

Abstract
Thyroid hormone (3,5,3′-triiodo-l-thyronine or T3) exerts a pleiotropic activity during central nervous system development. Hypothyroidism during the fetal and postnatal life results in an irreversible mental retardation syndrome. At the cellular level, T3 is known to act on neuronal and glial lineages and to control cell proliferation, apoptosis, migration, and differentiation. Oligodendrocyte precursor cells (OPC) found at birth in the optic nerves are self-renewing cells that normally differentiate during the first 3 weeks of rodent postnatal life into postmitotic myelinating oligodendrocytes. In vitro, the addition of T3 to OPC is sufficient to trigger their terminal differentiation. The present analysis of T3 receptor knockout mice reveals that the absence of all T3 receptor results in the persistence of OPC proliferation in adult optic nerves, in a default in myelination, and sometimes in the degeneration of the retinal ganglion neurons. Thus, T3 signaling is necessary in vivo to promote the complete differentiation of OPC.
Thyroid hormone (3,5,3′-triiodo-l-thyronine, T3) acts directly at the transcription level by binding to nuclear receptors (TRα1 and TRβ1, TRβ2, TRβ3) encoded by both TRα and TRβ genes to control a number of physiological and developmental processes. During the late embryonic postnatal period, T3 action is critical for brain development. However, although both hyperthyroidism and hypothyroidism are known to affect directly or indirectly the proliferation, apoptosis, migration, and differentiation of several neuronal and glial cell types (1, 2), only subtle brain defects have been reported to date in the nervous system of TR knockout mice (3, 4). Moreover, knock-in point mutations recently led to contrasting results that are difficult to interpret (5–7). In the rodent optic nerves, oligodendrocyte precursor cells (OPC) differentiate during the first postnatal weeks to give rise to the oligodendrocytes that synthesize myelin around the axons of the retinal ganglion cells (RGC). The in vitro culture of newborn optic nerve OPC (8–10) has offered a unique opportunity to study the precise influence of T3 on the differentiation of this category of glial cells (reviewed in ref. 10). This in vitro model has been extensively investigated, and a number of studies confirmed the direct effect of T3 on OPC differentiation. However, the fact that retinoic acid or glucocorticoids can substitute to T3 to trigger in vitro OPC differentiation (9, 11) raises the question of the respective importance of these factors in vivo. The respective contribution of TRα and TRβ in OPC hormonal response is also unclear (8, 10, 12). TRα is expressed at all stages of oligodendrocyte differentiation, whereas the status for TRβ remains controversial. Based on RNA and protein analysis, TRβ expression has been reported to be restricted to differentiated oligodendrocytes (13–16) or to occur in both OPC and oligodendrocytes (9, 17). To clarify the importance of T3 action in vivo and the respective functions of TRα and TRβ in OPC differentiation, we performed a detailed histological analysis of TR knockout mice lacking all TRα isoforms (18) (TRα0 allele), all TRβ isoforms (19), or both.
Materials and Methods
Knockout Mice.
All knockout mouse strains have been described. They are viable and kept in a mainly C57BL/129Sv background. Unlike TRα−/− mice used in previous studies (19, 20), the TRα0/0 mice used here do not express any of the TRα isoforms and display alterations in small intestine and bone development (18, 21). TRβ−/− mice are characterized mainly by an increase in the level of circulating T3 and thyroid-stimulating hormone (TSH), deafness, and color blindness (3, 4). TRα0/0 β−/− mice that lack all TR isoforms are also viable and display many features of congenital hypothyroidism including growth defect (18, 22). As they display very limited fertility, all of the TRα0/0β−/− animals used in the present studies were obtained by crossing TRα0/+β−/− animals. Control animals were from the same genetic background.
Preparation of Samples for Microscopy.
For immunofluorescence analysis, animals were perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.5). Optic nerves were dissected, fixed for 24 h, and then transferred to 30% sucrose/phosphate buffer (24 h), frozen (−50°C, 1 min), and sectioned by using a cryostat. Retina sections were performed after Bouin–Hollande fixation and paraffin embedding and were stained with hematoxylin and eosin according to conventional protocols. Glutaraldehyde (10% in phosphate buffer) fixation was used for electron microscopy analysis performed according to conventional protocols.
