

Abstract
CueO (YacK), a multicopper oxidase, is part of the copper-regulatory cue operon in Escherichia coli. The crystal structure of CueO has been determined to 1.4-Å resolution by using multiple anomalous dispersion phasing and an automated building procedure that yielded a nearly complete model without manual intervention. This is the highest resolution multicopper oxidase structure yet determined and provides a particularly clear view of the four coppers at the catalytic center. The overall structure is similar to those of laccase and ascorbate oxidase, but contains an extra 42-residue insert in domain 3 that includes 14 methionines, nine of which lie in a helix that covers the entrance to the type I (T1, blue) copper site. The trinuclear copper cluster has a conformation not previously seen: the Cu-O-Cu binuclear species is nearly linear (Cu-O-Cu bond angle = 170°) and the third (type II) copper lies only 3.1 Å from the bridging oxygen. CueO activity was maximal at pH 6.5 and in the presence of >100 μM Cu(II). Measurements of intermolecular and intramolecular electron transfer with laser flash photolysis in the absence of Cu(II) show that, in addition to the normal reduction of the T1 copper, which occurs with a slow rate (k = 4 × 107 M−1⋅s−1), a second electron transfer process occurs to an unknown site, possibly the trinuclear cluster, with k = 9 × 107 M−1⋅s−1, followed by a slow intramolecular electron transfer to T1 copper (k ∼10 s−1). These results suggest the methionine-rich helix blocks access to the T1 site in the absence of excess copper.
Multicopper oxidases (MCOs) are prominent in all kingdoms and play a critical role in iron metabolism and copper homeostasis. In mammals, ceruloplasmin functions as a ferroxidase, catalyzing the conversion of Fe(II) to Fe(III), and is the major copper-containing protein in plasma (reviewed in refs. 1 and 2). Animals deficient in ceruloplasmin are anemic, presumably because of the loss of ferroxidase activity. Copper is both essential and toxic, because of metal-catalyzed protein oxidation, and improper copper metabolism in humans is linked to several diseases, including Menkes and Wilson diseases (3), Alzheimer's disease, and other aging disorders (4). In yeast, the MCO Fet3p has ferroxidase activity and is required for iron uptake through generation of ferric iron, the form thought to be used by the yeast iron transporter Ftr1p (5, 6). In plants, ascorbate oxidase (AO) and laccases (also a fungal enzyme) oxidize various phenolic substrates that, in the case of laccase, lead to protective polymers (7).
In bacteria, the genes and proteins necessary for copper homeostasis are just now coming to light. Studies of copper toxicity in Escherichia coli have unveiled two regulatory pathways, the cus system (for Cu-sensing) and cue system (for Cu-efflux) (8). The cus operon codes for three proteins that form a multiunit transport complex that spans the periplasmic space and links the inner and outer membranes. The complex is proposed to function in copper efflux (8, 9) and is most active under anaerobic growth conditions (8). The cue operon codes for two proteins, CueO, a MCO residing in the periplasmic space (8, 10, 11), and CopA, a copper efflux P-type ATPase related to the proteins that, when dysfunctional, cause Menkes and Wilson diseases (12). The Wilson disease ATPase transports copper into the hepatocyte secretory pathway for incorporation into ceruloplasmin. Thus, the cue pathway in bacteria may be functionally similar to both copper and iron homeostasis activities in humans.
MCOs couple four one-electron substrate oxidation steps to the four-electron reduction of dioxygen to water (2). This reaction has been best studied in the proteins laccase (E.C. 1.10.3.2), AO (E.C.1.10.3.3), and ceruloplasmin (E.C. 1.16.3.1). MCOs typically contain four copper atoms, one type I (T1) or “blue” copper site, and three other atoms in a trinuclear cluster consisting of one type II (T2) or “normal” copper, and two type III (T3) or “binuclear” coppers (2). Molecular oxygen binds at the trinuclear site and is reduced to water through four single-electron transfer steps from the T1 site, located about 13 Å away. Some MCOs, such as laccase, have little specificity and can oxidize a large variety of substrates, whereas others, such as AO, are more specific.
The crystal structures of three enzymes in this family, laccase (13), AO (14), ceruloplasmin (15), and a fourth enzyme containing a T1 copper and a second copper center, nitrite reductase (16), have been determined at resolutions ranging from 1.9 to 3.1 Å. In laccase and AO, each polypeptide chain adopts a three-domain structure with the trinuclear copper center located in the interior of the protein between domains I and III. The T1 copper center is in domain III and somewhat buried. There is a shallow depression at the protein surface where substrates can approach the T1 copper site and where ligand discrimination occurs in AO. The T1 copper center and the trinuclear copper center are connected by the amino acid sequence HCH, where the two histidine residues ligate the two T3 copper atoms of the trinuclear cluster and the cysteine is a ligand of the T1 copper.
