

Abstract
As a direct simulation of a multistep proton transfer reaction involving protein residues, the proton relay shuttle between A and I forms of green fluorescent protein (GFP) is simulated in atomic detail by using a special molecular dynamics simulation technique. Electronic excitation of neutral chromophore in wild-type GFP is generally followed by excited-state proton transfer to a nearby glutamic acid residue via a water molecule and a serine residue. Here we show that the second and third transfer steps occur ultrafast on time scales of several tens of femtoseconds. Proton back-shuttle in the ground state is slower and occurs in a different sequence of events. The simulations provide atomic models of various intermediates and yield realistic rate constants for proton transfer events. In particular, we argue that the I form observed spectroscopically under equilibrium conditions may differ from the I form observed as a fast intermediate by an anti to syn rotation of the carboxyl proton of neutral Glu-222.
Because of its unique photophysical properties—strong green fluorescence without need for an additional cofactor—green fluorescent protein (GFP) has become a very powerful marker for gene expression, cellular localization, and dynamic intracellular events over the past 5 years (1). Its rich photophysical behavior was characterized quite well by a great variety of spectroscopic techniques (1–4), and its three-dimensional structure was determined by x-ray crystallography (5, 6). Wild-type GFP exhibits two absorption maxima “A” and “B” at 395 nanometers (nm) and at 475 nm (1) that are present roughly in a 6:1 ratio (1), and that correspond to forms of the protein with either neutral (A) or anionic (B) chromophores. Time-resolved fluorescence spectroscopy identified an additional form “I*” that is populated as a fast intermediate after excitation of the A form, A* → I* (2, 3). The mechanism of interconversion is likely caused by excited-state proton transfer from the chromophore to a nearby protein residue (2, 3) because the new form emits at a similar wave length as B* and because the transfer rate slows down upon deuteration (2); see Fig. 1 for an overview.
Figure 1.

Photophysical states of wild-type GFP as inferred from time-resolved fluorescence spectroscopy (2, 3). Thick and thin arrows indicate dominant and weak absorption and fluorescence processes. The thermal conversion between I and B is very slow.
Although “I form” originally denoted only this fast intermediate, it has become common to use “I form” as a term for GFP species that absorb at wavelengths red-shifted with respect to the B form. Surprisingly, recent hole-burning experiments demonstrated that an I form is already present in wild-type GFP samples at room temperature under equilibrium conditions (4). It is currently unclear whether this is the same I form observed in the time-resolved fluorescence experiments. Although the I form has so far not been described in atomic detail, a structural model of I was suggested on the basis of comparisons between the crystal structures of wild-type (A form) and mutant GFP proteins containing the B form (7, 8); see Fig. 2. In this model, Glu-222 is assumed to be protonated in the I form (7), thus requiring a rise of the pKa of Glu-222 by more than 4 pH units. Stabilization energy may be provided by the extensive hydrogen bond network in the chromophore environment, and this protonation assignment is supported by electrostatic continuum calculations indicating that the protonation states of the chromophore and nearby Glu-222 are tightly coupled (9). By shifting the proton from anti to syn position (see Fig. 2), Glu-222 may also play a crucial role in the transition from I to B considered to be slow (7). Despite this discussion, it is not known whether there exists more than one I form, the molecular structures of these I forms, the structural mechanism for transitions between the various A, I, and B forms, and the rates of interconversion.
Figure 2.

Schematic drawing of the chromophore (Cro) surrounding in GFP. From upper to lower left are shown the A form with a neutral chromophore, two putative intermediate I forms, and the B form with anionic chromophores. This model is closely related to the models of Brejc et al. (7) and Palm et al. (8). The most clearly visible change between A and B is a rotation of Thr-203 around its χ1 angle to cause the hydroxyl group to either point toward the anionic chromophore or point away from the neutral chromophore. In the Upper Left, 1–3 indicate the three proton transfer events and the sequence in which they occur on the transformation A* → I*. In the Upper Right , 1–3 denote the proton transfer events during the transformation transformation I → A in the ground state (see also Fig. 3).
