

Abstract
Amyloid fibrils are associated with a variety of neurodegenerative maladies including Alzheimer's disease and the prion diseases. The structures of amyloid fibrils are composed of β-strands oriented orthogonal to the fibril axis (“cross β” structure). We previously reported the design and characterization of a combinatorial library of de novo β-sheet proteins that self-assemble into fibrillar structures resembling amyloid. The libraries were designed by using a “binary code” strategy, in which the locations of polar and nonpolar residues are specified explicitly, but the identities of these residues are not specified and are varied combinatorially. The initial libraries were designed to encode proteins containing amphiphilic β-strands separated by reverse turns. Each β-strand was designed to be seven residues long, with polar (○) and nonpolar (●) amino acids arranged with an alternating periodicity (○●○●○●○). The initial design specified the identical polar/nonpolar pattern for all of the β-strands; no strand was explicitly designated to form the edges of the resulting β-sheets. With all β-strands preferring to occupy interior (as opposed to edge) locations, intermolecular oligomerization was favored, and the proteins assembled into amyloid-like fibrils. To assess whether explicit design of edge-favoring strands might tip the balance in favor of monomeric β-sheet proteins, we have now redesigned the first and/or last β-strands of several sequences from the original library. In the redesigned β-strands, the binary pattern is changed from ○●○●○●○ to ○●○K○●○ (K denotes lysine). The presence of a lysine on the nonpolar face of a β-strand should disfavor fibrillar structures because such structures would bury an uncompensated charge. The nonpolar → lysine mutations, therefore, would be expected to favor monomeric structures in which the ○●○K○●○ sequences form edge strands with the charged lysine side chain accessible to solvent. To test this hypothesis, we constructed several second generation sequences in which the central nonpolar residue of either the N-terminal β-strand or the C-terminal β-strand (or both) is changed to lysine. Characterization of the redesigned proteins shows that they form monomeric β-sheet proteins.
We previously described the design and characterization of combinatorial libraries of de novo proteins (1–3). The design of these libraries is based on a strategy in which the binary patterning of polar and nonpolar amino acids is designed explicitly, but the exact identities of these residues are not specified, and are varied combinatorially. Combinatorial diversity is made possible by the organization of the genetic code: Five nonpolar amino acids (Met, Leu, Ile, Val, and Phe) are encoded by the degenerate codon NTN; and six polar amino acids (Lys, His, Glu, Gln, Asp, and Asn) are encoded by the degenerate codon VAN. (N represents the DNA bases A, G, C, or T; V represents A, G, or C.)
Binary patterned sequences capable of burying hydrophobic side-chains (and exposing hydrophilic ones), while simultaneously forming secondary structure, are designed by constraining the sequence periodicity of polar and nonpolar residues to match the inherent structural periodicity of the desired secondary structure. For α-helices, the structural periodicity is 3.6 residues per repeat, and so de novo sequences targeted to form amphiphilic α-helices are designed with a binary pattern that places a nonpolar residue (●) every three or four positions (e.g., ○●○○●●○○●○○●●○). Conversely, β-strands have a structural periodicity of two residues per repeat, and so sequences targeted to form amphiphilic β-strands are designed with an alternating pattern (e.g., ○●○●○●○).
By using the binary code strategy, we constructed several libraries of de novo proteins. Initial efforts focused on the design of four-helix bundles (1). Characterization of proteins from the α-helical libraries demonstrated that the binary code strategy can generate proteins that (i) fold into native-like stable structures, (ii) bind cofactor, (iii) bind small molecules, and (iv) catalyze reactions (refs. 4–8; Y. Wei, T. Liu, I. Pelzer, S. Sazinsky, D. A. Moffet, and M.H.H., unpublished results).
More recently we used the binary code strategy to design two libraries of β-sheet proteins (2, 3, 9, 10). Sequences in the first library contained six repeats of the amphiphilic β-strand pattern, ○●○●○●○. Sequences in the second library contained eight repeats. In both libraries, successive β-strand patterns were separated by sequences designed to form four-residue turns (9). Biophysical characterization of proteins from both libraries demonstrated that they indeed folded into β-sheet structures. These structures, however, were not monomeric; they self-assembled into large oligomers. Electron microscopy of the oligomers revealed fibrils resembling those found in amyloid (2, 9–11). Moreover, the de novo proteins bound Congo red, a dye used as a diagnostic test for amyloid structures.
