply but the kinetics differ. Amyloid requires long times, harsh conditions or mutations to form, in a process that involves some degree of refolding: uncovering interior β strands, unfolding protective edge features, or even changing α into β structure. It would be interesting to find out whether mutants with regularized edge strands aggregate rapidly, but that would not reproduce the usual mechanism of fiber formation.
The implications of the current work for protein design are clear. It appears that for β-sheet at least, our natural inclination to make the design quite perfect and regular is actually a mistake. Those messy and complex edges in natural proteins are there for good reason rather than just tolerated, and our designs should follow suit. Protein redesigns, where sequence is changed on a known backbone, work well because the irregularities are kept in the redesign. De novo designs typically aim for regularity and simplicity, both for ease of design and for clarity in what they can teach us; therefore, de novo β structure typically gets in trouble with insolubility and aggregation. However, those designs have worked well in the sense of teaching us about an important aspect of protein folding we might not have noticed otherwise.
One recent successful β-hairpin design, the “TrpZip” (20), is especially interesting because it is well ordered and monomeric without using any of the edge-protection strategies described above. It is based both on a specific β-hairpin from the protein G domain (at the rear in Fig. 4) and also on experimental studies of the stability of amino acid substitutions at wide-pair H-bond positions in such a hairpin (21); a Trp–Trp pair was by far the most stabilizing combination despite its vanishingly small occurrence in natural proteins. Fig. 6a shows the consensus NMR model of TrpZip4, which adds two pairs of adjacent Trp to the protein G hairpin. All-atom contacts (14) show that the structure is both well packed and well determined (compact but with no bad clashes). The Trp rings pack extensively both with the backbone and with each other, and adopt favorable rotamers (22). TrpZip4 is very strongly twisted (36° per residue) and highly bent (20° bend along the strand axis), whereas the 1IGD hairpin is unusually straight (only 11° twist and 7° bend) even for a multistranded β-sheet. The high twist and bend are constrained by the Trp–Trp pairs, as shown by trying to model them onto a low-twist hairpin; the Trp rings actually interpenetrate, as indicated by the red contact spikes in Fig. 6b, and can be relieved only by large φ,ψ changes toward the polyPro conformation used in the TrpZips.
Figure 6.
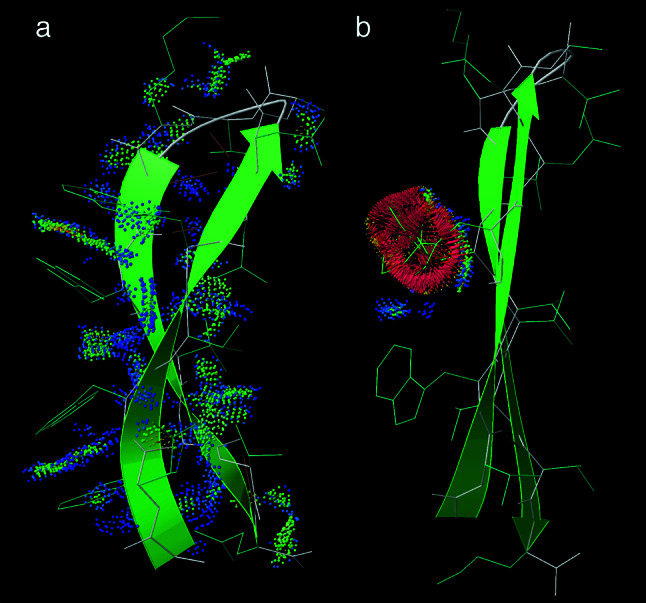
A comparison of the high-twist, designed 1HS0 TrpZip4 β-hairpin (20) with the 46–61 β-hairpin from the intact 1IGD protein G structure (33). (a) Model 1 of the TrpZip4 NMR structure, with β-strand ribbons to emphasize its high twist and bend, and with all-atom contacts shown as color-coded dots (14). The Trp–Trp pairs form good contacts (green dots) on the convex side of the β-hairpin. (b) The similar but low-twist β-hairpin from protein G, with a Trp–Trp pair modeled and optimized as well as possible without backbone motion. The red spikes of their all-atom contacts show their physically impossible interpenetration.
