

Abstract
The activity of the p53 tumor suppressor protein and the c-Jun protooncogene is regulated by posttranslational modifications, such as phosphorylation or ubiquitination. In addition, covalent attachment of the ubiquitin-like modifier SUMO appears to modulate their transcriptional activity. Sumoylation proceeds via an enzymatic pathway that is mechanistically analogous to ubiquitination, but requires a different E1-activating enzyme and Ubc9, a SUMO-specific E2-conjugating enzyme. Here, we show that two members of the PIAS family, PIAS1 and PIASxβ, act as specific E3-like ligases that promote sumoylation of p53 and c-Jun in vitro and in vivo. The PIAS proteins physically interact with both p53 and c-Jun. In addition, they bind to Ubc9, suggesting that they recruit the E2 enzyme to their respective substrate. The SUMO ligase activity requires the conserved zinc-finger domain, which is distantly related to the essential RING-finger motif, found in a subset of ubiquitin ligases. Furthermore, similar to RING-type ubiquitin ligases, PIASxβ can catalyze its own modification. Hence, these data further extend the analogy between the ubiquitin and SUMO pathway. Strikingly, PIAS proteins strongly repress the transcriptional activity of p53, suggesting that the PIAS–SUMO pathway plays a crucial role in the regulation of p53 and presumably other transcription factors.
Many cellular key functions are regulated by the covalent modification of proteins with the ubiquitin-like SUMO-1 modifier (1–3). Two important examples are the transcriptional activity of the c-Jun protooncogene and the p53 tumor suppressor, which seem to be regulated by sumoylation (4–6). Mammalian SUMO-1 and its close relatives, SUMO-2 and SUMO-3, are about 18% identical to ubiquitin and are conjugated via an enzymatic cascade that requires a SUMO-specific, heterodimeric E1-activating enzyme (Aos1/Uba2) and a single E2-type conjugating enzyme, Ubc9. In the ubiquitin pathway, at least one additional factor, called E3 or ubiquitin ligase, is required for substrate recognition and formation of an isopeptide bond between ubiquitin and the target protein. Because the SUMO-specific E2 enzyme Ubc9 has been shown to physically interact with almost all known SUMO substrates in yeast two-hybrid assays, Ubc9 was considered to be sufficient for substrate recognition.
Very recently, however, in the yeast Saccharomyces cerevisiae, the Siz1 and Siz2 proteins were identified as E3-like factors that promote SUMO conjugation to yeast proteins, in particular to proteins of the septin family (7–9). Siz proteins are homologous to members of the mammalian PIAS (protein inhibitor of activated Stat) family of proteins. In humans, this family consists of at least five members: PIAS1, PIAS3, the α and β splice variants of PIASx, and PIASy (10). PIAS proteins were initially identified as specific inhibitors of Stat transcription factors. PIAS1 and PIAS3 block the DNA binding activity of activated Stat1 and Stat3, respectively, and inhibit Stat-mediated transcription (11, 12). Subsequently, however, PIAS proteins were also reported to regulate the activity of several other transcription factors. For example, an NH2-terminal truncated version of PIASxβ (also known as Miz1) was reported to activate the DNA binding affinity of the Msx2 homeodomain-containing protein (13), and PIAS3 was shown to regulate the activity of the Gfi-1 zinc-finger transcription factor (14). Furthermore, several PIAS proteins were shown to act as either positive or negative coregulators of nuclear hormone receptors (15, 16). PIAS1, PIASxα, and PIASy had also been isolated as p53-interacting proteins in two-hybrid screens by using p53 as bait (17–19), but the functional significance of this interaction has remained unclear. Intriguingly, one of these screens identified both Ubc9 and PIAS1. Moreover, PIASxα has been reported to physically interact with SUMO-1 (18).
Here, we report that PIAS proteins can act as specific SUMO ligases that mediate sumoylation of p53 and c-Jun. In addition, we show that PIAS proteins exert a strong repressive effect on p53-dependent transactivation.
Materials and Methods
Cell Culture, Transfection, and Western Blotting.