The tendency of proteins from these initial β-sheet libraries to assemble into oligomeric structures is consistent with their design. Alternating sequences of polar and nonpolar residues are predisposed to form amphiphilic β-strands (12). Such strands would be expected to hydrogen bond to one another and assemble into a structure that maximizes hydrophobic burial. Such a structure is modeled in Fig. 1A, which shows a sequence from the six-stranded library modeled as a β-sandwich that oligomerizes into a long fibril with a continuous hydrophobic core. If the six-stranded β-sandwich did not oligomerize, but remained as an isolated monomer, then the hydrophobic side chains of the edge strands would be accessible to water (Fig 1B). By oligomerizing into fibrils, the protein can “hide” these nonpolar residues from solvent.
Figure 1.

(A) Schematic representation of a fibril formed by open ended oligomerization of a six-stranded β-sheet protein. β-strands are shown in green, and turns in silver. Polar side chains are shown in red and nonpolar side chains in yellow. The side chains depicted in this model (and in B–D) are those of protein 17-6 (see Fig. 1E for amino acid sequence). (B) Monomeric six-stranded β-sandwich (rotated 90° relative to A). In the monomer, the hydrophobic side chains (yellow) of the edge stands are accessible to water. For simplicity, flat β-sheets are depicted. In reality, a six-stranded β-sandwich would be twisted. (C) Monomeric six-stranded β-sandwich in which lysine side chains (shown in blue) are substituted in place of Ile-5 in the N-terminal β-strand and Val-60 in the C-terminal β-strand. In the monomeric structure, the charged ends of the lysine side chains on the edge strands are exposed to solvent. (D) Same as C, rotated by 90°. (E) Schematic representation of six-stranded (Upper) and eight-stranded (Lower) de novo β-sheet proteins. Residues mutated to lysine are highlighted in red. β-strands are shown as arrows. The alternating pattern of polar (○) and nonpolar (●) residues in the original binary code library is indicated with polar residues (Lys, His, Glu, Gln, Asp, and Asn) in white font on black background, and nonpolar residues (Leu, Ile, Val, and Phe) in black font on gray background. Combinatorial diversity is incorporated at positions marked ○, ●, and t (turn). (Combinatorial turn residues are Gly, Ser, Asp, and Asn—see refs. 2 and 9.) Fixed residues were incorporated at the termini and in some of the turns (2, 9). [Proteins in the original six-stranded library are denoted as “**-6”; those in the original eight-stranded library were denoted “**-8” (Ref. 9).]
After the observation of these fibrils, we became interested in the possibility of designing mutations that would favor monomeric β-sheet structures relative to the oligomeric fibrils. We reasoned that “negative design” (13, 14) might be used to disfavor the fibrillar structure and thereby favor monomers. In particular, based on a strategy suggested by Jane Richardson, we hypothesized that an amino acid substitution that buried a charge at the intermolecular interface would destabilize the fibril and prevent oligomerization.
The current work describes an explicit test of this hypothesis: We replaced the central nonpolar residue in the first and/or last β-strand of several of our de novo sequences. In the newly designed edge strands, the binary pattern was changed from ○●○●○●○ to ○●○K○●○ (where K denotes lysine). Several lysine-substituted proteins were prepared by directed mutagenesis, and the resulting proteins were purified. Biophysical characterization of the redesigned sequences demonstrates that they indeed form monomeric β-sheet proteins.
Materials and Methods
Construction of Mutant Proteins.
PCR was used to construct single site Npl → Lys mutations into either the N-terminal β-strand or the C-terminal β-strand (or both). Oligonucleotides were obtained from the Princeton Syn/Seq facility. PCR products were purified with a Qiagen (Chatsworth, CA) PCR purification kit, and were digested with restriction enzymes NcoI and BamHI (Promega). The doubly digested genes were purified using low melting agarose, and ligated into NcoI/BamHI cut plasmid pET3d. Plasmids were transformed into E. Coli BL21(DE3) for protein expression.
Protein Solubility in Vivo.