The TrpZip backbone conformation is a favorable one for two-strand β ribbons, which have long been known to twist and bend much more than larger β-sheets (23). However, most isolated β-hairpins are free to untwist to join a larger sheet, whereas the TrpZip could not untwist. Another unusual feature is that the TrpZip hydrophobic side chains are on the convex side of the β-hairpin bend, whereas other β-hairpins have their hydrophobics on the concave side (in the narrow-pair H-bond position) where they can form a more compact cluster (24). We believe the constrained high twist and bend are what keep TrpZips monomeric. Trp–Trp pairs are sure to be used in the future for designing unprecedentedly stable β-hairpins, but it should be kept in mind that they probably will not work in larger β-sheets except as protruding two-strand extensions.
There is another, very direct, method for achieving monomeric β-sheet designs, but it is applicable only for peptide synthesis and not for biological protein synthesis. Both Kelly (25) and Doig (26) have successfully used the methylation of exposed backbone NH groups in edge β-strands to prevent aggregation.
The results reported here enable a different approach to β-sheet design, of deliberately including edge-strand features that block aggregation. Table 1 summarizes the edge-strand protection features found, with their occurrence frequency and their suitability as deliberate design strategies. Most of the strategies used by natural proteins are beyond our current understanding to design de novo. Proline or β-bulge irregularities are designable (e.g., by offsetting the hydrophobic alternation and using good bulge residues), but they would require quite sophisticated modeling to propagate the backbone changes correctly. The inward-pointing charge strategy is the clear winner: it affects a large region with a single-residue change, it requires no backbone distortion, and it is very easy to design. Lys is usually the best choice, because it is the commonest charged residue and contributes the most to general solubility, as well as being used most often to protect the edge strands of natural proteins. The accompanying paper by Wang and Hecht (8) gives experimental confirmation of this proposal, by successfully using inward-pointing edge-strand lysines to transform a set of binary-patterned β-sandwich designs from large aggregates to soluble monomers.
Table 1.
Design strategies for β-sheet edges
| Common? | Designable? | |
|---|---|---|
| Continuous H-bonding | √√ | No |
| Covering loop | √√ | No |
| β bulge | √√ | ? |
| Proline | √√ | ? |
| Inward-pointing charge | √√ | Yes |
| Sheet edge rolls in | √ | No |
| Very short | √ | No |
| Very twisted | √ | Only for two-strand parts |
| LβGly bend, reverse twist | — | No |
| Switch between sheets | — | No |
Features marked with √√ are very common; √ are common; — are rare. For designability, ? means perhaps, subject to understanding effects on backbone conformation.
Acknowledgments
We thank Michael Hecht for sharing his protein design results with us, for suggesting that we expand our β-edge ideas into a detailed bioinformatic analysis, and for facilitating the side-by-side publication of both studies. This work was supported by National Institutes of Health Grant GM-15000.
References
- 1.Mattice W L. Annu Rev Biophys Biophys Chem. 1989;18:93–111. doi: 10.1146/annurev.bb.18.060189.000521. [DOI] [PubMed] [Google Scholar]
- 2.Sunde M, Blake C. Adv Prot Chem. 1997;50:123–159. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- 3.Mutter M. Trends Biochem Sci. 1988;13:260–265. doi: 10.1016/0968-0004(88)90159-4. [DOI] [PubMed] [Google Scholar]
- 4.Richardson J S, Richardson D C, Tweedy N B, Gernext K M, Quinn T P, Hecht M H, Erickson B W, Yan Y, McClain R D, Donlan M E, Surles M C. Biophys J. 1992;63:1186–1209. [PMC free article] [PubMed] [Google Scholar]
- 5.Pessi A, Bianchi E, Crameri A, Venturini S, Tramontano A, Sollazzo M. Nature (London) 1993;362:367–369. doi: 10.1038/362367a0. [DOI] [PubMed] [Google Scholar]