CueO (formerly YacK), identified as a 53.4-kDa homologue of laccase (17), has been shown to oxidize a wide variety of substrates including 2,6-dimethyoxyphenol (DMP), enterobactin (a catecholate siderophore produced by E. coli), and ferrous iron (10, 11), and depends on oxygen for activity. Intriguingly, very little oxidase activity occurs in the absence of excess copper. The mechanism by which CueO protects against copper stress in cells is not fully understood, but is unlikely to be solely because of copper binding (8, 9).
An unusual feature of the CueO sequence is a region of ≈45 aa at the beginning of domain III (355–400) that includes 14 methionines and could provide binding sites for exogenous copper ions. A similar methionine-rich region also is seen in PcoA and CopA (Pseudomonas syringae), two proteins also implicated in copper tolerance, leading to the suggestion that this methionine-rich region is important for copper tolerance in bacteria (18). Here, we report the crystal structure of CueO, which shows the methionine-rich region to be largely helical and to cover the substrate-binding region. We also report both steady-state oxidation and transient intramolecular electron transfer kinetics. The CueO structure, at 1.4 Å, is the highest resolution MCO structure determined for any species. The kinetic studies support a model where excess copper alters access to the T1 site and suggests that intramolecular electron transfer between the trinuclear cluster and the T1 site is about 16-fold slower than in AO.
Materials and Methods
CueO Preparations.
CueO protein was purified from an overproducing E. coli strain, as described (10). The protein was expressed with its 28-aa N-terminal signal for periplasmic translocation, which is removed during export through the TAT translocation pathway (10, 11, 19), and with a C-terminal fusion of the Strep–TagII epitope (SAWSHPNFEK) for purification, which was left intact. Protein quality was assayed by PAGE. Protein concentration was determined spectroscopically by using ɛ280 = 63,036 M−1. All spectroscopic measurements were performed on a Varian Cary 300.
Activity and Steady-State Kinetics.
DMP was used as a model substrate to assay the oxidase activity of CueO, as described (10). Oxidase activity was monitored by the addition of purified protein (250 nM, final concentration) to a reaction mixture containing DMP (2 mM) in a 1-ml volume of 0.1 M sodium phosphate or sodium acetate buffer and 250 μM CuCl2, except where copper concentration was varied. Oxidation of DMP was followed by an increase of absorbance at 468 nm. DMP autooxidation occurs at alkaline pH or at higher concentrations of CuCl2 and was measured in the absence of protein and subtracted from the total oxidation rate.
Electron Transfer Kinetics.
Laser flash photolysis experiments were performed as described (20). The reaction solution contained 100 μM 5-deazariboflavin in 100 mM sodium phosphate buffer (pH = 8.0). Semicarbazide (1 mM) was also present as a sacrificial electron donor to the flavin triplet state generated by the laser flash. Solutions were made anaerobic by bubbling with O2-sparged Ar for 45–60 min before the addition of a small aliquot of CueO. Trace amounts of oxygen incorporated in the solution by protein addition were removed by blowing Ar across the surface. Generation of the neutral semiquinone of 5-deazariboflavin (5-dRFH⋅) was accomplished by using a ns flash from a N2-pumped dye laser (emission wavelength = 395 nm). Kinetic traces were analyzed by using kinfit (OLIS, Jefferson, GA).
Crystallization and Data Collection.
Monoclinic crystals of CueO grew as rectangular blue plates by the hanging drop vapor diffusion method from 20% PEG 4000, 200 mM ammonium acetate, 100 mM sodium acetate, pH 4.6 at room temperature. The crystals belong to space group P21 with unit cell dimensions a = 49.8 Å, b = 90.5 Å, c = 53.1 Å, β = 102.9°, and one molecule in the asymmetric unit. Before data collection, a 0.4 × 0.3 × 0.05-mm crystal was transferred to 50% PEG 4000 and frozen in liquid nitrogen. The native data and three multiple anomalous dispersion data sets at the copper K absorption edge were collected at the Stanford Synchrotron Radiation Laboratory (Stanford, CA), beam line 9–2, at 100 K temperature. Data were processed with D*TREK (21) and scaled with scaleit (Table 1).
Table 1.
X-ray data measurement and phase determination
| Native | Peak | Remote | Inflection | |
|---|---|---|---|---|
| Data measurement | ||||
| Wavelength (Å) | 1.0000 | 1.3772 | 1.1922 | 1.3804 |
| Resolution (Å) | 26–1.40 | 26–2.14 | 26–2.14 | 26–2.14 |
| Observed reflections | 733,346 | 91,035 | 89,158 | 91,943 |
| Unique reflections | 88,826 | 24,997 | 24,462 | 25,191 |
| Completeness (%)* | 98.5/87.1 | 97.2/99.0 | 96.2/99.6 | 97.6/98.9 |
| Rsym* | 0.064/0.280 | 0.024/0.039 | 0.024/0.041 | 0.025/0.044 |
| I/σ(I)* | 7.0/2.3 | 19.0/13.6 | 16.5/12.2 | 18.5/12.4 |
| Phasing | ||||
| Phasing power† | 0.8/0.53 | 1.29/1.00 | ||
| Rcullis† | 0.88/0.88 | 0.76/0.67 | ||
| Rcullis anomalous | 0.76 | 0.82 |
Overall/outermost shell.