Immunofluorescence Assays.
Cryostat transversal sections of optic nerves were labeled with the anti-NG2 rabbit antibody (Chemicon) specific for OPC (23, 24). Astrocytes were stained with an antibody directed against the glial fibrillar acidic protein (Dako). RGC axons were detected by using an antibody directed against the phosphorylated 70-kDa neurofilament protein (Dako). Terminally differentiated oligodendrocytes were stained with an antibody directed against the myelin basic protein (25).
BrdUrd Labeling and Terminal Deoxynucleotidyltransferase-Mediated dUTP Nick End Labeling (TUNEL) Assay.
Intraperitoneal injection of BrdUrd (Sigma, 100 mg/kg) was performed at days 0 and 3, and optic nerves were dissected at day 7 for TRβ−/−, TRα0/0, and wild-type mice. For TRα0/0β−/− animals and other wild-type controls, BrdUrd was injected at days 0 and 1 before dissection at day 2. The presence of neosynthesized DNA was visualized by using a mouse anti-BrdUrd antibody (Amersham Pharmacia) and a secondary anti-mouse antibody (Jackson ImmunoResearch). Fragmented DNA characteristic of cell death was visualized by using the TUNEL assay (Roche Laboratories).
Results
Optic Nerve Developmental Characteristics 15 Days After Birth.
Because a delay of optic nerve myelination is observed in hypothyroid animals during the first postnatal weeks, an immunohistological analysis was first performed 15 days after birth in optic nerve to compare wild-type, TRα0/0, TRβ−/−, and TRα0/0β−/− mice. At this time, OPC were identified on optic nerve sections in all mice by using the NG2 antibody, confirming recent results obtained for TRβ−/− mice (16). Analysis of differentiated astrocytes and oligodendrocytes failed to detect major abnormalities in the optic nerve of any TR mutant mice (data not shown). Characterization of the optic nerve neuronal axons with an antineurofilament was also performed 15 days after birth for wild-type (Fig. 1A), TRα0/0, TRβ−/− (data not shown), and TRα0/0β−/− mice (Fig. 1B) and did not reveal any systematic difference between wild-type and TR mutant mice.
Figure 1.
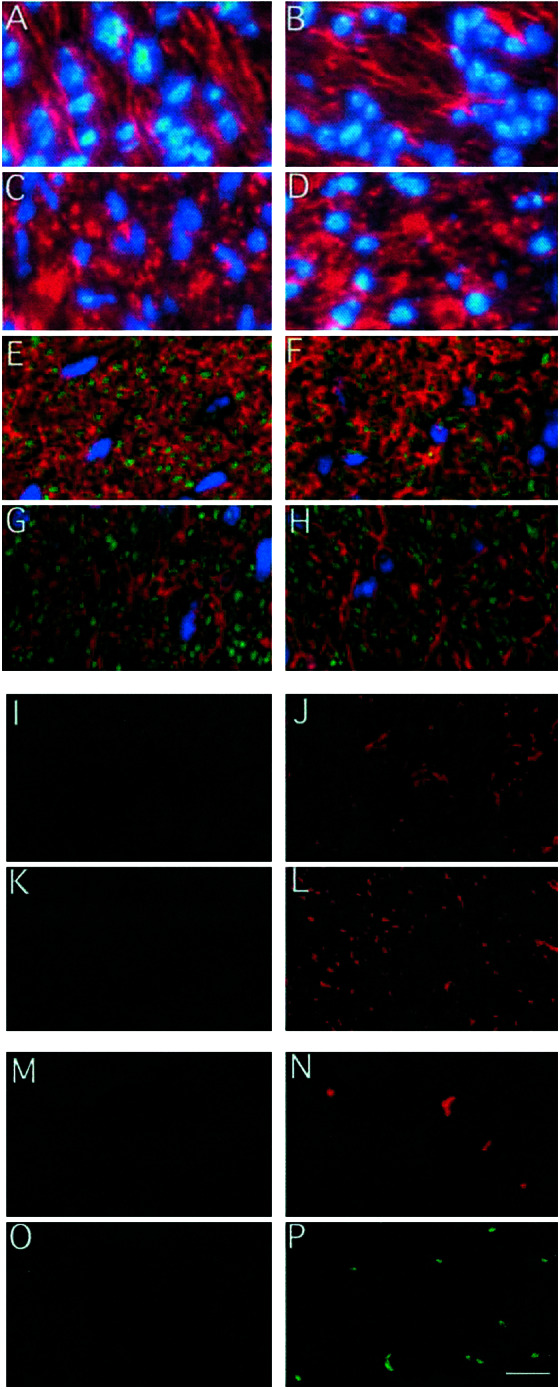
Phenotype of the optic nerve. Nuclei staining with bisbenzimide (in blue) and neurofilament immunostaining (in red) reveals the presence of neurones in wild-type (A) and TRα0/0β−/− (B) Fifteen-day-old optic nerve and 6-month-old wild-type (C) and TRα0/0β−/− (D). Double immunostaining of 6-month-old optic nerve sections with myelin basic protein (in red) and neurofilament (in green) reveals the presence