HeLa cells were grown under standard conditions and transfected by using Lipofectamine plus reagent (GIBCO/BRL). For 3.5-cm-diameter cell culture dishes, the amount of transfected plasmid was 0.3 μg for p53, 0.7 μg for SUMO-1, and 1 μg for each PIAS. To allow normalization of transfection efficiency, 0.3 μg of pEGFP control plasmid (CLONTECH) was cotransfected; 36 h after transfection, cells were directly lysed in SDS-sample buffer. Western Blotting was done by using the Western-Star immunodetection system (Tropix, Bedford, MA).
Antibodies, Plasmids, and Mutagenesis.
The monoclonal anti-p53 antibodies (Pab1801 and Do-1) were purchased from Santa Cruz Biotechnology, the monoclonal anti SV-5 and the polyclonal anti-GFP antibody from Invitrogen, and the anti-Flag antibody (M2) from Sigma. Plasmid p53 in pRC/CMV (Invitrogen) for eukaryotic expression and in vitro translation of p53 was provided by M. Oren and Y. Haupt, mouse c-Jun in pGEM (Promega) was provided by L. Bakiri and M. Yaniv (Pasteur Institute, Paris), the SV5-tagged PIASxβ clone in pcDNA3.1/GS was purchased from Invitrogen, and the cDNA for PIAS1 was isolated by reverse transcriptase–PCR from HeLa cells and cloned into pCMV-Tag2B (Stratagene). Cytomegalovirus IE-1 and the processed version of SUMO-1, SUMO-GG, which lacks the four C-terminal amino acids, were cloned into pSG5 (Stratagene). Site-directed mutagenesis was carried out by using the quick change site-directed mutagenesis kit (Stratagene). For bacterial expression of glutathione S-transferase (GST)-fusion proteins PIASxβ, PIASxβC362S, and PIAS1 were cloned into either pGex-2TK or pGex-4T1 (Amersham Pharmacia). For bacterial expression of the heterodimeric E1 enzyme, the Aos1 subunit was cloned into pGex-2TK and the Uba2 subunit into pET28a(+) (Novagen). To express the MBP-Ubc9 fusion, Ubc9 was subcloned into the pMal-c2E vector (New England Biolabs).
Expression of Recombinant Proteins and in Vitro Sumoylation.
GST-PIAS1, GST-PIASxβ, and GST-PIASxβC362S fusion proteins were expressed in Escherichia coli BL21, and the proteins were purified on glutathione-Sepharose 4B beads (Amersham Pharmacia) according to the manufacturer's protocol. MBP-Ubc9 was purified on an amylose resin (New England Biolabs) according to the manufacturer's protocol. The heterodimeric E1 was purified from bacteria expressing His-tagged Uba2 together with GST-tagged Aos1 as described (20). For in vitro sumoylation, the substrates were in vitro translated by using the TNT quick-coupled reticulocyte lysate system (Promega); 1.5 μl of translation product was incubated for 2 h at 30°C in a 20-μl reaction (50 mM Tris, pH 7.5/5 mM MgCl2/2 mM ATP) containing 100 ng E1 (Aos1/Uba2), 50 ng of Ubc9, and 2 μg of SUMO-GG. GST-PIAS proteins were added in a concentration range from 10 to 100 ng.
GST-Pulldown Assays.
[35S]Methionine-labeled in vitro translated proteins were incubated with the relevant GST-fusion proteins loaded on glutathione-Sepharose 4b beads for 2 h at 4°C in GST binding buffer (120 mM NaCl/50 mM Tris, pH 8/0.25% Nonidet P-40/1 mM PMSF/1 mM DTT). Beads were washed three times with binding buffer, and bound proteins were eluted with SDS sample buffer and analyzed by gel electrophoresis, followed by autoradiography.
Reporter Gene Assays.
Cells were transfected in six-well dishes as described above by using 200 ng of a firefly luciferase reporter gene plasmid (pRGC, provided by M. Oren) that harbors a p53 DNA binding site in its promoter. To normalize for transfection efficiency, 50 ng of a pRL-SV40 renilla luciferase control vector (Promega) were cotransfected. Twenty-four hours after transfection, cells were lysed in 500 μl of passive lysis buffer, and 20 μl of cell extract were analyzed with the Dual Luciferase Reporter Assay (Promega) by using a Berthold Lumat LB9507 apparatus.
