

Abstract
Small diffusible redox proteins facilitate electron transfer in respiration and photosynthesis by alternately binding to integral membrane proteins. Specific and transient complexes need to be formed between the redox partners to ensure fast turnover. In respiration, the mobile electron carrier cytochrome c shuttles electrons from the cytochrome bc1 complex to cytochrome c oxidase. Despite extensive studies of this fundamental step of energy metabolism, the structures of the respective electron transfer complexes were not known. Here we present the crystal structure of the complex between cytochrome c and the cytochrome bc1 complex from Saccharomyces cerevisiae. The complex was crystallized with the help of an antibody fragment, and its structure was determined at 2.97-Å resolution. Cytochrome c is bound to subunit cytochrome c1 of the enzyme. The tight and specific interactions critical for electron transfer are mediated mainly by nonpolar forces. The close spatial arrangement of the c-type hemes unexpectedly suggests a direct and rapid heme-to-heme electron transfer at a calculated rate of up to 8.3 × 106 s−1. Remarkably, cytochrome c binds to only one recognition site of the homodimeric multisubunit complex. Interestingly, the occupancy of quinone in the Qi site is higher in the monomer with bound cytochrome c, suggesting a coordinated binding and reduction of both electron-accepting substrates. Obviously, cytochrome c reduction by the cytochrome bc1 complex can be regulated in response to respiratory conditions.
Electron transfer processes are of great importance in many metabolic pathways of living organisms. They are essential for photosynthesis and respiration, in which energy gained from capturing light or by oxidation of nutrients is converted to the energy of the anhydride bond of ATP. Energy conversion is achieved by coupling the transfer of electrons to the translocation of protons across a lipid membrane. The generated electrochemical proton gradient is used for ATP synthesis.
The mitochondrial respiratory chain consists of four large multisubunit membrane protein complexes embedded in the inner mitochondrial membrane that are linked by the freely diffusible electron carriers ubiquinone (Q) and cytochrome c (CYC). The latter is a small water-soluble protein with a covalently attached heme c group that is located in the intermembrane space. CYC molecules shuttle electrons between ubiquinol/cytochrome c oxidoreductase (QCR, cytochrome bc1 complex) and cytochrome c oxidase (1). QCR (EC 1.20.2.2) is a multisubunit integral membrane protein complex. The catalytic core of the enzyme comprises cytochrome b with two noncovalently attached heme b groups, the so-called Rieske iron–sulfur protein (RIP1), with an [2Fe − 2S] iron–sulfur cluster and cytochrome c1 (CYT1), with a covalently attached heme c group. Mitochondrial QCRs contain up to eight additional subunits. The enzyme catalyses the electron transfer from ubiquinol (QH2) to CYC:
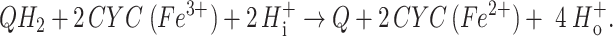 |
1 |
Ubiquinol oxidation at the Qo site involves a bifurcated electron transfer. One electron is transferred via the [2Fe − 2S] cluster to heme c1, the electron donor for CYC reduction. The second electron is used by the enzyme to reduce Q bound at the Qi site. The protons of QH2 are released to the outside, whereas Q is protonated from the mitochondrial matrix. After a second round of electron transfer, the fully reduced Q leaves the binding pocket. This mechanism, known as the Q cycle, couples the electron transfer to translocation of protons across the inner mitochondrial membrane, thereby contributing to the generation of the proton gradient (2, 3).
The proper encounter complex between CYC and QCR is critical for ubiquinol oxidation and full turnover of QCR. Stabilizing interactions of the complex must ensure fast complex formation, optimal orientation, and distance of the reaction partners for electron transfer as well as fast dissociation of the complex after electron transfer. CYC reduction by QCR is slowed down by an increase in ionic strength, indicating that the binding has an electrostatic component (4, 5). A ring of conserved lysine residues surrounding the heme cleft of CYC and negatively charged residues of the subunit CYT1 of QCR were identified by mutagenesis and chemical modifications to be involved in CYC binding (6–8). In addition, the acidic subunit of the bovine QCR, the so-called hinge protein, which is homologous to yeast subunit 6 of QCR (QCR6), was found to be essential for the formation of the CYT1/CYC complex (9) involving patches of acidic residues (10). In contrast, the first structure of a complex involving CYC bound to one of its redox partners, namely the soluble cytochrome c peroxidase (CCP), indicated that nonpolar forces are important for the stabilization of the CCP/CYC complex (11). Despite extensive studies that focused on the importance of electrostatic interactions, the nature of the electron transfer complex between CYC and QCR and the electron transfer pathway were not known until now. Electron transferring protein complexes form transiently, and their structures are difficult to obtain (11, 12). Structures of QCR (13–16) and CYC (17) have been separately determined, but the structure of their complex, critical for electron transfer, has remained undescribed. We here present the first structure, to our knowledge, of such a complex from the respiratory chain.