Fifty-milliliter cultures were grown to an OD600 of 0.6–0.7, induced with isopropyl-β-d-thiogalactoside (IPTG, final concentration 0.5 mM), and grown for an additional 3 h. Cells were harvested by centrifugation and resuspended in 5 ml of 10 mM Tris⋅HCl (pH 8.0), 100 mM NaCl. Lysozyme was added to a final concentration of 100 μg/ml. The resuspended cells were incubated at room temperature for 30 min. DNase I was then added to a final concentration of 2 μg/ml, and the resuspension was incubated at room temperature for another 30 min. The supernatant was separated from the pellet by centrifugation. A 10-μl portion of supernatant was mixed with 10 μl of 2× gel loading buffer, boiled for 5 min, and loaded onto an 8–25% acrylamide gradient PHAST gel (Amersham Pharmacia Biotech). Gels were stained with Coomassie Blue.
Protein Expression and Purification.
Cultures were grown to an OD600 of 0.6–0.7, induced with 0.5 mM IPTG, harvested by centrifugation, and subjected to three freeze-thaw cycles (freeze at −70°C, thaw at 10°C). The pellet was then resuspended in 20 ml of 10 mM Tris⋅HCl (pH 8.0). Lysozyme was added (final concentration 100 μg/ml), and the suspension was incubated at room temperature for 30 min. MgCl2 (final concentration of 1 mM), MnCl2 (final concentration of 0.1 mM), and DNase I (final concentration 2 μg/ml) were added, and the suspension was incubated at room temperature for another 30 min. Soluble and insoluble fractions were separated by centrifugation at 15,300 × g for 20 min. The supernatant was retained for purification of soluble proteins; otherwise, the pellet was used for protein purifications. Soluble proteins were purified on either a 20HQ (PerSeptive Biosystems, Framingham, MA) anion-exchange column (buffer A: 10 mM sodium phosphate, pH 8; buffer B, buffer A plus 1M NaCl) or a 20HS cation-exchange column (buffer A: 10 mM Na acetate, pH 4.5; buffer B, buffer A plus 1M NaCl). Insoluble proteins were purified from inclusion bodies as described (2). Purity was confirmed by SDS/PAGE.
Size-Exclusion Chromatography.
Size-exclusion chromatography (SEC) was preformed on a Sephadex 70 column (Amersham Pharmacia) in a buffer containing 10 mM sodium phosphate/100 mM NaCl, pH 7.8, except for sequence no. 4–6, which required a different buffer (pH 6.0, 10 mM sodium acetate buffer) to fold from inclusion bodies. In this case, the pH 6.0 buffer was used for SEC. In all cases, the concentration used for SEC is the same for the mutants and the parent. The concentrations for sequences 4-6, 17-6, and 45-8 were 120 μM, 95 μM, and 100 μM, respectively.
Circular Dichroism.
CD spectra were measured in the same buffer used for SEC. Spectra were measured at 4°C in a 1-mm cuvette by using a 62 DS spectropolarimeter (Aviv Associates, Lakewood, NJ). Protein concentrations were determined by quantitative amino acid analysis (Bioanalytical Center, Cornell University).
Modeling.
The schematic models shown in Fig. 1 were constructed using the insight/discover package of programs (Molecular Simulations, Waltham, MA). For the image shown in Fig. 1B, three seven-residue polyalanine β-strands were placed into the standard conformation for antiparallel β-structure. Constrained (to maintain a flat β-sheet) minimization brought the β-strands to a separation distance consistent with backbone hydrogen bonding. The three-stranded β-sheet was then replicated and rotated, and the replica was placed on top of the first sheet to generate a six-stranded β-sandwich. The β-strands were then joined by four-residue turns. Side chains for the sequence of protein 17-6 were appended onto the backbone, and the structure was minimized. In the simplified model shown in Fig. 1B, the β-sheets are flat (not twisted). In reality, a six-stranded β-sandwich would be twisted. The image shown in Fig. 1A was constructed by replicating the monomeric six-stranded β-sandwich to generate a continuous fibril.
Results
Design of Mutations.