Results
PIAS1 and PIASxβ Stimulate Sumoylation of p53 and c-Jun in Vitro.
The findings described above led us to investigate whether PIAS proteins could promote the sumoylation of p53. To address this question, we used a reconstituted in vitro sumoylation assay, where a 35S-labeled substrate, generated by in vitro translation, serves as a target for SUMO modification in the presence of recombinant E1 (the Aos1/Uba2 heterodimer), recombinant Ubc9, SUMO-1, and ATP. In this system, quite a robust sumoylation of p53 had been observed previously without any additional components (4–6, 21). However, these experiments were performed in the presence of relatively high amounts of Ubc9. When reducing the amount of Ubc9, only minimal sumoylation of p53 was observed (Fig. 1 A and B, lane 2). Strikingly, when recombinant, bacterially expressed GST-PIAS1 was added to the reaction, a drastic dose-dependent stimulation of p53 sumoylation was detected (Fig. 1A, lanes 3–5). Next, we investigated whether the related PIASxβ protein exerts similar activities. As shown in Fig. 1B, PIASxβ enhanced SUMO modification of p53 to an even slightly higher extent than PIAS1. Interestingly, at higher concentrations, PIASxβ induced the attachment of at least one additional SUMO molecule to p53. This could either represent SUMO–SUMO chains that are formed on lysine 386, the major SUMO attachment site in p53, or may result from SUMO conjugation to additional lysine residues of p53. To distinguish between these two possibilities, we used the p53K386R mutant that has lost the major sumoylation site as a substrate in our assay. As shown in Fig. 1C, p53K386R seems to be not sumoylated under basal conditions but undergoes a weak sumoylation at high PIASxβ concentrations. Hence, although we cannot formally exclude the possibility that PIAS proteins can promote chain formation, these data are more consistent with the interpretation that PIAS not only enhances sumoylation of p53 on lysine 386, it also catalyzes the attachment of SUMO to additional, but less potent acceptor lysine residues in p53.
Figure 1.

PIAS 1 and PIASxβ promote sumoylation of p53 in vitro. The indicated proteins were in vitro translated and incubated either in the absence (−) or presence (+) of the assay mix containing recombinant E1 (Aos1/Uba2), Ubc9, and SUMO-1. Where indicated, GST-PIAS1 (A), GST-PIASxβ (B and C), or GST-PIASxβC362S (D) was added at concentrations of 10, 50, and 100 ng.
A subfamily of E3 ubiquitin ligases is defined by the presence of the zinc binding RING-finger motif that is essential for their ligase function (22). Intriguingly, PIAS proteins harbor a zinc-finger region, designated as the Miz zinc-finger or SP-RING motif (23) that is distantly related to the classical RING motif. To test whether the integrity of the zinc finger is important for PIAS activity, we changed a cysteine residue at amino acid position 362 within this region in PIASxβ to serine (PIASxβC362S) and used this mutant in an in vitro sumoylation experiment. As shown in Fig. 1D, the PIASxβC362S mutant has almost completely lost its ability to promote the formation of p53-SUMO conjugates, strongly suggesting that the zinc-finger domain plays an essential role for the SUMO ligase function of PIAS.
Importantly, the SUMO ligase activity of PIAS toward p53 did not depend on proteins that were present in the reticulocyte lysate, as the modification of bacterially expressed recombinant GST-p53 was also drastically stimulated by PIASxβ (Fig. 2).
Figure 2.

PIAS induces sumoylation of recombinant p53. p53 expressed as a GST-fusion protein in E. coli was used for in vitro sumoylation as described in Fig. 1. GST-p53 was used at a concentration of 50 ng and GST-PIASxβ at 250 ng. p53 was detected by immunoblotting by using an anti-p53 antibody.