of myelinated axons in both wild-type (E) and TRα0/0β−/− animals (F). Double immunostaining of 6-month-old optic nerve sections with glial fibrillar acidic protein (in red) and neurofilament (in green) reveals the presence of astrocytes in wild-type (G) in TRα0/0β−/− animals (H). All of these analyses failed to reveal a significant anomaly in young or aged TRα0/0β−/− mice. NG2 immunostaining of 6-month-old optic nerve sections reveals the absence of OPC in wild-type (I) and TRα0/0 (K) and the persistence of OPC in TRβ−/− (J) and TRα0/0β−/− animals (L). In vivo labeling of neosynthesized DNA with BrdUrd (injection at days 1 and 2, fixation at day 3) reveals the absence of cell proliferation in 6-month-old wild-type (M) but persistence of cell proliferation in 6-month-old TRα0/0β−/− optic nerves (N). No cell death was detected in 6-month-old wild-type optic nerve (O). (P) Detection of cell death in 6-month-old TRα0/0β−/− optic nerve section with the in situ TUNEL assay. (Scale bar = 10 μm.)
Nuclei staining with bisbenzimide of wild-type, TRα0/0, TRβ−/−, and TRα0/0β−/− mice revealed, however, a slight increase in the average number of cell nuclei in TRβ−/− (Fig. 2A). Because an increase in cell density was not observed in double TRα0/0β−/− knockout mice, this feature is unlikely to reflect a defect in T3 response but may rather be a consequence of the high T3 level found in TRβ−/− animals acting by means of TRα1 (4). These initial observations led us to suspect a subtle defect in OPC differentiation and to address its possible long-term consequences.
Figure 2.

Cell density in optic nerve. Distribution of average nuclei number per section. All animals were from different litters. For each animal, two sections were counted in the median part of the optic nerve, and the average was reported. Variations were minimal within a given optic nerve. Each dot corresponds to one animal. Nuclei were counted after staining with bisbenzimide of wild-type, TRβ−/−, TRα 0/0, and TRα0/0β−/−. (A) Fifteen-day-old optic nerve. The distribution for wild type, TRα0/0, and TRα0/0β−/− seems to be similar. We thus calculated the average number of nuclei/section is 161 ± 36. The four values for the TRβ−/− optic nerve are located outside of this 5% confidence interval, which can occur by chance with a probability of 6 × 10−6. (B) Six-month-old optic nerve. Cell density (132 ± 31 nuclei/section) is not significantly changed by TR knockout (P = 5%).
Permanent Turnover of OPC in 6-Month-Old TRβ−/− and TRα0/0β−/− Optic Nerves.