The sequences mutated in the current study were chosen from two libraries of binary patterned β-sheet sequences described in earlier work (2, 9, 10). The first library was designed to form six amphiphilic β-strands punctuated by reverse turns, and the second library was designed to form eight amphiphilic β-strands punctuated by turns (9). A schematic representation of the designed binary patterns is shown in Fig. 1E. All of the β-strands were designed by using the identical seven-residue binary pattern, with polar and nonpolar residues arranged with the alternating periodicity that matches the structural repeat of amphiphilic β-strands (12, 15, 16). Combinatorial diversity was incorporated into the original β-sheet libraries by allowing polar residues to be His, Lys, Asn, Asp, Gln, or Glu, and nonpolar residues to be Leu, Ile, Val, or Phe. Proteins from both the six- and the eight-stranded libraries appeared as fibrils in electron microscope (EM) images (2, 10). High resolution structures of these de novo fibrillar structures are not available, nor are such structures available for natural amyloid. Therefore, we constructed a plausible model for the fibrillar structure of one of the six-stranded de novo sequences. The model in Fig. 1A shows six amphiphilic β-strands (green backbone) with polar side chains (red) exposed to solvent, and nonpolar side chains (yellow) buried in the hydrophobic core. Because all six β-strands share the identical polar/nonpolar pattern, they all have a hydrophobic face that requires burial. In the original collection, none of the β-strands were designed to form edge strands that would be accessible to solvent in a monomeric β-sandwich protein.
To encourage formation of edge strands, we designed new mutations
into representative sequences from both the six-stranded and the
eight-stranded libraries. In each sequence, the central residue on the
N-terminal strand or the C-terminal strand (or both) was changed from a
nonpolar amino acid to lysine. We chose lysine because this side chain
is charged at the terminus, but nonpolar near the backbone. On the
interior facing side of an edge β-strand, lysine could position its
methylene groups to protect the hydrophobic core, while its terminal
NH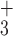 would be accessible to solvent. Accessibility,
however, would be possible only in monomeric structures (Fig. 1
C and D), and not in fibrillar structures (Fig.
1A).
would be accessible to solvent. Accessibility,
however, would be possible only in monomeric structures (Fig. 1
C and D), and not in fibrillar structures (Fig.
1A).
Two sequences from the six-stranded library (sequences 4-6 & 17-6) and one sequence from the 8-stranded library (sequence 45-8) were targeted for mutagenesis (Fig. 1E). In sequence 4-6, the mutations were Leu-5 → Lys, Phe-60 → Lys, and the double mutant. In 17-6, the mutations were Ile-5 → Lys, Val-60 → Lys, and the double mutant. In sequence 45-8, the mutations were Val-5 → Lys, Phe-82 → Lys, and the double mutant. In total, nine mutant proteins were constructed.
Solubility in Vivo of the Redesigned Proteins.
As described in earlier work (2, 9, 10), proteins from the original β-sheet library expressed in Escherichia coli as insoluble inclusion bodies. Although the structures of these inclusion bodies are not known, it seems likely that the intermolecular interactions that cause the purified proteins to associate into fibrils in vitro would also favor aggregation into inclusion bodies in vivo.
The effects of the newly designed nonpolar → lysine mutations on solubility in vivo are summarized in Table 1. In some cases, a single nonpolar → lysine substitution is sufficient to convert an insoluble inclusion body into a soluble protein; both 17-6 and 45-8 are rendered soluble by a single lysine incorporated into the nonpolar face of their N-terminal strands. Even for protein 4-6, where a single nonpolar → lysine mutation is not sufficient to solubilize the protein in vivo, the double mutant, Leu-5 → Lys + Phe-60 → Lys, reduced the percentage of protein in inclusion bodies by ≈50%. Although formation of inclusion bodies in vivo is difficult to interpret in terms of precise sequence/structure relationships, it is nonetheless clear from Table 1 that the redesigned sequence modifications significantly reduce the tendency of these de novo proteins to self-associate.
Table 1.
Solubility in vivo
| Protein | Original sequence | Lys in N-term. β-strand | Lys in C-term. β-strand | Double mutant |
|---|---|---|---|---|
| 4-6 | Inclusion body | Inclusion body | Inclusion body | 50% soluble |
| 50% incl. body | ||||
| 17-6 | Inclusion body | Soluble | Inclusion body | Soluble |
| 45-8 | Inclusion body | Soluble | Inclusion body | Soluble |
Oligomeric State of Redesigned Proteins.