Proton diffusion in aqueous solution is crucial for acid-base reactions and is known to proceed extremely fast. According to the so-called Grotthus mechanism, proton transport should not be viewed as transport of one proton particle through solution but as the net result of many bond-forming and bond-breaking processes between the bare proton and neighboring water molecules. Simulating proton diffusion in bulk water has also spurred the development of classical and quantum mechanical simulation techniques and can nowadays be well understood from first principles (see, for example, refs. 10–12). Furthermore, proton transfer in proteins is a very important process in enzymatic reactions and bioenergetic processes. However, compared with bulk solution, our understanding of this mechanism is far less complete, and only individual transfer steps (13–15) or transfer through chains of internal water molecules (16, 17) have been considered by simulation methods.
We have recently developed the method Q-HOP molecular dynamics (MD) to simulate proton transfer events in biomolecular systems by classical MD simulations augmented by instantaneous proton hopping (18). In this method, reaction barriers for proton transfer reactions between typical functional groups in a protein and to water are carefully parametrized against quantum mechanical calculations as a function of the donor–acceptor distance R(DA) and the energy difference between donor- and acceptor-bound states E12, which includes the chemical nature of donor and acceptor, and the relative electrostatic stabilization of both states by the environment (19, 20). Hopping probabilities are obtained either by transition-state theory including zero point energy and tunneling corrections or, for transfer over low barriers, by parametrizing the solutions of the time-dependent Schrödinger equation in one dimension (21). When these barriers are precomputed, the simulations require only a small computational overhead of less than 50% compared with standard MD simulations. The proton transfer rates obtained and the proton diffusion coefficient of an excess proton in a box of water molecules (18) are in excellent agreement with more elaborate quantum chemical simulations (10, 11) and with experimental data. Here, this method is applied to a protein system.
In general, our simulations agree well with the model of Brejc et al. (7) and Palm et al. (8), where Glu-222 protonated in anti position is assigned as an I form, and where transition from I to B may proceed by means of an intermediate with Glu-222 protonated in syn position. The key findings of this study are kinetic estimates for the various proton transfer steps leading from A* to I* and from I to A. The short lifetime of the fast intermediate I form that we found indicates that the intermediate with Glu-222 protonated in anti position cannot be the I form observed in spectroscopic studies under equilibrium.
Methods
The MD simulations were started from a previously equilibrated
system of a single GFP molecule in a rectangular solvent box with
periodic boundary conditions (22). Precisely, the coordinates and
velocities were taken from the configuration after heating up and 80 ps
of MD simulation at room temperature. To save computation time, the
system size was reduced to 16,993 atoms, keeping at each side a minimum
distance of 4 Å between GFP and the wall of the simulation box. Twenty
picoseconds of additional equilibration was performed to adapt the
system to the smaller box size. The simulations were performed with a
locally modified sequential version of the argos program
(23) using the Amber95 force field (24). Fifteen
picoseconds could be simulated per day on a 1-GHz desktop Linux PC. By
use of separate Berendsen thermostats for protein atoms and solvent
atoms and a barostat, the system temperature was kept at 293 K and the
pressure at 105 Pa. The Q-HOP MD procedure was
used as described in refs. 18–21. Missing proton transfer barriers
E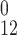 were parameterized as described
previously (20). In the simulations, proton transfer was allowed only
between chromophore, Wat-22, Ser-205, and Glu-222. Wat-22 was
considered in the three protonation states
H3O+,
H2O, and OH−. Partial
atomic charges of neutral and anionic chromophores in their relaxed
ground and first excited singlet states were computed by electrostatic
potential fit and were taken from ref. 25. Although we and others have
shown that the excited state potential energy surface of the neutral
and anionic chromophores is torsionally unstable (26, 27), it was
assumed for the present study that the excited neutral chromophore
remains planar during the time scale of less than 12 ps (2) for the
proton shuttle A* → I* studied here. As noted by one referee, this
assumption is supported by the observed fluorescence (A* → A), which
suggests that the chromophore is constrained to near planar geometries
by close packing with protein residues and by the hydrogen bonding
network with surrounding groups. Except for the partial atomic charges,
the same parametrization was used for ground-state and excited-state
chromophores. A more careful parametrization would certainly be
necessary if one is interested in the dynamics of the excited
chromophore during its full lifetime of 3.3 ns. According to
Hartree–Fock calculations of the torsional barrier of a glutamic acid
residue for rotation of Hδ from an
anti to a syn position, the syn
position is more favorable than the anti position by 36
kJ/mol, and the energy barrier for anti → syn
transition amounts to 27 kJ/mol. To model this barrier in our
simulations, the dihedral force constants for rotation around
O–C–O–H and C–C–O–H were modified to barrier heights of 20
kJ/mol.