To further examine whether the SUMO ligase activity of PIAS proteins is restricted to p53, we tested various other SUMO targets in this in vitro system. PIAS did not exert any effects on sumoylation of the IκBα protein (Fig. 3A Left), the IE1 protein from cytomegalovirus (Center), or the Sp100 protein (Right). By contrast, SUMO modification of the c-Jun transcription factor was induced by both PIASxβ and PIAS1. As shown in Fig. 3B, under conditions of low Ubc9 concentration, no basal sumoylation of c-Jun was observed in this assay. However, addition of either PIASxβ or PIAS1 greatly stimulated the conjugation of SUMO to c-Jun. Similarly to what was observed for p53, at higher PIAS concentrations at least two SUMO molecules were added to c-Jun. The c-JunK229R mutant, which has lost the lysine residue 229 functioning as the major SUMO attachment site, could still undergo “monosumoylation” (data not shown), strengthening the idea that in the presence of PIAS, alternative lysine residues of substrate proteins are targeted by SUMO.
Figure 3.

PIAS 1 and PIASxβ promote sumoylation of c-Jun in vitro. The indicated proteins were in vitro translated and incubated either in the absence (−) or presence (+) of the assay mix as described in Fig. 1. In A, GST-PIASxβ was added at a concentration of 100 ng, where indicated. In B, GST-PIASxβ and GST-PIAS1 were used at concentrations of 50 and 100 ng.
PIAS Interacts with Substrate and Ubc9.
E3 ubiquitin ligases are considered to act as adaptor molecules that are able to recruit ubiquitin-conjugating enzymes to their substrate proteins. This activity is brought about by the ability of the ligase to bind both the substrate and the E2. To investigate whether PIAS functions analogously, we tested for PIAS–substrate and PIAS–Ubc9 interaction by performing in vitro GST-pulldown assays. Consistent with the p53–PIAS1 interactions found in yeast-two-hybrid interaction assays, in vitro translated 35S-labeled p53 bound GST-PIAS1. In addition, we observed binding to GST-PIASxβ but not the GST-control beads or beads loaded with a fragment of PIASxβ containing only the zinc-finger domain (amino acids 336–397) (Fig. 4A). The mutant PIASxβC362S is still able to bind p53, indicating that its inability to promote sumoylation of p53 is not because of the loss of interaction. Similarly to p53, c-Jun could also bind specifically to PIAS1, PIASxβ, and PIASxβC362S (Fig. 4B). The unrelated luciferase protein did not bind to any of the GST-fusion proteins, demonstrating the specificity of the observed interactions (Fig. 4C).
Figure 4.

p53 and c-Jun bind to PIAS1 and PIASxβ. 35S-labeled in vitro translated p53 (A), c-Jun (B), and luciferase (C), serving as a negative control, were incubated with the indicated GST fusion proteins bound to glutathione-Sepharose beads. Beads were washed, eluted, and subjected to electrophoresis and autoradiography to reveal the bound radiolabeled proteins. The input representing 20% for each radiolabeled protein is shown.
A putative PIAS–Ubc9 interaction was tested by incubating in vitro translated 35S-labeled PIASxβ with GST-Ubc9, GST-p53 as a positive control, and GST as a negative control. As shown in Fig. 5A, PIASxβ strongly and specifically bound to both GST-p53 and GST-Ubc9 but not to the GST protein, demonstrating that PIAS may indeed act as an adaptor between substrate and E2.
Figure 5.

PIASxβ binds to Ubc9. 35S-labeled in vitro translated PIASxβ (A) or luciferase (B), serving as a negative control, was incubated with the indicated GST fusion proteins as in Fig. 4.
PIAS Catalyzes Its Own Modification.
Another characteristic feature of RING-type E3 ubiquitin ligases is their ability to catalyze their own ubiquitination in the absence of added substrates. Because of the apparent mechanistic analogy between RING-type ligases and PIAS proteins, we wanted to investigate whether PIASxβ could undergo “autosumoylation.” To this end, in vitro translated 35S-labeled PIASxβ was used in the in vitro sumoylation assay without additional substrate proteins. As shown in Fig. 6, PIASxβ is strongly sumoylated when incubated with E1, Ubc9, and SUMO-1 indicated by the appearance of at least three higher molecular weight SUMO conjugates. The identity of these bands was verified by immunoblotting with an anti-SUMO antibody (not shown). Sumoylation of PIASxβC362S was severely compromised, as only minimal conjugation occurs. Intriguingly, addition of recombinant wild-type PIASxβ protein could at least partially restore the modification of PIASxβC362S. This indicates that PIAS proteins not only mediate the transfer of SUMO to heterologous substrates but also to themselves. Consistent with the data from these in vitro experiments, we found that PIASxβ could undergo sumoylation in vivo when expressed together with SUMO (data not shown).