Methods
Crystallization.
QCR from the yeast Saccharomyces cerevisiae was prepared and complexed with an antibody Fv fragment, as described (16). Iso-1 CYC from S. cerevisiae was purchased from Sigma. Dimerization of yeast CYC via a single cysteine close to the C terminus is known to take place in solution. This reaction was prevented by treating the protein with methylthiomethane–sulfonate, as described (18). The QCR/Fv fragment complex and CYC were mixed in a molar ratio of 1:1.3 in buffer [250 mM NaCl/20 mM Tris⋅HCl, pH 7.5/0.05% Undecylmaltoside (UM)/1 μM Stigmatellin]. To crystallize the ternary complex, ionic strength was adjusted to yield a final value of about 120 mM, presumably close to the physiological value in yeast. The protein solution was rapidly mixed with a solution of 0.75–2% polyethylene glycol 4000 in 20 mM Tris⋅HCl, pH 7.5/0.05% UM. Crystal growth was very fast; the first visible crystals appeared after 20 min, and nucleation was extremely difficult to control. Only a few crystals could be grown to a size suitable for x-ray diffraction studies.
X-Ray Data Collection and Data Processing.
X-ray diffraction data were collected at synchrotron beamline ID14EH3 at the European Synchrotron Radiation Facility, Grenoble, France, at 4°C by using a charge-coupled device detector (mar CCD, mar USA, Evanston, IL). Data were processed with the programs denzo and scalepack (19) from the HKL package (HKL Research, Charlottesville, NC). The crystals belong to the space group P21 with unit cell parameters of a = 147.2 Å, b = 165.5 Å, c = 195.9 Å, and β = 104.2°.
Structure Solution and Refinement.
The structure was solved by molecular replacement by using the program amore (20). The refined structure of the yeast QCR/Fv fragment complex (coordinates, PDB ID code 1EZV) was used as a search model after omitting all water molecules and quinone. The rotational and translational search resulted in two very pronounced and equivalent solutions representing the two halves of the dimeric molecule with a correlation coefficient of 0.42 and an R factor of 49.5%. The dimer is present in the asymmetric unit. The mobile extrinsic domain of subunit RIP1 containing the iron–sulfur cluster is present in the b position, i.e., the metal center is in close contact to heme bL of cytochrome b. The b position is caused by the binding of Stigmatellin at the Qo site, which resembles an intermediate step of quinol oxidation (16). The position of the model was adjusted by rigid-body refinement by using the cns program package (version 1.0) (21), treating the two monomers as separate entities. The R factor and free R factor decreased to 30.8 and 31.2%, respectively. The electron-density maps after rigid-body refinement allowed the placement of a CYC molecule close to CYT1 of monomer B. On the basis of the 2Fobs − Fcalc electron-density map, a model of the yeast iso-1-CYC (PDB ID code 1YCC) was positioned manually by using the program o (22). The entire complex was subjected to rigid-body refinement by adding CYC as third entity. The R factor and free R factor were lowered to 27.8 and 28.0%, respectively. Residues Gly-23, Gly-24, and Pro-25 of CYC had to be repositioned according to the electron-density map by using the program o. Minor repositioning of residues within the CYC structure was carried out. The distance between the heme iron atom of CYC and its ligating sulfur atom of Met-80 was taken from the x-ray structure of the oxidized yeast CYC (23) and included in the refinement parameters. The model was refined by applying positional and B factor refinement by using the program package cns (21) to a final R factor and free R factor of 22.8 and 26.7%, respectively. Noncrystallographic symmetry constraints were not applied during the entire procedure of structure solution and refinement, to allow separate analysis of the two CYC-binding sites of the homodimeric QCR. Data collection and refinement statistics are summarized in Table 1. The QCR/Fv fragment model includes 4,356-aa residues with no major differences to the recent 2.3-Å resolution structure (16). CYC consists of 108 residues. The main and side chain atoms of its three N-terminal amino acid residues are disordered, as found previously in the high-resolution structure determination (17).