The histological studies described above were repeated with 6-month-old animals at a time where OPC, which normally disappear from optic nerve within 4 weeks after birth, are very scarce in wild-type animals. Immunohistological characterization of neuronal axons (Fig. 1 C and D), myelinating oligodendrocytes (Fig. 1 E and F), and astrocytes (Fig. 1 G and H) in the optic nerves was performed for wild-type (Fig. 1 C, E, and G), TRα0/0β−/− (Fig. 1 D, F, and H), TRα0/0, and TRβ−/− (data not shown) mutant mice. This analysis failed to detect major anomalies. However, immunostaining with the OPC specific antibody (NG2) revealed a clearly abnormal situation in the 6-month-old mice optic nerves. It has been reported in rat that few OPC are present in adult optic nerve (26). In 6-month-old wild-type mice, most optic nerve sections do not contain any OPC (Fig. 1I), although one can be occasionally observed. We also did not identify OPC in TRα0/0 optic nerves (Fig. 1K), whereas some were detected in all TRβ−/− sections (Fig. 1J). This number was much higher in TRα0/0β−/− mice (Fig. 1L). The proliferation of the OPC was demonstrated by in vivo BrdUrd incorporation (Fig. 1 M and N). BrdUrd incorporation was completely absent in wild-type (Fig. 1M) and TRα0/0 optic nerves even 7 days after the beginning of BrdUrd treatment. Proliferating cells were observed in TRα0/0β−/− mice within 48 h (5 ± 3/section; Fig. 1N) and only after 7 days in TRβ−/− mice (data not shown). This difference suggests a possible cooperation of the two TR gene products in the arrest of wild-type OPC proliferation.
Nuclei staining in the optic nerves showed that the persistence of OPC proliferation was not linked to any significant increase in cell density (Fig. 2B). Indeed, if apoptosis was not observed in wild-type adult optic nerve (Fig. 1O), OPC proliferation was concomitant to an increased rate of cell death in TRα0/0β−/− adults, as shown by the presence of pycnotic nuclei (data not shown) and by using the in situ TUNEL assay (Fig. 1P). Because this assay can also label the Okazaki fragments present in cycling cells during the S phase, we controlled that only a fraction (50%) of TUNEL-positive cells are BrdUrd positive by double immunostaining (data not shown). The existence of double-labeled cells reflects the presence of S phase cells and apoptotic cells that underwent DNA synthesis during the labeling period. Thus, although the presence of apoptotic cells in TRα0/0β−/− adults optic nerves is ascertain, a precise quantitation of apoptosis is difficult.
In conclusion, a permanent turnover of OPC takes place in TRα0/0β−/− and to a lesser extent in TRβ−/− adult optic nerves with little or no effect on cell density.
Myelination Defects in TRα0/0β−/− Optic Nerves.
The partial differentiation of OPC in adult TRβ−/− and TRα0/0β−/− optic nerves might result into myelination defects that would not be visible with conventional histological methods. We thus performed an ultrastructural analysis of optic nerve section by electron microscopy. This failed to reveal any obvious abnormality in 15-day-old TRα0/0β−/− (data not shown) and 6-month-old wild-type mice (Fig. 3 A and C). TRα0/0β−/− optic nerve images (Fig. 3 B and D) were clearly different. Although image analysis (National Institutes of Health image software) showed that the average ratio between the surface of compacted myelin and the surface of axons is not affected, the axonal shape was obviously abnormal in TRα0/0β−/−, the contours of myelin bundles being irregular with concave portions (Fig. 3B). High magnification observation revealed that the periaxonal collar (27, 28), which normally consists in only one turn of myelin in wild type (Fig. 3C), is often replaced by several turns of noncompacted myelin in TRα0/0β−/− optic nerves (arrow, Fig. 3D) and that myelin folds occasionally enter the central axonal area (not shown).
Figure 3.

Electron microscopy of ultra-thin cross sections of the 6-month-old TRα0/0β−/− mice optic nerve. At low magnification, the abnormal shape of axons is clearly visible in the TRα0/0β−/− mice optic nerve (B) compared with wild type (A). High magnification micrograph (C and D) reveals the presence of several turns of noncompacted myelin (arrow) in the majority of the TRα0/0β−/− axons (D). (Scale bar = 2 μm.) The average myelin/axon section surface ratio is not significantly affected in mutants animals (0.69 ± 0.26 for wild type and 0.73 ± 0.26 in TRα0/0β−/− mutants estimated from the measure of 20 axons sections for each genotype).