Figure 6.
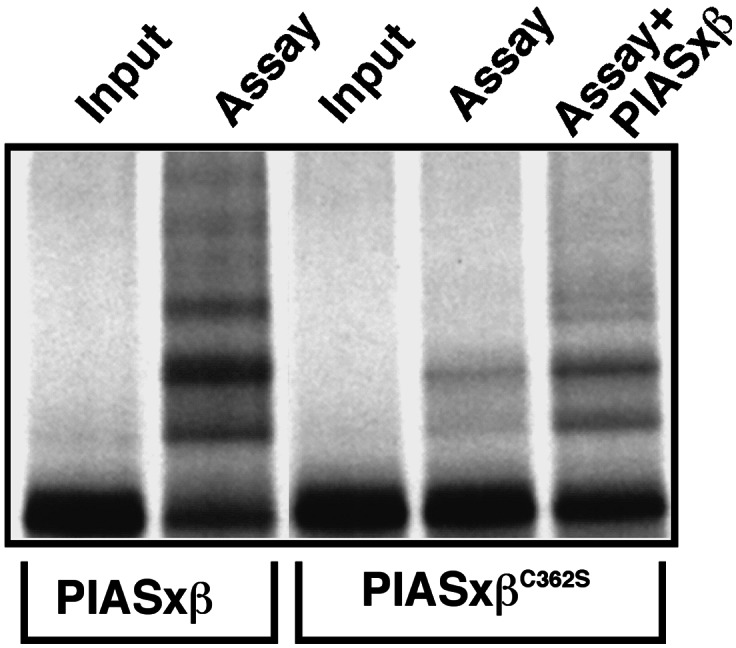
PIASxβ catalyzes its own modification.35S-labeled in vitro translated PIASxβ or PIASxβC362S was incubated either in the absence (−) or presence (+) of the assay mix containing recombinant E1 (Aos1/Uba2), Ubc9, and SUMO-1. GST-PIASxβ (100 ng) was added where indicated.
PIASxβ and PIAS1 Stimulate Sumoylation of p53 in Vivo.
After having established the stimulating effect of PIAS on SUMO-1 modification in vitro, we wanted to test whether PIAS proteins could mediate sumoylation in vivo. To this end, we first examined the effect of PIAS1 expression on sumoylation of p53. As shown previously, a small fraction of exogenously expressed p53 undergoes covalent modification when coexpressed together with SUMO-1 in HeLa cells (Fig. 7A Upper, lane 2). Strikingly, sumoylation of p53 was drastically increased when a Flag-tagged version of PIAS1 was coexpressed (lane 4). When we carried out analogous experiments by expressing SV5-tagged PIASxβ, a strong enhancement of p53 sumoylation was detected (Fig. 7B). Similar to what was observed in vitro, upon longer exposure, a second sumoylated p53 species was detected in the presence of PIAS (data not shown), indicating that in vivo PIAS cannot only induce the formation of the mono-sumoylated p53, but it promotes the attachment of additional SUMO molecules to p53. Consistent with the in vitro experiments, the zinc-finger mutant PIASxβC362S has completely lost the capacity to stimulate the formation of p53-SUMO conjugates (Fig. 7C). This apparent loss of activity was not dependent on PIASxβC362S protein levels as revealed by Western blotting (Fig. 7 B and C, Middle).
Figure 7.

PIAS 1 and PIASxβ enhance sumoylation of p53 in vivo. HeLa cells were transfected with p53 alone or together with SUMO-1 (SUMOGG) as indicated. Plasmids expressing a Flag-tagged PIAS1 (A), an SV5-tagged PIASxβ (B), and the SV5-tagged PIASxβC362S mutant (C) were cotransfected where indicated. Cells were directly lysed in SDS-sample buffer, and samples were analyzed by Western blotting with antibodies as indicated. All samples had been cotransfected with plasmids expressing green fluorescent protein to verify equal transfection efficiency by immunoblotting with an anti-GFP antibody.