Acentric/centric.
Structure Determination and Refinement.
A consistent solution for four copper sites was obtained from anomalous and dispersive Patterson syntheses. Refinement of the copper sites was done with mlphare, yielding phases to 2.2 Å and a mean figure of merit of 0.55. After solvent flattening with dm, the mean figure of merit increased to 0.76, and the resulting electron density map was readily interpretable.
The initial protein model was built by using ARP/WARP (22). Data to a resolution of 1.5 Å were included in the procedure, and copper atoms were included at the positions located from the Patterson map. After 300 refinement cycles and 30 building cycles, 443 protein residues had been found and placed in six chains (CueO is monomeric). The side chains built by the automatic procedure in ARP/WARP had a confidence factor of 0.91. Manual intervention was required at this point and four of five missing loops were built into 2 Fo − Fc electron density maps using o (23). This model, which also contained nearly all of the ordered water molecules, was input to shelx (24), and initial refinement resulted in Rwork = 0.19 and Rfree = 0.23 (5% of reflections set aside) for all data to 1.5 Å. After rebuilding with o and refinement with shelx, using all data between 30 and 1.4 Å, Rwork = 0.185 and Rfree = 0.221. The final model is lacking residues 29–30 at the N terminus (numbering starts with the signal peptide, residues 1–28); residues 380–403, which lie in an apparently disordered loop; and the C-terminal purification tag (residues 517–526). One cis peptide bond exists, at Pro-309. The final model displayed excellent geometry (rms deviation bond length = 0.01 Å, rms deviation bond angle = 0.026 Å, 88% residues with most favorable phi/psi angles, no residues with disallowed phi/psi angles). All programs not otherwise referenced are part of the CCP4 package (25).
Results
Steady-State Kinetics.
Recombinant CueO required excess Cu(II) for activity, as reported (10, 11). Maximal activity was found for CuCl2 concentrations in the range of 0.1 to 1 mM, regardless of the protein concentration, whereas at 10 μM, only 10% of the maximal activity was observed. At concentrations greater than 1 mM, activity was reduced to less than 40% of the maximal value. CueO activity was strongly pH dependent, with maximal activity observed at pH 6.5 and dropping to 36% and 27% maximal activity at pH 5.5 and pH 7, respectively. This pH optimum is higher then that typically associated with fungal laccases and is more suitable for conditions encountered in the E. coli periplasm. Protein activity increased as the temperature was raised, up to a maximum at 55°C.
Steady-state kinetic measurements performed at room temperature, pH 6.5, in the presence of 250 nM protein, and 250 μM CuCl2, yielded values for the apparent Km for DMP of 70.5 μM, Vmax = 178 μM⋅min−1, and kcat = 120 s−1. This value for the Km is ∼30 times smaller than that reported by Kim et al. (11), which was determined at pH 5.
Electron Transfer Kinetics.
We used laser flash photolysis to measure both intermolecular substrate oxidation kinetics and intramolecular electron transfer between the trinuclear copper center and the T1 copper site. Fig. 1 shows the time-resolved spectral changes observed at 610 nm after laser flash excitation of a solution containing CueO, 5-dRFH⋅, and semicarbazide. The decrease in absorbance at 610 nm, corresponding to the reduction of the T1 Cu of CueO, occurs in two distinct kinetic phases. The slower phase in Fig. 1A can be fit with a single exponential function (solid curve through the data), corresponding to a rate constant of ≈10 s−1. The observed rate constant for this reduction phase is independent of CueO concentration up to 70 μM (data not shown). This finding is consistent with a first-order reduction of the T1 Cu, presumably by means of intramolecular electron transfer from some unknown site on the enzyme, which has been directly reduced by 5-dRFH⋅ in a bimolecular reaction. This bimolecular reaction was measured by monitoring the decay of 5-dRFH⋅ through a change in absorption at 500 nm (k = 9 × 107 M−1⋅s−1).
Figure 1.