were parameterized as described
previously (20). In the simulations, proton transfer was allowed only
between chromophore, Wat-22, Ser-205, and Glu-222. Wat-22 was
considered in the three protonation states
H3O+,
H2O, and OH−. Partial
atomic charges of neutral and anionic chromophores in their relaxed
ground and first excited singlet states were computed by electrostatic
potential fit and were taken from ref. 25. Although we and others have
shown that the excited state potential energy surface of the neutral
and anionic chromophores is torsionally unstable (26, 27), it was
assumed for the present study that the excited neutral chromophore
remains planar during the time scale of less than 12 ps (2) for the
proton shuttle A* → I* studied here. As noted by one referee, this
assumption is supported by the observed fluorescence (A* → A), which
suggests that the chromophore is constrained to near planar geometries
by close packing with protein residues and by the hydrogen bonding
network with surrounding groups. Except for the partial atomic charges,
the same parametrization was used for ground-state and excited-state
chromophores. A more careful parametrization would certainly be
necessary if one is interested in the dynamics of the excited
chromophore during its full lifetime of 3.3 ns. According to
Hartree–Fock calculations of the torsional barrier of a glutamic acid
residue for rotation of Hδ from an
anti to a syn position, the syn
position is more favorable than the anti position by 36
kJ/mol, and the energy barrier for anti → syn
transition amounts to 27 kJ/mol. To model this barrier in our
simulations, the dihedral force constants for rotation around
O–C–O–H and C–C–O–H were modified to barrier heights of 20
kJ/mol.
Results and Discussion
Excitation A → A* Leading to Excited-State Proton Transfer A* → I*.
The first proton transfer after excitation A → A* will protonate the nearby water molecule 22, see 1 in the upper left of Fig. 2. The MD simulations were started from a carefully equilibrated system of a GFP molecule in its electronic ground state solvated in a rectangular solvent box (22). To model the proton shuttle after electronic excitation of the chromophore, the partial atomic charges of the chromophore were replaced by appropriate charges for the excited state (25). Because it is currently still very hard to accurately compute the energy barrier for proton release from the excited chromophore by quantum mechanical methods, we simply shifted the proton from the chromophore (Cro) to Wat-22 and started the MD simulation after this first step at the situation Cro−⋅H3O+. To our great surprise, the next two transfer events simulated (2) Ser-205-OH⋅Glu-222-COO− → Ser-205-O−⋅Glu-222-COOH and (3) H3O+⋅Ser-205-O− → H2O⋅Ser-205-OH occur within 10–80 fs and within 10 fs (see Table 1 and Fig. 2). Ten femtoseconds is actually the shortest transfer interval that we allow in our Q-HOP MD approach (18). The two transfer steps are strongly coupled to each other because the deprotonated Ser-205-O− is a very unstable intermediate in the reaction. Moreover, both steps are ultrafast barrierless processes leading to a much more favorable product state. The reason for this “downhill process” seems to come from the modified charge distribution of the chromophore in its excited state, because no transfer occurs from Ser-205 to Glu-222 when simulations are started from the Cro−⋅H3O+ situation with ground state charges of the chromophore, while not allowing back-transfer from H3O+ to the chromophore.