PIAS Down-Regulates p53-Dependent Transactivation.
To examine the effect of PIAS on the biological properties of p53, we performed reporter gene assays by using a luciferase reporter gene, which harbors a synthetic p53 DNA binding site in its promoter (24). HeLa cells were transfected with this reporter alone or together with p53, p53/SUMO, p53/PIAS, or a combination of p53/PIAS/SUMO. The experimental conditions were identical to those described above for the in vivo modification assays, assuring a strong up-regulation of p53 sumoylation. The results are summarized in Fig. 8; data represent the mean (±SD) of four independent experiments with triplicate transfections. As shown, the expression of p53 induces luciferase activity by about 10-fold. The addition of SUMO did not significantly alter the transcriptional capacity of p53 on this promoter, whereas PIAS1 and PIASxβ slightly reduced p53 transactivation. Strikingly, coexpression of SUMO with either PIAS1 or PIASxβ strongly enhanced the repression of p53 activity. When compared with p53 expression alone, the coexpression of PIAS1/PIASxβ and SUMO together with p53 down-regulated p53-mediated transcription about 3–4-fold. By contrast, the PIASxβC362S mutant slightly induced the transactivation potential of p53, indicating that it may even exert a dominant negative effect (Fig. 8). To test whether the repressive effect of PIAS was because of the enhancement of p53 sumoylation, we performed the same set of experiments by replacing the wild-type p53-expressing vector with the p53K386R mutant that lacks the major sumoylation site. In agreement with previous reports, p53K386R shows a slightly higher basal transactivation activity when compared with the wild-type protein (4). Surprisingly, similar to what was observed for the wild-type protein expression of PIAS1/PIASxβ and SUMO reduced the transactivation potential of p53K386R. Thus, the repressive effect of PIAS on p53 does not seem to depend on sumoylation of lysine 386.
Figure 8.

PIAS down-regulates the transcriptional activity of p53. HeLa cells were transiently transfected with the pRGC luciferase reporter plasmid that harbors a p53 DNA binding site in its promoter, together with the plasmids as indicated. Values represent the average of four independent experiments with triplicate transfections after normalization for the internal control renilla luciferase activity. Activities are expressed relative to p53-induced promoter activity, which was set at 100.
Discussion
Taken together, our data strongly suggest that members of the PIAS family function as specific E3-like SUMO ligases for p53 and c-Jun. Together with recent data from other laboratories, they extend the view that Siz/PIAS proteins can function as SUMO ligases in yeast and mammals (7–9, 25). Taken together, these data further substantiate the concept that sumoylation is mechanistically highly related to ubiquitination. However, ubiquitin and SUMO ligases may differ in some important aspects. In contrast to E3 ubiquitin ligases, PIAS/Siz proteins do not seem to be essentially required for the sumoylation of substrates but rather function as factors that stimulate SUMO conjugation, suggesting that at higher concentrations Ubc9 may be sufficient for substrate recognition. One possible interpretation is that PIAS plays a role in stabilizing the interaction between Ubc9 and substrate.
An important question concerns the substrate specificity of PIAS proteins and the specific role of the distinct PIAS forms. We could show that PIASxβ and PIAS1 do not stimulate transfer of SUMO to any substrate, suggesting that they indeed play a role in substrate selection. However, the fact that at least two substrates, p53 and c-Jun, are targeted by the same ligase and recent data reporting an interaction of PIAS1 with two other SUMO substrates, the androgen receptor (16, 26) and the cytomegalovirus IE2 protein (27) may indicate that PIAS proteins are less important determinants for the substrate specificity in the SUMO pathway than are ubiquitin ligases in the ubiquitin pathway. However, an alternative possibility is that PIAS modifications or interactions with auxiliary factors may extend the repertoire of substrate specificity.