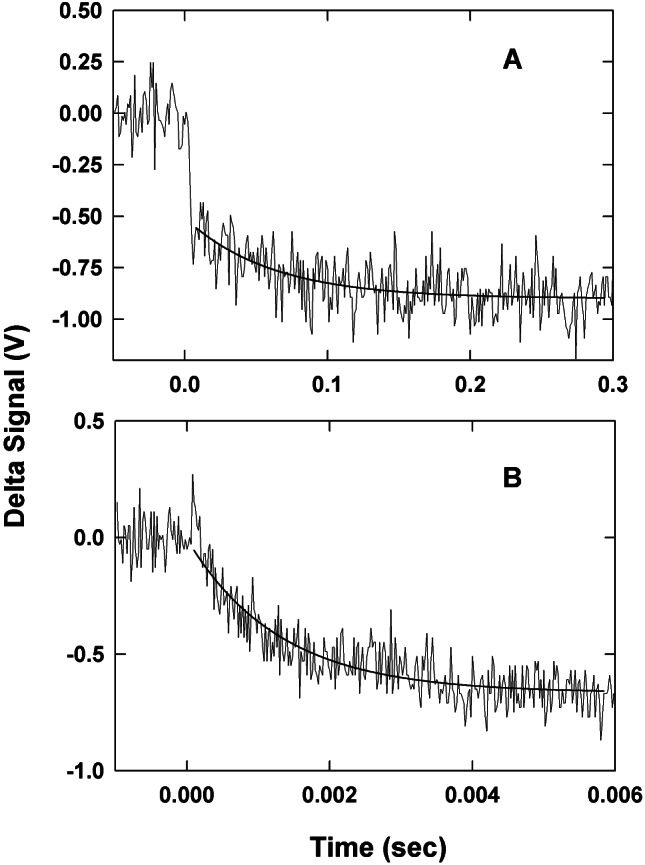
Transient absorbance changes obtained at 610 nm for anaerobic laser flash reduction of 35 μM CueO by 5-deazariboflavin semiquinone in 100 mM sodium phosphate buffer at pH = 8.0. (A) Kinetic trace for the intramolecular reduction of the T1 Cu. The solid line represents a single exponential fit to the data with a rate constant of 12 s−1. (B) Kinetic trace for the bimolecular (direct) reduction of the T1 Cu by 5-dRFH⋅. The solid line represents a single exponential fit to the data with a kobs = 180 s−1.
The more rapid observed reduction process can also be adequately fit with a single exponential equation (Fig. 1B). In this case, however, the rate constants show a linear dependence on enzyme concentration (data not shown). We conclude that the second-order rate constant calculated for this reaction (4 × 107 M−1⋅s−1) corresponds to direct reduction of the T1 Cu by 5-dRFH⋅. Note that this value is approximately one-half of that obtained by directly monitoring 5-dRFH⋅ decay, implying that there are two competing electron input reactions leading to CueO reduction. Because these two rate constants are similar, and because 5-dRFH⋅ decay also occurs by means of a competing disproportionation reaction, we are not able to deconvolute these directly from the 5-dRFH⋅ kinetics. Thus, the second-order rate constant determined at 500 nm must be considered an approximate value.
When equimolar CuCl2 is added to the reaction solution, the rapid phase (i.e., direct) reduction of T1 Cu is lost, whereas the intramolecular phase is not affected.
Crystal Structure of CueO.
The crystal structure of CueO was determined by using multiple anomalous dispersion phasing and solvent flattening. The resulting map was easily interpretable and the structure was built with the automated procedure of ARP/WARP (described in Materials and Methods), resulting in an excellent initial model (Fig. 2). The final model displayed excellent geometry except for the first two amino acids (amino acids 29–30, starting after the signal sequence) and a loop near the T1 Cu (380–403), which were disordered in the crystal.
Figure 2.

Representative electron density (2 Fo − Fc αc) after automatic model building and one round of refinement, before manual intervention. The model shown is in the region surrounding the trinuclear copper center and was built automatically with ARP/WARP.
The structure of CueO (Fig. 3) is similar to that of the MCOs laccase and AO, containing three similar azurin-like domains connected by linker peptides. The CueO copper sites are in the same positions, relative to the polypeptide chains, as in laccase and AO, and are ligated by the same types of amino acids. The most obvious difference in CueO is the presence of a methionine-rich helix (residues 356–371, seven methionines) and loop (residues 372–379, two methionines) lying over the T1 copper site. Unfortunately, residues 380–403, which link the end of this helix to His-404, located near the top of this helix and near the T1 copper center, and which contain an additional five methionines, are disordered and not visible in the electron density map. This region of MCOs has been called the “tower” (26) and is thought to be responsible for substrate specificity and binding in AO (14), and to control the interaction of nitrite reductase with its electron transfer partner pseudoazurin (27). Here, methionines 355, 364, 368, 376, and 379 from the CueO insert, and methionines 440 and 441 from a neighboring β-strand, form a seven-methionine cluster at the entrance to the T1 copper site that may be important for copper-dependent activity (Fig. 3C).
Figure 3.