Table 1.
Data collection and refinement statistics
| Resolution range, Å | 30–2.97 |
| Rsym*, % | 10.6 |
| I/σI overall (3.08–2.97 Å) | 6.8 (2.3) |
| Completeness, % overall (3.08–2.97 Å) | 92.8 (76.5) |
| Multiplicity | 3.2 |
| No. of reflections used in working set, % | 158,002 (84.4) |
| No. of reflections used in test set, % | 17,442 (9.3) |
| No. of nonhydrogen atoms with occupancy > 0 in the model | 35,647 |
| Protein atoms | 35,264 |
| Heterog. atoms | 383 |
| Solvent atoms | 0 |
| B factor from Wilson plot (Å2) | 43.7 |
| Rfree†, % | 26.7 |
| Rcryst‡, % | 22.8 |
| rms bond length, Å | 0.008 |
| rms bond angles, ° | 1.42 |
Diffraction data were collected from a single crystal at the beamline ID14EH3 (European Synchrotron Radiation Facility, Grenoble, France) at 4°C by using a wavelength of 0.931 Å.
Rsym = ∑i,hkl|〈I(hkl)〉 − Ii(hkl)|∑i,hklIi(hkl).
Rfree = ∑(hkl)ɛT∥Fobs| − |Fcalc∥/∑(hkl)ɛT|Fobs|, where T is the test set (39).
Rcryst = ∑(hkl)∥Fobs| − |Fcalc∥/∑(hkl)|Fobs|.
Results and Discussion
Overall Structure of the QCR/CYC/Fv Fragment Complex.
Crystallization of the yeast QCR/CYC complex was achieved at approximately physiological ionic strength and with an antibody Fv fragment bound to subunit RIP1. The Fv fragments are essential for crystal packing, as is the case for the QCR/Fv fragment crystals, which were used for the 2.3-Å resolution structure of yeast QCR (16). The attached fragments provide spacious packing of the QCR molecules, thus allowing unhindered binding of CYC.
The structure was solved by molecular replacement. The dimeric complex is present in the asymmetric unit. 2Fobs − Fcalc electron-density maps after rigid-body refinement and energy minimization of the QCR/Fv fragment input model clearly revealed the presence of one CYC molecule bound to one QCR monomer (monomer B). Distinct electron density for most of the polypeptide main chain of the CYC molecule was visible. A clear-cut density for heme c and the adjacent α-helix (residues 61–69) was apparent. The highest-density peak was present at the position of the iron atom. Furthermore, side chains could be specifically identified in helical regions. These features clearly indicate the tight binding of CYC to QCR and allow the unambiguous placement of the CYC molecule in the structure (Fig. 1 A and B). Refinement of the structure resulted in Rcryst = 22.8% and Rfree = 26.7%. Some additional electron density was present after final refinement at the presumed CYC-binding site of monomer A. It was very weak and restricted mainly to the direct vicinity of the potential position of the heme iron. This result suggests binding with very low occupancy or disordered binding of CYC at the second binding site. It is unlikely that the binding of a second CYC molecule is prevented by crystal packing. Superimposition of monomer B and its bound substrate molecule with monomer A reveals no steric hindrance for a CYC molecule bound in equivalent orientation to the recognition site of monomer A.
Figure 1.

(A) Half-of-the-sites binding of CYC to the homodimeric QCR. The overall structure of the complex between the redox partners CYC and QCR with bound Fv fragment is shown. Protein subunits are depicted in ribbon representation with respective colors: CYC (yellow), CYT1 (red), cytochrome b (blue), RIP1 (green), QCR6 (cyan), and Fv fragment (orange). All other subunits are colored in gray. Redox cofactors (ball-and-stick representation) are colored in black. The complex is viewed parallel to the plane of the inner membrane (IM) that separates the intermembrane space (IMS) from the matrix (MA). The position of the inner membrane is indicated as gray boxes. (B) Close-up view of the recognition site (indicated by a black frame in A) showing the experimental electron-density map before inclusion of CYC to the model. The 2Fobs − Fcalc electron-density map (blue) is contoured at 1σ, and the corresponding part of the refined model (ball-and-stick presentation) is superimposed. The orientations of the CYC polypeptide (yellow) and its cofactor heme c (green) are unambiguously defined by distinct electron density. Protein residues of CYT1 and heme c1 are colored in red and magenta, respectively. The figure was generated by using the programs molscript (36) and bobscript (37).