In our experiments, we did not observe major differences between PIASxβ and PIAS1 with regard to their ligase activity toward p53 and c-Jun, suggesting that a given substrate can be targeted by different PIAS family members. Consistent with this idea, p53 has also been reported to interact with PIASxα and PIASy, possibly indicating that they can also mediate sumoylation of p53 (18, 19). Similarly, the androgen receptor has been shown to interact with several PIAS members (15, 16, 28, 29). Although it remains to be determined whether PIAS proteins can indeed promote sumoylation of the androgen receptor, these observations also seem to argue against the restricted use of a single PIAS protein on a defined substrate. Because for some PIAS proteins a highly tissue-specific expression has been reported, one possible interpretation might be that the different PIAS members are required for substrate sumoylation in specific tissues (15).
The functional consequence of p53 sumoylation has remained a matter of discussion. Although in two reports overexpression of SUMO correlated with the activation of p53-mediated transactivation in reporter gene assays (4, 5), these results could not be confirmed by others (18, 21). We also did not observe any changes in p53 activity by simply overexpressing SUMO in our transactivation experiments by using a reporter gene that harbors a synthetic p53 response element. This is consistent with the finding that even upon overexpression of SUMO only a very small fraction of p53 undergoes sumoylation. However, a striking observation was that the addition of PIAS drastically down-regulated p53-mediated transactivation. This repression was at least partially SUMO-dependent and required the integrity of the zinc finger, suggesting that it is linked to the SUMO ligase activity of PIAS. Surprisingly, however, the p53K386R mutant that has lost the major sumoylation site was also repressed by PIAS/SUMO. One possibility might thus be that the repression is not because of the enhanced sumoylation of p53 itself but results from the PIAS-mediated modification of another SUMO substrate. Alternatively, the weak residual sumoylation of the p53K386R mutant that we still detect in the presence of PIAS might be sufficient to reduce its transactivation potential. To clarify this issue, we are currently trying to identify alternate SUMO acceptor sites in p53 to create mutants that are deficient for sumoylation even in the presence of PIAS.
Intriguingly, PIASy has also been shown very recently to inhibit p53-mediated transactivation by inhibiting the DNA binding activity of p53 in nuclear extracts (19). Hence, it is tempting to propose a model where sumoylation of p53 or an associated regulatory factor may negatively regulate DNA binding activity of p53. This would also explain the finding that the p53K386R that is severely compromised in sumoylation shows a slightly higher basal transactivation activity when compared with the wild-type protein (4). Interestingly, we have shown previously that the SUMO-deficient c-JunK229R mutant similarly shows an increased transactivation potential on an AP-1 containing promoter when compared with the wild-type protein (6). Considering that PIAS proteins have been found to control the activity of a variety of transcriptional regulators, these findings may point to a crucial role of this new SUMO–PIAS pathway in transcriptional regulation.
Acknowledgments
We greatly thank Erica Johnson for communicating unpublished data. We thank Stefan Jentsch for stimulating discussions, critical reading of the manuscript, and support.
Abbreviation
- GST
glutathione S-transferase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jentsch S, Pyrowolakis G. Trends Cell Biol. 2000;10:335–342. doi: 10.1016/s0962-8924(00)01785-2. [DOI] [PubMed] [Google Scholar]
- 2.Melchior F. Annu Rev Cell Dev Biol. 2000;16:591–626. doi: 10.1146/annurev.cellbio.16.1.591. [DOI] [PubMed] [Google Scholar]