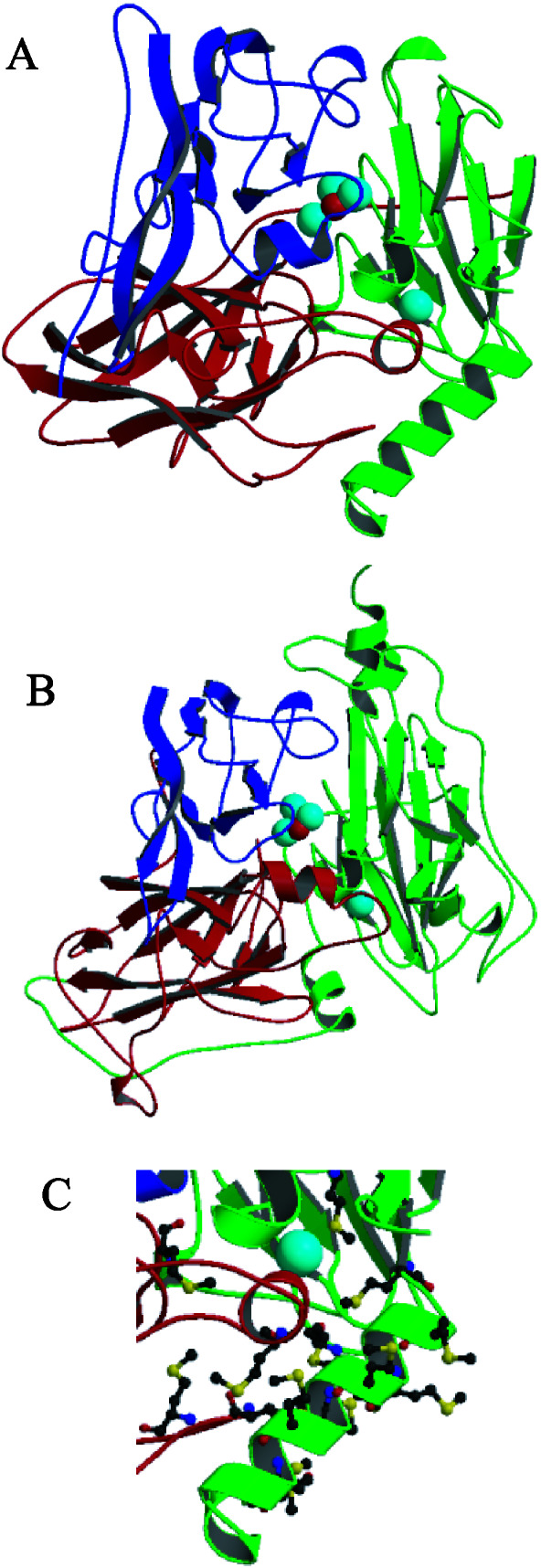
(A) Ribbon drawing of CueO, with the T1 copper and Cu3O cluster shown as space-filling spheres. The three related domains are colored separately, and the methionine-rich helix is located in the lower right. (B) Ribbon drawing of AO (PDB entry 1AOZ), an MCO with a more accessible T1 site. (C) Close-up of the methionine-rich region.
Close-ups of the copper sites are shown in Fig. 4. The T1 and trinuclear sites are connected by the familiar HCH motif that mediates electron transfer between the copper sites. T1 Cu sites have a coordination geometry that is usually described as distorted tetrahedral, but, in this case, can be better described as a trigonal bipyramidal structure with a missing axial ligand, as in azurin (28). The axial Cu-Met 510 SD distance is 3.23 Å, similar to the 3.1 Å observed in azurin. In place of the missing axial ligand, there is a long contact between the copper atom and the carbonyl oxygen of Leu-442 (3.73 Å).
Figure 4.

Stereoview showing the geometry of the T1 and trinuclear copper sites. Shown are the copper atoms (cyan), oxygen atoms (red), HCH residues (499–501), nitrogens (blue), and sulfurs (yellow) from ligating histidines, cysteines, and methionines. T1 Cu is ligated to His-443, His-503, Cys-500, and Met-510. T2 Cu is ligated to His-101, His-446, and a water molecule. Cu2 is ligated to His-103, His-141, His-501, and the bridging oxygen. Cu3 is ligated to His-143, His-448, His-499, and the bridging oxygen.
The trinuclear cluster has a geometry that differs from that seen in either fully oxidized or fully reduced AO (14, 29) and from that of laccase (13). Although the copper-histidine bond distances and bonding geometries for all three Cu atoms of the trinuclear cluster are the same as in AO, the copper–copper and copper–oxygen distances differ. In CueO, the Cu-O-Cu bridge is nearly linear (170°), is symmetric (Cu-O distances of 2.3 and 2.4 Å), and displays a longer Cu-Cu distance (4.7 Å) than in AO (3.7 Å). T2 Cu depleted laccase has a similar Cu-Cu distance (4.9 Å), but the oxo bridge is asymmetric (Cu-O distances of 2.1 and 3.1 Å) and bent (130°) (13). The T2 copper in CueO is 3.1 Å from the bridging oxygen, closer than in AO, but about the same distance to the T3 coppers as in AO, although positioned in a slightly asymmetric arrangement (3.6 Å from Cu3 and 4.0 Å from Cu2). The temperature factor of the T2 copper is twice that of the other atoms in the trinuclear cluster, but there is no evidence of disorder in the vicinity of this atom. Refinement of the occupancy led to a value of 75%, indicating that the T2 Cu site is not significantly depleted.