CYC is bound to subunit CYT1 of monomer B of the dimeric QCR and is located close to the neighboring complex in the crystal lattice. There is no evidence that the orientation of the substrate molecule is influenced by crystal contacts. Opposed residues of CYC and subunit QCR2 of the neighboring QCR dimer are 5–7 Å apart. Furthermore, the side chains of the three N-terminal amino acid residues of CYC, which are located in the vicinity, are disordered, as is the case in the high-resolution structure of CYC (10); they are therefore unlikely to contribute to specific interactions. Therefore, we conclude that the binding between enzyme complex and substrate molecule is specific.
Binding Interactions Between the Redox Partners QCR and CYC.
The architecture of the complex results in close proximity between the heme c1 and heme c groups; their pyrrole C rings are pointing toward each other (Fig. 1). Remarkably, the CBC atoms of the two respective vinyl groups are only 4.5 Å apart. The distance between the two iron centers is 17.4 Å, the closest reported distance between the redox centers of CYC and its redox partners. The interplanar angle of the heme groups is 55°. Interestingly, this angle is close to the value of approximately 60° found for the CCP/CYC complex structure (11). This geometry might be generally favorable for electron transfer complexes between c-type cytochromes and their heme group containing redox partners.
Binding of CYC to QCR is stabilized by interactions of atoms surrounding the heme crevices, thus forming a tightly interacting and complementary contact area of trapezoid shape (Fig. 2A). All interactions of atoms between the redox partners with a distance below 4 Å are listed in Table 2. Mainly nonpolar interactions contribute to the binding (Fig. 2B). Most pronouncedly, Arg-13 of CYC and Phe-230 of CYT1 form a stable planar stacking interaction. Stacking pairs located at molecular surfaces of proteins are known to be involved in formation of enzyme substrate complexes (24). In the yeast CCP/yeast CYC complex, there is only a single van der Waals contact between Arg-13 of CYC and Tyr-39 of CCP. A positively charged amino acid residue is conserved at position 13 of CYC (17). In mammalian CYC, Lys-13 has been shown to be important for binding to QCR (6). The structure indicates that for the binding of yeast CYC, nonpolar interactions of Arg-13 with aromatic residues of the redox partner seem to be more important than the conserved positive charge.
Figure 2.

The complementary recognition sites in the QCR/CYC complex. Surface representations of CYC and CYT1 are shown on Left and Right, respectively. (A) Residues that are involved in CYC binding and have intermolecular contacts of less than 4 Å are colored in green. (B) Residues, which are hydrophobic, are colored in orange. (C) Side chains, which have positive or negative full charges, are colored in blue or red, respectively. Color maxima correspond to +25 and −25 kBT. The figure was generated by using grasp (38).
Table 2.
Distances and binding forces relevant for the interaction between the redox partners CYC and QCR
| Cytochrome c1 | Cytochrome c | Distance, Å |
|---|---|---|
| Distances between heme groups | ||
| Heme c1 Fe | Heme c Fe | 17.4 |
| Heme CBC | Heme CBC | 4.5* |
| Nonpolar interactions | ||
| Ala-103 CB | Ala-81 CB | 3.4 |
| Phe-230 CD1 | Arg-13 CD | 3.6 |
| Phe-230 CD1 | Arg-13 NE | 3.3 |
| Phe-230 CE1 | Arg-13 CG | 3.8 |
| Phe-230 CE1 | Arg-13 CD | 3.8 |
| Phe-230 CE1 | Arg-13 NE | 3.8 |
| Phe-230 CG | Arg-13 NE | 4.0 |
| Phe-230 CG | Arg-13 CD | 4.0 |
| Phe-230 CZ | Arg-13 CG | 4.0 |
| Phe-230 CZ | Thr-12 O | 3.3 |
| Met-233 CE | Arg-13 NH2 | 3.8 |
| Ala-168 O | Val-28 CG1 | 4.0 |
| Potential polar interactions | ||
| Glu-235 OE2 | Lys-86 NZ† | 3.3 |
| Ala-164 O | Lys-79 NZ† | 3.8 |
Minimum distance between heme groups.