The three proteins from the original library and the nine redesigned proteins were purified and dialyzed into a non-denaturing buffer (see Materials and Methods). At concentrations of ≈100 μM, the proteins from the original library, although visible as amyloid-like fibrils in EM images (2), remain soluble in native buffers (see Discussion). The nine redesigned proteins were also soluble in vitro. The oligomeric states of the proteins in native buffer were determined by SEC. For all three sequences, a single nonpolar → Lys mutation in the N-terminal β-strand was sufficient to disfavor high order oligomers (Table 2). For protein 17-6, the Ile-5 → Lys substitution converted the protein entirely to monomers. For the other two proteins, 4-6 and 45-8, the N-terminal mutation had a significant effect, and the double mutation (nonpolar → Lys in both the N- and C-terminal strands) converted the entire sample to monomers. Size exclusion data for the monomeric proteins are compared with one of the parental proteins in Fig. 2. The parental protein, 17-6, elutes in the void volume of the column, indicating on oligomeric state of >100. In contrast, the monomeric nonpolar → Lys mutants elute at approximately the same volume as plastocyanin, a natural β−sandwich of similar size. Thus, a single nonpolar → Lys mutation—or in some cases, two such mutations—is sufficient to prevent fibrils and thereby favor monomeric structures.
Table 2.
Oligomeric state of purified proteins
| Protein | Original sequence | Lys in N-term. β-strand | Lys in C-term. β-strand | Double mutant |
|---|---|---|---|---|
| 4-6 | Oligomer | Monomer & olig. | Oligomer | Monomer |
| 17-6 | Oligomer | Monomer | Oligomer | Monomer |
| 45-8 | Oligomer | Monomer & olig. | Oligomer | Monomer |
Oligomeric state was determined by SEC. Proteins eluting in the void volume of the column were designated as high order oligomers. Those eluting at volumes similar to that of plastocyanin were designated as monomers. (Poplar plastocyanin is a 100-residue β-sheet protein.) Examples of chromatograms are shown in Fig. 2.
Figure 2.

Size exclusion chromatography. Protein 17-6 from the original library is soluble and elutes in the excluded volume of the column, consistent with an oligomerization state of >100. In contrast, the newly designed nonpolar → lysine mutants elute at times similar to that of plastocyanin, a monomeric β-sheet protein with a mass of 10.6 kDa.
β-Sheet Secondary Structure of the Redesigned Proteins.
The rationale of our design was that introduction of a lysine onto the hydrophobic face of a β-strand would favor monomers by forcing the mutated β-strand to be a solvent-exposed edge in the β-sheet. Implicit in this design was the assumption that the mutated proteins would retain their β-sheet structures. To assess the veracity of this assumption, we measured the CD spectra of the mutant proteins. CD spectra of a monomeric variant of each of the three test proteins are shown in Fig. 3. The spectra of all proteins display a single minimum at ≈217 nm, demonstrating that the mutated sequences maintain β-structure. As summarized in Table 3, all six of the single mutants form β-sheet structures. In two of three cases, even the double mutants maintained β-structure (Table 3).
Figure 3.

Circular dichroism spectra. A single minimum at ≈217 nm indicates β-sheet secondary structure. (Upper) The CD spectra of the newly designed nonpolar → lysine mutants of the six-stranded proteins 4-6 and 17-6. These spectra are similar to those of the parental proteins 4-6 and 17-6 (2). (Lower) Comparison between the CD spectrum of the eight-stranded protein 45-8 and that of the newly designed nonpolar → lysine mutants of this sequence. The spectra are similar, indicating that, although the mutations prevent oligomerization, they do not prevent formation of β-sheet secondary structure.
Table 3.
Secondary structure determined by circular dichroism spectroscopy
| Protein | Original sequence | Lys. in N-term. β-strand | Lys in C-term. β-strand | Double mutant |
|---|---|---|---|---|
| 4-6 | β | β | β | β |
| 17-6 | β | β | β | Disordered |
| 45-8 | β | β | β | β |
A single minimum at 217 nm indicates β-sheet secondary structure. Sample spectra are shown in Fig. 3.
Monomeric β-Sheet Proteins.
For all three test proteins, we were able to isolate a second generation protein that is monomeric and retains β-sheet structure. [In one case (sequence 17-6) a single mutation was sufficient to yield a fully monomeric sample; in the other cases (sequences 4-6 and 45-8), two mutations were required.] The sequences of the monomeric β-sheet proteins are [17-6 + Ile-5 → Lys], [4-6 + Leu-5 → Lys + Phe-60 → Lys], and [45-8 + Val-5 → Lys + Phe-82 → Lys]. At moderate concentrations, these sequences neither aggregated into insoluble precipitates, nor assembled into soluble oligomers. This finding stands in marked contrast to the parental sequences from which they were derived, which (at the same concentrations) assembled into high order oligomers that eluted in the void volume of a SEC column, and appeared in EM images as amyloid-like fibrils. (Because the three second generation proteins showed no evidence of assembly—either as insoluble aggregates or as soluble oligomers—they were not analyzed by EM.) Relative to their parental sequences, the propensities of the newly designed proteins to assemble into amyloid-like fibrils are reduced substantially.