Discussion
The MCO CueO from E. coli is expressed in response to excess copper. By analogy to other MCO proteins, CueO is thought to reduce molecular oxygen to water through sequential oxidation of four substrate molecules. CueO will oxidize a variety of molecules in aerated solutions; however, the identity of the true in vivo substrate(s) has not yet been determined. We find the optimal activity for this protein to occur at pH 6.5 and in the presence of at least 100 μM CuCl2. The latter fact is intriguing in that μM levels of copper are required for induced CueO expression (8). Further, a methionine-rich region of the protein is common to several proteins implicated in copper homeostasis in bacteria, and we show that this region lies over the T1 Cu site in CueO, in an arrangement that likely interferes with substrate binding (Fig. 3), but which may provide for copper regulation. That substrate binding is indeed affected by the binding of exogenous copper is suggested by transient kinetic experiments, as described below.
Electron Transfer.
The spectral changes observed for reduction of the T1 Cu of CueO by 5-dRFH⋅, conducted anaerobically and in the absence of exogenous copper, indicate that this process occurs by two distinct mechanisms. In one, a bimolecular reduction of the enzyme occurs at some undetermined site (most likely the trinuclear copper cluster) with a second-order rate constant of ≈108 M−1⋅s−1. This reduction reaction is followed by a slow first-order intramolecular electron transfer to the T1 Cu (rate constant ≈10 s−1). As the protein concentration is increased, a second bimolecular reaction between the flavin semiquinone and the protein becomes more readily apparent. This results in a direct second-order reduction of the T1 Cu, with a rate constant of ≈4 × 107 M−1⋅s−1. However, addition of equimolar CuCl2 results in a loss of this direct reduction of T1 Cu, but not of the indirect (intramolecular) phase. The difference in the two bimolecular rate constants indicate that the steric accessibilities of the two reduction sites differ, i.e., that reduction of the T1 site is hindered to a greater degree than the reduction of the spectrally silent site, a result consistent with the CueO structure. The loss of the direct T1 reduction on adding copper indicates that access to this site has somehow changed.
These kinetic results are in sharp contrast to those obtained from the more extensively studied MCO, AO from zucchini squash (20, 30–34). Upon reduction of AO by laser flash photolysis, a rapid bimolecular reaction with the T1 Cu occurs with a rate constant of 5 × 108 M−1⋅s−1 (20), about 10-fold larger than that for CueO. This may reflect a greater degree of steric hindrance of the T1 Cu site in CueO as compared with that of AO, an interpretation supported by the CueO and AO structures (Fig. 3). Furthermore, a biphasic reduction of the T1 Cu of AO does not occur, unlike with CueO.
Subsequent to the initial reduction of the T1 Cu in AO, a slower, first-order reoxidation of the T1 Cu occurs, presumably by electron transfer to the copper atoms of the trinuclear cluster. This second kinetic process results in an ≈50% return of the absorbance at 610 nm caused by Cu2+ regeneration at the T1 site, implying that the two copper centers have similar reduction potentials, an interpretation further supported for AO by using pulse radiolytic reduction and monitoring of absorption by the trinuclear cluster at 330 nm (31, 32). The rate constant for this intramolecular electron transfer reaction in AO, which is the sum of the forward and reverse rate constants, has a value of ≈160 s−1 (20, 33), 16-fold larger than that for CueO (≈10 s−1). It is important to note that the intramolecular rate constant measured for CueO has not yet been confirmed to involve the trinuclear cluster copper sites.
Curiously, unlike with AO, an intramolecular oxidation of the T1 Cu in CueO is not observed. One possible explanation for this is that the difference in the reduction potentials of the T1 Cu and the Cu atoms of the trinuclear cluster may be too large to allow appreciable reoxidation of the T1 Cu. A second possibility is that reoxidation is in fact occurring but that the intramolecular reduction process obscures it. A careful determination of the midpoint potentials for the different Cu atoms in CueO will be required to resolve this issue.
CueO Structure.
CueO is composed of three repeated pseudoazurin domains with a T1 copper site in domain 3 and a trinuclear copper cluster between domains 1 and 3, much like the arrangement found in AO and laccase (Fig. 3). However, domain 3 of CueO has a methionine-rich insert that apparently hinders access to the T1 Cu site.