- 6.West M W, Wang W, Patterson J, Mancias J D, Beasley J R, Hecht M H. Proc Natl Acad Sci USA. 1999;96:11211–11216. doi: 10.1073/pnas.96.20.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hecht M H, Richardson J S, Richardson D C, Ogden R C. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Hecht M H. Proc Natl Acad Sci USA. 2002;99:2760–2765. doi: 10.1073/pnas.052706199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman H M, Westbrook J, Feng Z, Gilliland G, Bhat T N, Weissig H, Shindyalov I N, Bourne P E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murzin A, Brenner S E, Hubband T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 11.Richardson D C, Richardson J S. Protein Sci. 1992;1:3–9. doi: 10.1002/pro.5560010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merritt E A, Bacon D J. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 13.Word J M, Lovell S C, Richardson J S, Richardson D C. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- 14.Word J M, Lovell S C, LaBean T H, Taylor H C, Zalis M E, Presley B K, Richardson J S, Richardson D C. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 15.Word J M, Bateman R C, Jr, Presley B K, Lovell S C, Richardson D C. Protein Sci. 2000;9:2251–2259. doi: 10.1110/ps.9.11.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richardson J S, Getzoff E D, Richardson D C. Proc Natl Acad Sci USA. 1978;75:2574–2578. doi: 10.1073/pnas.75.6.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salemme F R. Prog Biophys Mol Biol. 1983;42:95–133. doi: 10.1016/0079-6107(83)90005-6. [DOI] [PubMed] [Google Scholar]
- 18.Sternberg M J E, Thornton J M. J Mol Biol. 1977;115:1–17. doi: 10.1016/0022-2836(77)90242-x. [DOI] [PubMed] [Google Scholar]
- 19.Aggeli A, Bell M, Boden N, Keen J N, Knowles P F, McLeish T C B, Pitkeathly M, Radford S E. Nature (London) 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 20.Cochran A G, Skelton N J, Starovasnik M A. Proc Natl Acad Sci USA. 2001;98:5578–5583. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell S J, Cochran A G. J Am Chem Soc. 2000;122:12600–12601. [Google Scholar]
- 22.Lovell S C, Word J M, Richardson J S, Richardson D C. Proteins Struct Funct Genet. 2000;40:389–408. [PubMed] [Google Scholar]
- 23.Richardson J S. Adv Prot Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 24.Richardson D C, Richardson J S. In: Prediction of Protein Structure and the Principles of Protein Conformation. Fasman G D, editor. New York: Plenum; 1989. pp. 1–98. [Google Scholar]
- 25.Nesloney C L, Kelly J W. J Am Chem Soc. 1996;118:5836–5845. [Google Scholar]
- 26.Doig A J. Chem. Commun. 1997. 2153–2154. [Google Scholar]
- 27.Iverson T M, Alber B E, Kisker C, Ferry J G, Rees D C. Biochemistry. 2000;39:9222–9231. doi: 10.1021/bi000204s. [DOI] [PubMed] [Google Scholar]
- 28.Beisel H G, Kawabata S, Iwanaga S, Huber R, Bode W. EMBO J. 1999;18:2313–2322. doi: 10.1093/emboj/18.9.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deacon A, Gleichmann T, Kalb A J, Price H, Raftery J, Bradbrook G, Yariv J, Helliwell J R. J Chem Soc Faraday Trans. 1997;93:4305–4312. [Google Scholar]
- 30.Liao D I, Kapadia G, Ahmed H, Vasta G R, Herzberg O. Proc Natl Acad Sci USA. 1994;91:1428–1432. doi: 10.1073/pnas.91.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley J, White H E, O'Hara B P, Oliva G, Srinivasan N, Tickle I J, Blundell T L, Pepys M B, Wood S P. Nature (London) 1994;367:338–345. doi: 10.1038/367338a0. [DOI] [PubMed] [Google Scholar]
- 32.Crennell S, Garman E, Laver G, Vimr E, Taylor G. Structure (London) 1994;2:535–544. doi: 10.1016/s0969-2126(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 33.Derrick J P, Wigley D B. J Mol Biol. 1994;243:906–918. doi: 10.1006/jmbi.1994.1691. [DOI] [PubMed] [Google Scholar]
- 34.Hamilton J A, Steinrauf L K, Braden B C, Liepnieks J, Benson M D, Holmgren G, Sandgren O, Steen L. J Biol Chem. 1993;268:2416–2424. [PubMed] [Google Scholar]