Orientation of side chain not unambiguously defined.
Additional van der Waals interactions are present between Phe-230 CZ and Thr-12 O, Met-233 CE and Arg-13 NH2, Ala-168 O and Val-28 CG1, and Ala-103 CB and Ala-81 CB of CYT1 and CYC, respectively (Table 1). Ala-103 is in the third position of the heme-binding motif CXXCH of CYT1. This residue is as close as 3.4 Å to Ala-81 of CYC. The preceding residue Met-80 is one of the axial heme ligands in CYC. This interaction could be involved in electron transfer between the redox partners, as will be discussed below.
Chemical labeling, crosslinking, and mutagenesis studies have identified two regions of acidic residues in CYT1, which are important for CYC binding (7, 8), by interaction with a ring of lysine residues around the heme cleft of CYC (6). In the present structure, only weak polar interactions in the recognition site are possible between Glu-235 OE2 and Lys-86 NZ (3.3 Å) as well as between Ala-164 O and Lys-79 NZ (3.8 Å) of CYT1 and CYC, respectively (Table 1; Fig. 2C). The charge of Glu-235 is conserved in mitochondrial CYT1 (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). Lys-86 is completely conserved in CYC, and chemical labeling studies have indicated that this residue is very important for binding of CYC (6). However, the orientation of the lysine side chains cannot be unequivocally defined. Additional pairs of charged and often conserved residues are found surrounding the described recognition site. They are not close enough for polar interactions (e.g., 4.8 Å between Lys-87 of CYC and Asp-232 of CYT1). It cannot be excluded that water molecules, which are not resolved at the given resolution, mediate stabilizing forces. Because the orientation of the respective side chains at the protein surface is often disordered, a stabilizing effect seems unlikely. The conserved charged residues, which surround the actual recognition site, may be involved in electrostatic interactions that modulate intermediate states of binding and unbinding of CYC to QCR, similar to the proposed model of plastocyanin binding to cytochrome f (25).
Subunit QCR6, a small acidic protein known as hinge protein, is thought to be involved in CYC binding via its N-terminal peptide, which contains 79% negatively charged residues (8). The N terminus seems to be mobile; the first 48-aa residues are disordered, as previously reported for the structure of yeast QCR (16). The first ordered residues are in close vicinity to Lys-86 and Lys-87 of CYC. However, no direct interactions are visible between QCR6 and CYC in the structure. CYC of yeast, in comparison to CYC from other eukaryotic species, has a relatively high dipole moment of 554 debye (26). Interestingly, the positive dipole portion of CYC bound to QCR points toward that part of the N-terminal acidic patch of QCR6 that is ordered and therefore resolved in the structure, which might indicate that the subunit QCR6 directs the orientation of CYC to be optimal for binding and electron transfer.
Formation of transient protein complexes is required for the CYC-mediated electron shuttling between QCR and cytochrome c oxidase. For rapid electron transfer, weak protein complexes with short lifetimes are expected. An extensive study of a variety of protein–protein recognition sites has shown that low-stability complexes exhibit interfacial areas <1,600 Å2 (27). The recognition site in the QCR/CYC complex was determined (28) to be 880 Å2. This site is smaller than the 1,150 Å2 for the CCP/CYC complex interface and is the smallest recognition site reported so far (27). The small interface of the QCR/CYC complex is well suited for a fast turnover. In addition, no significant conformational changes are present, compared with the structures of the noncomplexed molecules, as is expected for low-stability complexes (27).
In conclusion, there is a tight and specific interaction between CYC and QCR, which is mediated by a small compact contact site dominated by nonpolar forces. We propose that the structure resembles the physiological electron transfer complex. The reduction of CYC could occur via a single productive complex rather than multiple productive conformations.
Electron Transfer.