The high resolution for the CueO structure allows for a particularly clear description of the copper sites. In AO, the trinuclear copper center has usually been described as a Cu2O dimer with a T2 copper site in close proximity, an arrangement consistent with spectroscopic observations (2). The three copper atoms located in the AO structure were partitioned among those sites assuming that the magnetically coupled copper atoms were those connected by means of the bridging oxygen ligand. The copper atoms in CueO are better described as belonging to a trinuclear Cu3O cluster (Fig. 4). The oxidation states of the copper atoms are not yet known and difficult to predict without knowing the protonation state of the bridging oxygen atom; however, the differences between the present structure and that of fully oxidized AO suggest they may be partially reduced. The T1 Cu site also differs with AO in that the ligation state is better described as trigonal bipyramidal with a missing axial ligand (Fig. 4) than as distorted tetrahedral. It is tempting to speculate that these differences give rise to the very different intramolecular electron transfer rates displayed by the two proteins, because the distances between sites, as well as the ligating ligands, are nearly identical.
Copper and Iron Homeostasis.
CueO is able to protect periplasmic enzymes such as alkaline phosphatase from copper-mediated toxicity (10), but the mechanism for this protection is not yet known. It has been suggested by us (9, 10) and others (8) that CueO may in fact oxidize Cu(I), which is more toxic, to Cu(II), thereby directly protecting the organism. However, in preliminary experiments, Kim et al. (11) were unable to detect CueO oxygen consumption in the presence of Cu(I), suggesting it is not a substrate for CueO (11). Alternatively, CueO oxidation of siderophores, which releases iron, may balance copper and iron transport under conditions of high copper concentrations, or oxidation of phenolic metabolites may yield protective polymers (10, 11).
The molecular basis for copper stimulation remains unknown because our attempts to obtain diffraction-quality crystals with bound exogenous copper have not yet been successful. That direct binding occurs is suggested by copper-dependent changes in the CueO electron paramagnetic resonance and absorption spectra (11), and by the apparent change in T1 site accessibility revealed by our flash photolysis experiments. The methionine-rich region in concert with the disordered portion of the CueO structure (residues 380–403), which contains five methionines and five histidines, may provide additional sites for copper binding. Binding of copper here could lead to ordering of this loop and formation of a substrate-specific binding site. Such a mechanism could also explain the seemingly contradictory effects of copper on our steady-state kinetic measurements (increased activity) and flash photolysis kinetic measurements (decreased bimolecular T1 reduction), because the larger flavin molecule used in the photolysis experiments may be precluded from binding to a well-formed binding site, whereas the smaller DMP may not. Alternatively, three histidines (145 and 405–406) and three methionines (417, 497, and 511) near the trinuclear cluster are positioned such that copper binding might be possible, as previously suggested from modeling studies (11), although it is not clear how binding in this region would alter access to the T1 site.
Finally, CueO, like the related mammalian proteins ceruloplasmin and Fet3, is also a ferroxidase (11), and, like these other proteins, may have a role in iron homeostasis. Whether this similarity extends to function, or is simply a coincidence based on broad substrate specificity, awaits further studies.
Acknowledgments
We thank Dr. Irimpan Mathews of the Stanford Synchrotron Radiation Laboratory for invaluable assistance. This work was supported in part by National Institutes of Health Grants HL62969 and GM58727 (to W.R.M.) and DK15057 (to G.T.), and Hatch Project 136713 and National Institute on Environmental Health Sciences Grant ES04940 with funds from the Environmental Protection Agency (to C.R.). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences.
Abbreviations
- MCO
multicopper oxidase
- AO
ascorbate oxidase
- T1
type I
- T2
type II
- T3
type III
- DMP
dimethyoxyphenol
- 5-dRFH⋅
5-deazariboflavin
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1KV7).