Table 1.
Data for proton transfer events from 14 MD simulations of steps 2 and 3 of the proton shuttle connecting states A* and I* in wild-type GFP
| Run no. |
(2) Ser-H →
Glu-COO−
|
(3)
H3O+ → Ser−
|
||||
|---|---|---|---|---|---|---|
| Δt2, fs | R(DA), Å | E12, kcal/mol | Δt3, fs | R(DA), Å | E12, kcal/mol | |
| 1 | 10 | 2.55 | 0.0 | 10 | 2.50 | −36.3 |
| 2 | 10 | 2.59 | 1.8 | 10 | 2.54 | −33.4 |
| 3 | 10 | 2.47 | 5.8 | 10 | 2.60 | −42.1 |
| 4 | 80 | 2.40 | 1.4 | 10 | 2.57 | −27.0 |
| 5 | 80 | 2.50 | −0.1 | 10 | 2.59 | −31.2 |
| 6 | 40 | 2.65 | 2.7 | 10 | 2.46 | −25.4 |
| 7 | 10 | 2.54 | 0.0 | 10 | 2.63 | −28.7 |
| 8 | 30 | 2.52 | 2.1 | 10 | 2.54 | −27.8 |
| 9 | 80 | 2.54 | 3.5 | 10 | 2.50 | −24.0 |
| 10 | 80 | 2.51 | 6.3 | 10 | 2.44 | −18.8 |
| 11 | 20 | 2.51 | 6.5 | 10 | 2.35 | −14.8 |
| 12 | 70 | 2.53 | −0.8 | 10 | 2.57 | −26.4 |
| 13 | 20 | 2.66 | 3.3 | 10 | 2.59 | −34.6 |
| 14 | 10 | 2.57 | 2.0 | 10 | 2.64 | 38.9 |
Different snapshots from the equilibration run were used as starting configurations, and different random number seeds were used for the generation of hopping probabilities. Δt2 is the time interval between the start of the simulation in the excited state of the chromophore (with the first proton being transferred from Cro− to H3O+) and the second proton transfer between Ser-205 and Glu-222. Δt3 is the time interval between second and third proton transfer from H3O+ to Ser-205. R(DA) and E12 are the donor–acceptor distance and the relative energy difference between donor- and acceptor-bound states at the moment of transfer, respectively.
After three subsequent transfer steps, the proton ends up at Glu-222 in anti-position. When the reparametrized barrier between anti and syn positions was used (see Methods), the proton remained in anti position during typical simulation times of 100 ps.
The second and third proton transfer events A → A* → I* occur ultrafast, whereas the two main decay constants for the fluorescence from A* were measured as 3.6 ps (50%) and 12 ps (40%) (2). These must therefore be the decay constants for the first excited-state proton transfer from the electronically excited chromophore to the nearby water molecule, and it can be expected that the chromophore will have structurally relaxed during this time. Assigning a lifetime of ≈5–10 ps to the excited neutral chromophore is also in agreement with the experimentally observed weak fluorescence between 440 and 460 nm that originates from the neutral form of the chromophore.
Proton Back-Shuttle I → A in the Ground State.
After emission from I* after 3.3 ns on average (1), the proton on Glu-222 is expected to reprotonate the chromophore according to the current model (7, 8). Consequently, a second set of simulations was started from the final configurations obtained previously, where the partial atomic charges of the chromophore were set back from electronic excited to the ground state (25). The sequence of transfer events observed is shown in Fig. 2 Upper Right and in Fig. 3, and Table 2 lists typical transfer times. Simulations started with the Hδ on Glu-222 in syn-position did not lead to reprotonation of the chromophore because, as noted before, transitions between anti and syn conformation are unlikely on simulation time scales of 100 ps.