Discussion
Binary Patterning in Protein Design.
Binary patterning can be used to design either α-helical or β-sheet libraries. At first consideration, it would seem that design strategies for these two structural types would be similar: One must simply ensure that the periodicity of polar and nonpolar residues in the designed sequences matches the structural periodicity of the desired secondary structure. Construction and characterization of several binary patterned libraries has—to some extent—supported this expectation: Libraries based on the α-helical binary pattern (○●○○●●○○●○○●●○) indeed yield α-helical proteins; and libraries based on the β-sheet pattern (○●○●○●○) indeed yield β-sheet proteins (1, 2, 6). However, the properties of proteins from the two types of libraries differ dramatically: The α-helical proteins fold intramolecularly into monomeric (or occasionally dimeric) structures. In contrast, the β-sheet proteins assemble intermolecularly into large oligomeric structures resembling amyloid fibrils. These findings demonstrated that the binary code strategy cannot be applied in the identical way for the two different structural types.
It is not surprising that binary patterned libraries of β-strands favor intermolecular aggregation over intramolecular folding. Indeed, the tendency of de novo β-strands to aggregate is probably the main reason that, with a few exceptions (17–23), most successful protein design projects have focused on all α structures (23). The contrast between α and β structures stems from the fundamental difference in the hydrogen bonding patterns of the two types of secondary structures (15). In the α-helix, backbone hydrogen bonding is intra-segmental. It connects the C⩵O of residue i to the N—H of residue i + 4. Thus, an α-helix can satisfy most of its backbone hydrogen bonding requirements without help from a partner. Hence, the α-helix is relatively self-contained, and open-ended uncontrolled aggregation of designed sequences typically is not a serious problem.
The situation for β-strands is quite different. The C⩵O and N—H groups form hydrogen bonds to N—H and C⩵O groups on neighboring strands. A β-strand going into the page (Fig. 1A) can form hydrogen bonds with neighboring β-strands on its right and left. Side chains point up and down, and are available to interact with neighbors above and below the strand. Thus, a typical β-strand can interact with neighbors in four directions (left, right, up, and down). Because of this neighborliness, β-strands are inherently “gregarious” and prone to self-assembly. This result led to several early reports of β-sheet designs that aggregated into large oligomeric structures (24–26).
Despite the inherent tendency of β-strands to seek neighbors, and despite the difficulties protein designers have had in preventing β-strand aggregation, nature has no trouble generating a wide range of monomeric β-sheet proteins. Evolution apparently has selected sequences that disfavor open-ended aggregation. How does nature avoid β-strand aggregation?
One strategy used by nature is the avoidance of alternating patterns. A recent survey of natural protein sequences revealed that alternating patterns of polar and nonpolar residues occur significantly less frequently than other binary patterns with similar compositions (27). For example, for seven-residue lengths, there are 35 different ways of arranging four polar (○) and three nonpolar (●) residues. Among these, the alternating pattern ○●○●○●○ ranks 35th. Similar results were found for “windows” shorter or longer than seven residues. Apparently, evolution has selected against sequences that have an inherent tendency to form deleterious aggregates.
A more structurally oriented analysis of how nature avoids β-strand aggregation was recently completed by Richardson and Richardson (28). They report that natural β-sheet proteins avoid aggregation by “negative design”: Edge strands in natural β−sheet proteins often have sequences that disfavor intermolecular aggregation. One example is the incorporation of a lysine side chain onto the otherwise hydrophobic face of an edge β-strand (14, 28). Presumably, polar side chains at these sites were favored by natural selection because proteins with nonpolar residues at these loci are prone to assemble into deleterious aggregates.
Explanations for why evolution favors one option relative to another are necessarily speculative; nonetheless, the relative merits of alternative outcomes can be compared by constructing artificial systems in the laboratory to mimic both the observed (evolutionarily selected) outcome, and the unobserved (presumably deleterious) outcome. One such test was our earlier experiments designing novel β-sheet proteins by using the ○●○●○●○ pattern disfavored by nature (2, 27). The non-biological proteins based on this pattern formed amyloid-like fibrils. In contrast, the second generation proteins described in the current study were explicitly redesigned to test whether a pattern found in the edge strands of naturally selected proteins could alter the oligomerization state of our de novo proteins. The results described herein demonstrate that incorporation of a lysine into the nonpolar face of a β-strand can indeed prevent aggregation and favor monomeric β-sheet proteins (Tables 1–3).