Binding of CYC to QCR results in a very short distance of the two heme groups, which are facing each other with their solvent exposed edges (Fig 3). The architecture of the QCR/CYC complex leaves both heme groups solvent exposed. This orientation allows a direct electron transfer from heme c1 to heme c involving a through-space jump. To calculate the electron transfer rate, the electron-tunneling model for redox proteins of Dutton and colleagues (29) was used. The edge-to-edge distance of the heme groups (C3C/C3C) is determined to be 9.4 Å, and the packing density ρ is 0.65. The free-energy optimized electron-transfer rate based on the structure was calculated to be 1.2 × 109 s−1. Applying a midpoint potential of 270 mV for both CYT1 (30) and CYC (31), the driving force ΔG for electron transfer is zero. With a ΔG = 0 eV and a reasonable estimate of the reorganization energy λ of between 0.7 eV and 1 eV, an electron transfer rate of between 8.3 × 106 s−1 and 9.7 × 105 s−1 is calculated. This result compares to a measured transfer rate of 6 × 104 s−1 for bovine QCR and horse heart CYC determined by using an artificially introduced electron donor, which affects CYC binding (32). This discrepancy suggests a significantly higher transfer rate in the natural environment than has been measured with the artificial donor.
Figure 3.

Direct heme-to-heme electron transfer from CYT1 (Lower) of QCR to CYC (Upper). The short edge-to-edge distance (indicated as dashed line) between solvent exposed atoms of the pyrrole C rings of the cofactors allows rapid CYC reduction at a calculated rate of up to 8.3 × 106 s−1. The polypeptides are represented as ribbon model with helices, β-strands, and loops colored in cyan, green, and yellow, respectively, and the heme groups are shown in ball-and-stick representation. The heme carbons are colored in gray, all other atoms according to standard coloring. The figure was generated by using the programs molscript (36) and bobscript (37).
An alternative treatment for assessing the rate of electron transfer relies on tracking structural pathways through the intervening protein (33). A short relay of covalent bonds connects the pyrrole C of heme c1 via Cys-104 to Ala-103. The latter residue is in unusually close van der Waals contact to Ala-81 of CYC. Ala-81 is the neighboring residue to Met-80, which is the axial ligand to the heme c iron. A pathway of 16 covalent bonds and one van der Waals contact can be drawn to connect the two redox centers. On the basis of experimentally determined protein-mediated electron transfer rates in CYC (33), this pathway would presumably result in a significantly slower electron transfer rate compared with the above-described heme-to-heme transfer. In addition, the connecting alanines are not conserved, further indicating that the protein-mediated pathway is not of general importance.
On the basis of the orientation and close proximity of the heme groups in the protein environment and the calculated electron transfer rate resulting therefrom, we conclude that reduction of CYC by CYT1 takes place via direct heme-to-heme electron transfer.
Binding Stoichiometry.
Remarkably, CYC binds specifically only to one (monomer B) of the two possible recognition sites of the dimeric QCR, although CYC is present in the crystallization setup in slight molar excess. The observed binding stoichiometry is termed half-of-the-sites binding. The CYC-binding sites of monomer A and B are identical in conformation and do not show major displacements (larger than 1 Å) of main chain atoms as analyzed by superimposition of the two molecules. A group of negatively charged residues on the surface of CYT1 and QCR6 showed different side chain orientations in the two monomers, but these residues are outside the CYC contact area. As outlined above, the crystal packing does not prevent CYC binding to monomer A. In addition, there is no evidence that binding of CYC to monomer A is prevented by a higher degree of global disorder in monomer A. The averaged B factors for each subunit of monomer A and B are very similar. The average B factor of CYT1 in monomer A (45%) is 7% higher than in monomer B. The lower flexibility of CYT1 in monomer B might promote CYC binding; on the other hand, the bound CYC molecule might stabilize its binding partner and therefore could be the reason for the lower B factor. It has to be expected that subtle differences, which are not resolved at the given resolution of the structure, cause different binding properties of the two recognition sites for CYC.
The homodimeric QCR consists of two operational units. Each of them can, in principle, perform catalysis according to the Q cycle. It is not known whether these units function in a parallel, sequential, or independent mode. The specific binding of one CYC molecule to one monomer of QCR indicates that QCR might be able to reduce CYC with the second functional unit not being active, thereby supporting a sequential or independent mode. We propose that reversible “silencing” of one functional unit might be a tool to regulate QCR activity in response to the respiratory condition. The possibility of a so-called half-of-the-sites mechanism was previously suggested on the basis of a mutagenesis study, in which QCR6 deletion led to decrease in QCR activity by 50% (34).
Coordination of the Reduction Sites of CYC and Quinone.