Figure 3.

Snapshots from Q-HOP MD simulations to illustrate the proton back-shuttle between putative I and A form. (a) The starting situation with protonated Glu-222 and Ser-205 pointing toward Wat-22. (b) A first proton hopped from Wat-22 to reprotonate the chromophore. The next two steps in c and d seem to occur as one concerted process: transfer from Ser-205 to Wat-22 and transfer from Glu-222 to reprotonate Ser-205.
Table 2.
Data from four MD simulations of steps 1 to 3 of the proton back-shuttle connecting states I and A in wild-type GFP
| Run no. |
(1) H2O →
Cro−
|
(2) Ser-H →
OH−
|
(3) Glu-COOH →
Ser−
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Δt1, ps | R(DA), Å | E12, kcal/mol | Δt2, fs | R(DA), Å | E12, kcal/mol | Δt3, fs | R(DA), Å | E12, kcal/mol | |
| 1 | 3 in 3.77 | 2.54 | −5.5 | 440 | 2.47 | 9.1 | 10 | 2.59 | −10.1 |
| 2 | 6 in 46.54 | 2.51 | 0.7 | 10 | 2.59 | 0.3 | 10 | 2.62 | −13.3 |
| 3 | 1 in 0.94 | 2.48 | 2.3 | 330 | 2.50 | 8.4 | 10 | 2.65 | −12.6 |
| 4 | 1 in 2.07 | 2.52 | 3.5 | 530 | 2.41 | 6.8 | 20 | 2.60 | −6.1 |
In step 1, the proton may shuttle several times between H2O and Cro before the second transfer occurs. The listing “3 in 3.77” means that the proton hopped back and forth three times in 3.77 ps before step 2 took place. In four additional simulations (not shown), proton transfer did not occur during 200 ps. The conversion rate I → A is therefore estimated between 1 ps and 1 ns.
Based on our observed proton transfer rates and in agreement with the crystallographic model (7, 8), the configuration with Glu-222 protonated in anti position is confirmed as the long-sought I form when I is not defined in a spectroscopic sense (4) but as the fast intermediate after excited-state proton transfer from A* (2). Its relatively short lifetime (1 ps to 1 ns, compare Δt1 in Table 2) clearly rules out the possibility that this form may be visible under continuous illumination by laser light (4). On the other hand, the same molecular configuration with Hδ on Glu-222 relaxed into syn position should have a considerably longer lifetime and therefore be visible (Fig. 2 Lower Right). We therefore suggest the existence of two photophysically relevant I forms, a “nonrelaxed” I form with Hδ on Glu-222 in anti position, and a “relaxed” I form with Hδ on Glu-222 in syn position. The latter form may structurally be converted into the B form by rotation of Thr-203 and a side-movement of His-148. Different positions of His-148, either hydrogen bonding to the chromophore or not, may actually give rise to additional spectroscopic I forms, e.g., observed in low-temperature studies (28).
The Q-HOP MD simulation method used in this study seems a very powerful addition to the arsenal of biomolecular simulation tools. The major strengths of this approach compared with quantum chemical methods are its computational efficiency and the relative ease to parametrize hopping probabilities between arbitrary donor–acceptor pairs. Further work on proton equilibria in GFP on longer time scales and on proton transfer reactions in bioenergetics needs to be done. Significant progress in our understanding of proton transfer in proteins can be expected in the near future by combining data from simulation and experiments.
Acknowledgments
We thank Prof. Peter W. Langhoff, Dr. Peter Schellenberg, and the anonymous referees for valuable suggestions.