The image in Fig. 1A shows the first and last β-strands of the linear sequence as the edge strands of the structure. However, because none of the β-strands in the original library were explicitly designed to be edge strands, alternative structures could have been modeled equally well (e.g., a Greek key structure with the first and last strands as interior strands). In the second generation sequences, mutation of the ○●○●○●○ pattern to ○●○K○●○ explicitly marks the first and last β-strands as edge strands, and thereby disfavors open-ended aggregation. It would be interesting to see whether other β-strands (e.g., the second or third strands) could also be made into edge strands by nonpolar → polar mutations. Recent studies in which a library of random mutations was screened for monomeric structures suggest that this is indeed the case (unpublished results).
Comparison of de Novo Proteins and Natural Proteins.
The properties of proteins from our original library (2) resemble those of natural amyloid proteins in the following ways: (i) both assemble into large oligomers; (ii) both are dominated by β-sheet secondary structure; (iii) both form oligomers that appear in EM images as fibrils; and (iv) both bind the diagnostic dye, Congo red. In one significant respect, however, the de novo amyloid-like proteins differ from natural amyloid proteins: Whereas natural amyloid proteins are insoluble, the de novo binary code proteins formed soluble oligomers that elute as large species (molecular weight > 1 million) in the void volume of an SEC column. The solubility of these large oligomers presumably derives from the designed binary patterning, which specifies protein surfaces containing an abundance of polar residues and a total absence of nonpolar residues.
In contrast, the properties of the redesigned second generation proteins resemble those of natural globular β-sheet proteins: (i) both are soluble; (ii) both are monomeric; and (iii) both are dominated by β-sheet secondary structure. Moreover (iv), they do not oligomerize at moderate concentrations, and (v) they show no evidence of forming amyloid-like fibrillar structures.
At this point, however, we cannot say whether these de novo β-sheet proteins recapitulate fully the properties of natural proteins. Well-folded native proteins display thermodynamic and structural properties that distinguish them from molten globule folding intermediates (29). Thermodynamically, native proteins differ from molten globules by undergoing cooperative thermal denaturations with relatively large enthalpy changes. We measured the thermal denaturations of the monomeric β-sheet proteins, and they had denaturation midpoints between 45°C and 55°C. However, because the denaturations were irreversible, thermodynamic properties could not be determined from these experiments. [Chemically induced denaturation can also be monitored. However, we have found that most de novo sequences yield cooperative chemical denaturation profiles—whether they are native-like or molten globule-like (6, 30). Therefore, chemically induced denaturation is not a stringent method for determining whether a novel protein is native-like.] Native proteins can also be distinguished from molten globules by structural properties. Native proteins tend to form more-or-less rigid structures with unique side chain interactions, whereas molten globules tend to form dynamic structures capable of sampling multiple packing arrangements. These dynamic properties can be assessed by a variety of NMR experiments (31). Unfortunately, however, at conditions required for these experiments—high protein concentration and low salt—our de novo proteins are not soluble. Therefore, at this point, we cannot assess whether our monomeric β-sheet proteins are more similar to molten globules or native-like structures. Future work will focus on incorporating additional amino acid substitutions to further enhance the solubility of these proteins and thereby facilitate more detailed studies.
Acknowledgments
The strategy of using nonpolar → lysine mutations to disfavor aggregation was based on discussions with Jane Richardson. This work was supported by the Biological Sciences Directorate of the Army Research Office.