- 3.Muller S, Hoege C, Pyrowolakis G, Jentsch S. Nat Rev Mol Cell Biol. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez M S, Desterro J M, Lain S, Midgley C A, Lane D P, Hay R T. EMBO J. 1999;18:6455–6461. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz S E, Scheffner M, Del Sal G. EMBO J. 1999;18:6462–6471. doi: 10.1093/emboj/18.22.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller S, Berger M, Lehembre F, Seeler J S, Haupt Y, Dejean A. J Biol Chem. 2000;275:13321–13329. doi: 10.1074/jbc.275.18.13321. [DOI] [PubMed] [Google Scholar]
- 7.Johnson E S, Gupta A A. Cell. 2001;106:735–744. doi: 10.1016/s0092-8674(01)00491-3. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi Y, Kahyo T, Toh E A, Yasuda H, Kikuchi Y. J Biol Chem. 2001;276:48973–48977. doi: 10.1074/jbc.M109295200. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi Y, Toh-e A, Kikuchi Y. Gene. 2001;275:223–231. doi: 10.1016/s0378-1119(01)00662-x. [DOI] [PubMed] [Google Scholar]
- 10.Shuai K. Oncogene. 2000;19:2638–2644. doi: 10.1038/sj.onc.1203522. [DOI] [PubMed] [Google Scholar]
- 11.Chung C D, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 12.Liu B, Liao J, Rao X, Kushner S A, Chung C D, Chang D D, Shuai K. Proc Natl Acad Sci USA. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu L, Wu H, Ma L, Sangiorgi F, Wu N, Bell J R, Lyons G E, Maxson R. Mech Dev. 1997;65:3–17. doi: 10.1016/s0925-4773(97)00032-4. [DOI] [PubMed] [Google Scholar]
- 14.Rodel B, Tavassoli K, Karsunky H, Schmidt T, Bachmann M, Schaper F, Heinrich P, Shuai K, Elsasser H P, Moroy T. EMBO J. 2000;19:5845–5855. doi: 10.1093/emboj/19.21.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moilanen A M, Karvonen U, Poukka H, Yan W, Toppari J, Janne O A, Palvimo J J. J Biol Chem. 1999;274:3700–3704. doi: 10.1074/jbc.274.6.3700. [DOI] [PubMed] [Google Scholar]
- 16.Kotaja N, Aittomaki S, Silvennoinen O, Palvimo J J, Janne O A. Mol Endocrinol. 2000;14:1986–2000. doi: 10.1210/mend.14.12.0569. [DOI] [PubMed] [Google Scholar]
- 17.Gallagher W M, Argentini M, Sierra V, Bracco L, Debussche L, Conseiller E. Oncogene. 1999;18:3608–3616. doi: 10.1038/sj.onc.1202937. [DOI] [PubMed] [Google Scholar]
- 18.Minty A, Dumont X, Kaghad M, Caput D. J Biol Chem. 2000;275:36316–36323. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- 19.Nelson V, Davis G E, Maxwell S A. Apoptosis. 2001;6:221–234. doi: 10.1023/a:1011392811628. [DOI] [PubMed] [Google Scholar]
- 20.Long X, Griffith L C. J Biol Chem. 2000;275:40765–40776. doi: 10.1074/jbc.M003949200. [DOI] [PubMed] [Google Scholar]
- 21.Kwek S S, Derry J, Tyner A L, Shen Z, Gudkov A V. Oncogene. 2001;20:2587–2599. doi: 10.1038/sj.onc.1204362. [DOI] [PubMed] [Google Scholar]
- 22.Joazeiro C A, Weissman A M. Cell. 2000;102:549–552. doi: 10.1016/s0092-8674(00)00077-5. [DOI] [PubMed] [Google Scholar]
- 23.Hochstrasser M. Cell. 2001;107:5–8. doi: 10.1016/s0092-8674(01)00519-0. [DOI] [PubMed] [Google Scholar]
- 24.Kern S E, Pietenpol J A, Thiagalingam S, Seymour A, Kinzler K W, Vogelstein B. Science. 1992;256:827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 25.Kahyo T, Nishida T, Yasuda H. Mol Cell. 2001;8:713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 26.Poukka H, Karvonen U, Janne O A, Palvimo J J. Proc Natl Acad Sci USA. 2000;97:14145–14150. doi: 10.1073/pnas.97.26.14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahn J H, Xu Y, Jang W J, Matunis M J, Hayward G S. J Virol. 2001;75:3859–3872. doi: 10.1128/JVI.75.8.3859-3872.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Junicho A, Matsuda T, Yamamoto T, Kishi H, Korkmaz K, Saatcioglu F, Fuse H, Muraguchi A. Biochem Biophys Res Commun. 2000;278:9–13. doi: 10.1006/bbrc.2000.3753. [DOI] [PubMed] [Google Scholar]
- 29.Gross M, Liu B, Tan J, French F S, Carey M, Shuai K. Oncogene. 2001;20:3880–3887. doi: 10.1038/sj.onc.1204489. [DOI] [PubMed] [Google Scholar]