Abbreviations
- GFP
green fluorescent protein
- MD
molecular dynamics
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Tsien R Y. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 2.Chattoraj M, King B A, Bublitz G U, Boxer S G. Proc Natl Acad Sci USA. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lossau H, Kummer A, Heinecke R, Pollingerdammer F, Kompa C, Bieser G, Jonsson T, Silva C M, Yang M M, Youvan D C, Michel-Beyerle M E. Chem Phys. 1996;213:1–16. [Google Scholar]
- 4.Creemers T M H, Lock A J, Subramaniam V, Jovin T M, Voelker S. Nat Struct Biol. 1999;6:557–560. doi: 10.1038/9335. [DOI] [PubMed] [Google Scholar]
- 5.Yang F, Moss L G, Phillips G N., Jr Nat Biotechnol. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 6.Ormö M, Cubitt A B, Kallio K, Gross L A, Tsien R Y, Remington S J. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 7.Brejc K, Sixma T K, Kitts P A, Kain S R, Tsien R Y, Ormö M, Remington S J. Proc Natl Acad Sci USA. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palm G J, Zdanov A, Gaitanaris G A, Stauber R, Pavlakis G N, Wlodawer A. Nat Struct Biol. 1997;4:361–365. doi: 10.1038/nsb0597-361. [DOI] [PubMed] [Google Scholar]
- 9.Scharnagl C, Raupp-Kossmann R, Fischer S F. Biophys J. 1999;77:1839–1857. doi: 10.1016/S0006-3495(99)77028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vuilleumier R, Borgis D. Chem Phys Lett. 1998;284:71–77. [Google Scholar]
- 11.Schmitt U W, Voth G A. J Chem Phys. 1999;111:9361–9381. [Google Scholar]
- 12.Marx D, Tuckermann M E, Hutter J, Parrinello M. Nature (London) 1999;397:601–604. [Google Scholar]
- 13.Bala P, Grochowski P, Nowinski K, Lesyng B, McCammon J A. Biophys J. 2000;79:1253–1262. doi: 10.1016/S0006-3495(00)76379-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Röthlisberger U, Carloni P, Doclo K, Parrinello M. J Biol Inorg Chem. 2000;5:236–250. doi: 10.1007/s007750050368. [DOI] [PubMed] [Google Scholar]
- 15.Billeter S R, Webb S P, Iordanov T, Aggarwal P K, Hammes-Schiffer S. J Chem Phys. 2001;114:6925–6936. doi: 10.1021/ja011384b. [DOI] [PubMed] [Google Scholar]
- 16.Pomes R, Roux B. J Phys Chem. 1996;100:2519–2527. [Google Scholar]
- 17.Brewer M L, Schmitt U W, Voth G A. Biophys J. 2001;80:1691–1702. doi: 10.1016/S0006-3495(01)76140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lill M A, Helms V. J Chem Phys. 2001;115:7993–8005. [Google Scholar]
- 19.Lill M A, Hutter M C, Helms V. J Phys Chem A. 2000;104:8283–8289. [Google Scholar]
- 20.Lill M A, Helms V. J Chem Phys. 2001;114:1125–1132. [Google Scholar]
- 21.Lill M A, Helms V. J Chem Phys. 2001;115:7985–7992. [Google Scholar]
- 22.Helms V, Straatsma T P, McCammon J A. J Phys Chem B. 1999;103:3263–3269. [Google Scholar]
- 23.Straatsma T P, McCammon J A. J Comp Chem. 1990;11:943–951. [Google Scholar]
- 24.Cornell W D, Cieplak P, Bayly C I, Gould I R, Merz K M, Ferguson D M, Spellmeyer D C, Fox T, Caldwell J W, Kollman P A. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 25.Helms V, Winstead C, Langhoff P W. J Mol Struct. 2000;506:179–189. [Google Scholar]
- 26.Weber W, Helms V, McCammon J A, Langhoff P W. Proc Natl Acad Sci USA. 1999;96:6177–6182. doi: 10.1073/pnas.96.11.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voityuk A A, Michel-Beyerle M E, Rösch N. Chem Phys Lett. 1998;296:269–276. [Google Scholar]
- 28.Seebacher C, Deeg F W, Bräuchle C, Wiehler J, Steipe B. J Phys Chem B. 1999;103:7728–7732. [Google Scholar]