Abbreviations
- SEC
size-exclusion chromatography
- EM
electron microscope
References
- 1.Kamtekar S, Schiffer J M, Xiong H, Babik J M, Hecht M H. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 2.West M W, Wang W, Patterson J, Mancias J D, Beasley J R, Hecht M H. Proc Natl Acad Sci USA. 1999;96:11211–11216. doi: 10.1073/pnas.96.20.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu G, Wang W, Groves J T, Hecht M H. Proc Natl Acad Sci USA. 2001;98:3652–3657. doi: 10.1073/pnas.071400098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rojas N R, Kamtekar S, Simons C T, McLean J E, Vogel K M, Spiro T G, Farid R S, Hecht M H. Protein Sci. 1997;6:2512–2524. doi: 10.1002/pro.5560061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenbaum D M, Roy S, Hecht M H. J Am Chem Soc. 1999;121:9509–9513. [Google Scholar]
- 6.Roy S, Hecht M H. Biochemistry. 2000;39:4603–4607. doi: 10.1021/bi992328e. [DOI] [PubMed] [Google Scholar]
- 7.Moffet D A, Certain L K, Smith A J, Kessel A J, Beckwith K A, Hecht M H. J Am Chem Soc. 2000;122:7612–7613. [Google Scholar]
- 8.Moffet D A, Case M A, House J C, Vogel K, Williams R D, Spiro T G, McLendon G L, Hecht M H. J Am Chem Soc. 2001;123:2109–2115. doi: 10.1021/ja0036007. [DOI] [PubMed] [Google Scholar]
- 9.West M W. Ph.D. dissertation. Princeton: Princeton Univ.; 1997. [Google Scholar]
- 10.Wang W. Ph.D. dissertation. Princeton: Princeton Univ.; 2001. [Google Scholar]
- 11.Sunde M, Blake C. Adv Protein Chem. 1997;50:123–159. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- 12.Xiong H, Buckwalter B L, Shieh H M, Hecht M H. Proc Natl Acad Sci USA. 1995;92:6349–6353. doi: 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hecht M H, Richardson J S, Richardson D C, Ogden R C. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 14.Richardson J S, Richardson D C, Tweedy N B, Gernert K M, Quinn T P, Hecht M H, Erickson B W, Yan Y, McClain R D, Donlan M E, et al. Biophys J. 1992;63:1185–1209. [PMC free article] [PubMed] [Google Scholar]
- 15.Creighton T E. Proteins: Structures and Molecular Properties. 2nd Ed. New York: Freeman; 1993. [Google Scholar]
- 16.West M W, Hecht M H. Protein Sci. 1995;4:2032–2039. doi: 10.1002/pro.5560041008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kortemme T, Ramirez-Alvarado M, Serrano L. Science. 1998;281:253–256. doi: 10.1126/science.281.5374.253. [DOI] [PubMed] [Google Scholar]
- 18.De Alba E, Santoro J, Rico M, Jimenez M A. Protein Sci. 1999;8:854–865. doi: 10.1110/ps.8.4.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharman G J, Searles M S. J Am Chem Soc. 1998;120:5291–5300. [Google Scholar]
- 20.Schenck H L, Gellman S H. J Am Chem Soc. 1998;120:4869–4870. [Google Scholar]
- 21.Dahiyat B I, Mayo S L. Science. 1997;278:82–86. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 22.Struthers M D, Cheng R P, Imperiali B. Science. 1996;271:342–345. doi: 10.1126/science.271.5247.342. [DOI] [PubMed] [Google Scholar]
- 23.Degrado W F, editor. Chem Rev (Protein Design) 2001;101:3025–3232. doi: 10.1021/cr000663z. [DOI] [PubMed] [Google Scholar]
- 24.Osterman D G, Kaiser E T. J Cell Biochem. 1985;29:57–72. doi: 10.1002/jcb.240290202. [DOI] [PubMed] [Google Scholar]
- 25.Richardson J S, Richardson D C. In: Protein Engineering. Oxander D L, Fox C F, editors. New York: Liss; 1987. pp. 149–163. [Google Scholar]
- 26.Zhang S, Holmes T, Lockshin C, Rich A. Proc Natl Acad Sci USA. 1993;90:3334–3338. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broome B M, Hecht M H. J Mol Biol. 2000;296:961–968. doi: 10.1006/jmbi.2000.3514. [DOI] [PubMed] [Google Scholar]
- 28.Richardson J S, Richardson D C. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Betz S F, Raleigh D P, DeGrado W F. Curr Opin Struct Biol. 1993;3:601–610. [Google Scholar]
- 30.Roy S. Ph.D. dissertation. Princeton: Princeton Univ.; 1998. [Google Scholar]
- 31.Walsh S T R, Lee A L, DeGrado W F, Wand A J. Biochemistry. 2001;40:9560–9569. doi: 10.1021/bi0105274. [DOI] [PubMed] [Google Scholar]