References
- 1.Crichton R R, Pierre J-L. BioMetals. 2001;14:99–112. doi: 10.1023/a:1016710810701. [DOI] [PubMed] [Google Scholar]
- 2.Solomon E I, Sundaram U M, Machonkin T E. Chem Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 3.Culotta V C, Gitlin J D. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C R, Beaudet A L, Child B, editors. New York: McGraw–Hill; 2001. pp. 3105–3126. [Google Scholar]
- 4.Requena J R, Groth D, Legname G, Stadtman E R, Prusiner S B, Levine R L. Proc Natl Acad Sci USA. 2001;98:7170–7175. doi: 10.1073/pnas.121190898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dancis A, Yuan D S, Haile D, Askwith C, Eide D, Moehle C, Kaplan J, Klausner R D. Cell. 1994;76:393–402. doi: 10.1016/0092-8674(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 6.Askwith C, Eide D, Van Ho A, Bernard P S, Li L, Davis-Kaplan S, Sipe D M, Kaplan J. Cell. 1994;76:403–410. doi: 10.1016/0092-8674(94)90346-8. [DOI] [PubMed] [Google Scholar]
- 7.Henson J M, Butler M J, Day A W. Annu Rev Phytopathol. 1999;37:447–471. doi: 10.1146/annurev.phyto.37.1.447. [DOI] [PubMed] [Google Scholar]
- 8.Outten F W, Huffman D L, Hale J A, O'Halloran T V. J Biol Chem. 2001;276:30670–30677. doi: 10.1074/jbc.M104122200. [DOI] [PubMed] [Google Scholar]
- 9.Grass G, Rensing C. J Bacteriol. 2001;183:2145–2147. doi: 10.1128/JB.183.6.2145-2147.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grass G, Rensing C. Biochem Biophys Res Commun. 2001;286:902–908. doi: 10.1006/bbrc.2001.5474. [DOI] [PubMed] [Google Scholar]
- 11.Kim C, Lorenz W W, Hoopes J T, Dean J F D. J Bacteriol. 2001;183:4866–4875. doi: 10.1128/JB.183.16.4866-4875.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rensing C, Fan B, Sharma R, Mitra B, Rosen B P. Proc Natl Acad Sci USA. 2000;97:652–656. doi: 10.1073/pnas.97.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ducros V, Brzozowski A M, Wilson K S, Brown S H, Ostergaard P, Schneider P, Yaver D S, Pedersen A H, Davies G J. Nat Struct Biol. 1998;5:310–316. doi: 10.1038/nsb0498-310. [DOI] [PubMed] [Google Scholar]
- 14.Messerschmidt A, Ladenstein R, Huber R, Bolognesi M, Avigliano L, Petruzzelli R, Rossi A, Finazzi-Agro A. J Mol Biol. 1992;224:179–205. doi: 10.1016/0022-2836(92)90583-6. [DOI] [PubMed] [Google Scholar]
- 15.Zaitseva I, Zaitsev V, Card G, Moshkov K, Bax B, Ralph A, Lindley P. J Biol Inorg Chem. 1996;1:15–23. [Google Scholar]
- 16.Murphy M E P, Turley S, Adman E T. J Biol Chem. 1997;272:28455–28460. doi: 10.1074/jbc.272.45.28455. [DOI] [PubMed] [Google Scholar]
- 17.Alexandre G, Zhulin I B. Trends Biotechnol. 2000;18:41–42. doi: 10.1016/s0167-7799(99)01406-7. [DOI] [PubMed] [Google Scholar]
- 18.Cooksey D A. FEMS Microbiol Rev. 1994;14:381–386. doi: 10.1111/j.1574-6976.1994.tb00112.x. [DOI] [PubMed] [Google Scholar]
- 19.Stanley N R, Palmer T, Berks B C. J Biol Chem. 2000;275:11591–11596. doi: 10.1074/jbc.275.16.11591. [DOI] [PubMed] [Google Scholar]
- 20.Hazzard J T, Maritano S, Tollin G, Marchesini A. Arch Biochem Biophys. 1997;339:24–32. doi: 10.1006/abbi.1996.9860. [DOI] [PubMed] [Google Scholar]
- 21.Pflugrath J W. Acta Crystallogr D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 22.Perrakis A, Morris R, Lamzin V S. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 23.Jones T A, Zou J Y, Cowan S W, Kjelgard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 24.Sheldrick G M, Schneider T R. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 25.Collaborative Computational Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 26.Murphy M E P, Lindley P F, Adman E T. Protein Sci. 1997;6:761–770. doi: 10.1002/pro.5560060402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kukimoto M, Nishiyama M, Tanokura M, Adman E T, Horinouchi S. J Biol Chem. 1996;271:13680–13683. doi: 10.1074/jbc.271.23.13680. [DOI] [PubMed] [Google Scholar]
- 28.Baker E N. J Mol Biol. 1988;203:1071–1095. doi: 10.1016/0022-2836(88)90129-5. [DOI] [PubMed] [Google Scholar]
- 29.Messerschmidt A, Luecke H, Huber R. J Mol Biol. 1993;1993:997–1014. doi: 10.1006/jmbi.1993.1215. [DOI] [PubMed] [Google Scholar]
- 30.Meyer T E, Marchesini A, Cusanovich M A, Tollin G. Biochemistry. 1991;30:4619–4623. doi: 10.1021/bi00232a037. [DOI] [PubMed] [Google Scholar]
- 31.Farver O, Pecht I. Proc Natl Acad Sci USA. 1992;89:8283–8287. doi: 10.1073/pnas.89.17.8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farver O, Wherland S, Pecht I. J Biol Chem. 1994;269:22933–22936. [PubMed] [Google Scholar]
- 33.Hazzard J T, Marchesini A, Curir P, Tollin G. Biochem Biophys Acta. 1994;1208:166–170. doi: 10.1016/0167-4838(94)90174-0. [DOI] [PubMed] [Google Scholar]
- 34.Tollin G, Meyer T E, Cusanovich M A, Curir P, Marchesini A. Biochem Biophys Acta. 1993;1183:309–314. doi: 10.1016/0005-2728(93)90232-5. [DOI] [PubMed] [Google Scholar]