The CYC half-of-the-sites binding coincides with differences in the two ubiquinone reduction sites (Qi sites) of QCR (Fig. 4). The presence and orientation of a bound coenzyme Q6 (UQ6) molecule in the Qi site of monomer B are clearly indicated by continuous electron density (2Fobs − Fcalc) after refinement of the structure for the head group and the adjacent seven carbon atoms of the isoprenoid chain. The positions of the carbonyl groups are well defined. In contrast, in the Qi site of monomer A, there is only residual electron density for the benzoquinone moiety present, indicating a low occupancy and/or higher mobility of the molecule. Furthermore, the bent propionate A of heme bH is slightly displaced in monomer A. This substituent is stabilizing the quinone ring plane by nonpolar interaction (16). Two UQ6 molecules were introduced in the respective sites in an orientation taken from the yeast QCR structure (16). Refinement of this model confirmed the orientation of the side chains and propionate groups in the binding pocket. The B factor of the side chain of the UQ6 carbonyl ligand Asp-229 is 25% lower in monomer B compared with monomer A, whereas B factors of further residues in the binding pocket are very similar. This difference indicates a weaker interaction with UQ6 in monomer A, thereby supporting the importance of residue Asp-229 in stabilization of quinone binding and its presumable role as proton donor for quinone reduction (16).
Figure 4.

Differences in the occupancy of the quinone reduction sites (Qi sites) of the homodimeric QCR. The Qi site of (A) monomer A and (B) monomer B, the latter has CYC bound, is shown with part of the quinone-ligating sphere including Ser-206, Met-221, and propionate A (propA) of heme bH. The electron-density map after final refinement is shown in blue (2Fobs − Fcalc; contoured at 1σ) and in green (Fobs − Fcalc; contoured at 3σ). Protein and cofactors are shown as stick drawings with atoms displayed in standard colors. The figure was prepared by using the program o (22).
It is intriguing to speculate that the state of the Qi site affects CYC binding. We propose a coordinated binding model in which both electron acceptors for quinol oxidation are present in their respective sites at the same time. Coordinated binding implies long-range interactions within the QCR molecule. A kinetic study of yeast QCR, in which specific binding of the inhibitor antimycin to the Qi site impedes CYT1 reduction by 10-fold (35), provides a hint that communication between high- and low-potential redox components exists. Qi site occupancy and CYC binding might depend on the redox state of the respective redox centers. QCR preparations used for crystallization have been shown to be partially reduced (unpublished data). Coordination between the two binding sites could be communicated by redox-induced subtle conformational changes, alterations in the protonation state of side chains in the protein interior, or long-range electrostatic interactions. To test our assumption, further experiments are required to analyze whether the redox state of the different QCR cofactors affects CYC binding and how coordination of the two reduction sites, as well as between the two monomers, is achieved. Both coordinated and half-of-the-sites binding suggest that CYC reduction by QCR can be regulated in response to the respiratory condition.
Supplementary Material
Acknowledgments
We thank B. L. Trumpower and L. Dutton for critically reading the manuscript. We acknowledge assistance by the beamline staff of ID14EH3, European Synchrotron Research Facility, Grenoble, France, support with the data collection by N. Hanekop, and technical assistance by D. Vinzenz. We are grateful to H. Michel for continuous support. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 472) and the Max-Planck-Gesellschaft.
Abbreviations
- CYC
cytochrome c
- CCP
cytochrome c peroxidase
- CYT1
cytochrome c1
- QCR
cytochrome bc1 complex
- QCR6
subunit 6 of QCR
- RIP1
Rieske iron–sulfur protein
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1KYO).
References
- 1.Saraste M. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell P. J Theor Biol. 1976;62:327–367. doi: 10.1016/0022-5193(76)90124-7. [DOI] [PubMed] [Google Scholar]
- 3.Brandt U, Trumpower B L. Crit Rev Biochem Mol Biol. 1994;29:165–197. doi: 10.3109/10409239409086800. [DOI] [PubMed] [Google Scholar]
- 4.Yu C-A, Yu L, King T E. J Biol Chem. 1973;248:528–533. [PubMed] [Google Scholar]
- 5.Speck S H, Margoliash E. J Biol Chem. 1984;259:1064–1072. [PubMed] [Google Scholar]
- 6.Rieder R, Bosshard H R. J Biol Chem. 1980;255:4732–4739. [PubMed] [Google Scholar]
- 7.Broger C, Salardi S, Azzi A. Eur J Biochem. 1983;131:349–352. doi: 10.1111/j.1432-1033.1983.tb07269.x. [DOI] [PubMed] [Google Scholar]
- 8.Nakai M, Endo T, Hase T, Tanaka Y, Trumpower B L, Harudo I, Asada A, Bogaki M, Matsubara H. J Biochem. 1993;114:919–925. doi: 10.1093/oxfordjournals.jbchem.a124277. [DOI] [PubMed] [Google Scholar]
- 9.Kim C H, King T E. J Biol Chem. 1983;258:13543–13551. [PubMed] [Google Scholar]
- 10.Stonehuerner J, O'Brien P, Geren L, Millet F, Steidl J, Yu L, Yu C-A. J Biol Chem. 1985;260:5392–5398. [PubMed] [Google Scholar]
- 11.Pelletier H, Kraut J. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Durley R C, Mathews F S, Davidson V L. Science. 1994;264:86–90. doi: 10.1126/science.8140419. [DOI] [PubMed] [Google Scholar]
- 13.Xia D, Yu C A, Kim H, Xia J Z, Kachurin A M, Zhang L, Yu L, Deisenhofer J. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z L, Huang L S, Shulmeister V M, Chi Y I, Kim K K, Hung L W, Crofts A R, Berry E A, Kim S H. Nature (London) 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 15.Iwata S, Lee J W, Okada K, Lee J K, Iwata M, Rasmussen B, Link T A, Ramaswamy S, Jap B K. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 16.Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. Structure (London) 2000;8:669–684. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 17.Louie G V, Brayer G D. J Mol Biol. 1990;214:527–555. doi: 10.1016/0022-2836(90)90197-T. [DOI] [PubMed] [Google Scholar]
- 18.Moench S J, Satterlee J D. J Prot Chem. 1995;14:567–582. doi: 10.1007/BF01886883. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 21.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Jones A, Zou J-Y, Cowan S, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Berghuis A M, Brayer G D. J Mol Biol. 1992;223:959–976. doi: 10.1016/0022-2836(92)90255-i. [DOI] [PubMed] [Google Scholar]
- 24.Flocco M M, Mowbray S L. J Mol Biol. 1994;235:709–717. doi: 10.1006/jmbi.1994.1022. [DOI] [PubMed] [Google Scholar]
- 25.Ubbink M, Ejdebäck M, Karlsson B G, Bendall D S. Structure (London) 1998;6:323–335. doi: 10.1016/s0969-2126(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 26.Tiede D M, Vashista A-C, Gunner M R. Biochemistry. 1993;32:4515–4531. doi: 10.1021/bi00068a006. [DOI] [PubMed] [Google Scholar]
- 27.Lo Conte L, Chothia C, Janin J. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 28.Hubbard S J, Campbell S F, Thornton J M. J Mol Biol. 1991;220:507–530. doi: 10.1016/0022-2836(91)90027-4. [DOI] [PubMed] [Google Scholar]
- 29.Page C C, Moser C C, Chen X, Dutton P L. Nature (London) 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 30.T'sai A-L, Palmer G. Biochim Biophys Acta. 1983;722:349–363. doi: 10.1016/0005-2728(83)90083-x. [DOI] [PubMed] [Google Scholar]
- 31.Cutler R L, Pielack G J, Mauk A G, Smith M. Protein Eng. 1987;1:95–99. doi: 10.1093/protein/1.2.95. [DOI] [PubMed] [Google Scholar]
- 32.Tian H, Sadoski R, Zhang L, Yu C-A, Yu L, Durham B, Millet F. J Biol Chem. 2000;275:9587–9595. doi: 10.1074/jbc.275.13.9587. [DOI] [PubMed] [Google Scholar]
- 33.Gray H B, Winkler J R. Annu Rev Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt M E, Trumpower B L. J Biol Chem. 1990;265:17005–17011. [PubMed] [Google Scholar]
- 35.Snyder C H, Gutierrez-Cirlos E B, Trumpower B L. J Biol Chem. 2000;275:13535–13541. doi: 10.1074/jbc.275.18.13535. [DOI] [PubMed] [Google Scholar]
- 36.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 37.Esnouf R M. Acta Crystallogr D. 1999;55:938–940. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls A, Sharp K, Honig B H. Proteins Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 39.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}