Degeneration of TRα0/0β−/− RGC Layer in Retina.
The axons protected by optic nerve oligodendrocytes belong to RGC. The possible consequences of the myelination defect on retina neuron survival were addressed by observing histological sections of adults wild-type (Fig. 4A) and TRα0/0β−/− knockout mice (Fig. 4B) retinas. No significant changes in cell density were observed in the retina neuronal outer and inner neuronal layer. By contrast, a significant reduction of cell density was found in the RGC layer of several TRα0/0β−/− retinas (four of eight TRα0/0β−/− mice) (Fig. 4C). This alteration corresponds to patches where cells were totally absent (arrows on Fig. 4B). As it was not observed in five younger TRα0/0β−/− mice (between three 3-week-old and 4-month-old mice, data not shown), the low cell density in RGC layer is likely to result from cell death. This assumption was strengthened by the occasional observation of TUNEL-positive cells in this layer for TRα0/0β−/− retina (data not shown).
Figure 4.

Degeneration of the RGC layer in the retina of 6-month-old TRα0/0β−/− mice. (A) Wild-type retina section. (B) TRα0/0β−/− retina section stained with hematoxylin and eosin. ONL, outer neuronal layer; INL, inner neuronal layer; GCL, RGC layer. (C) Distribution of average nuclei number in the RGC layer (number of cells/retina section) of wild type, TRα00, TRβ−/−, and TRα0/0β−/−. For each retina, two median sections, including the optic nerve, were used. The average is plotted for each animal. All animals were from different litters. The wider distribution of the cell counts apparently for TRα0/0β−/− retina prevents the use of conventional statistical tests. We thus performed the statistical analysis as follows. We made the assumption that the theoretical distribution is identical for the three other genotypes and calculated a confidence interval (318 ± 25 RGC/section). This allowed us to define a threshold (293, P < 5%), under which the number of RGC is lower than normal. With this criterion, four of eight TRα0/0β−/− retinas showed clear signs of RGC degeneration.
Discussion
Early and Late Function of T3 in OPC Differentiation.
The present study provides genetic evidences that the TR gene products are necessary in vivo to ensure the complete terminal differentiation of all OPC into myelinating oligodendrocytes. However, the phenotype that we have found in TRα0/0β−/− animals is not the delay of postnatal OPC differentiation reported for hypothyroid rats but the persistence of OPC proliferation. Although a slight delay in TRα0/0β−/− optic nerve myelination may have been overlooked, a more likely interpretation for this discrepancy is that the TRα0/0β−/− mice do not fully recapitulate hypothyroidism. Unliganded TRs present in hypothyroid but not TRα0/0β−/− knockout mice are indeed able to bind DNA and to repress T3 target genes expression in vivo (29), and this aporeceptor activity may be responsible for the early delay in OPC differentiation observed in hypothyroid animals only. Our results mainly confirm that at least a fraction of OPC possesses a long-term proliferation capacity (30, 31) and does not use a cell-intrinsic timer to differentiate during the postnatal weeks (9, 17, 32–34). According to our observations, T3, acting mainly through TRβ but also through TRα, stops the self-renewal and induces the terminal differentiation of these cells. OPC seems to be more abundant in adult rats (26), suggesting that the control of their proliferation by T3 is more stringent in this species than in wild-type mice. Although undistinguishable in vivo, these late OPC proliferate more slowly in vitro and might constitute a distinct cell population (30). We might expect that the permanent proliferation of OPC would result in a constant increase in optic nerve cell density. However, oligodendrocytes and OPC survival depend on axon contacts (9, 35). It is therefore likely that a competition occurring between the cells for these contacts is responsible for the increased rate of cell death, which compensates for sustained proliferation.
Respective Function of the TRα and TRβ Gene.
The maintenance of OPC mainly in double knockout mice is a strong indication that both genes are able to mediate T3 signaling in OPC and that the T3 target genes responsible for OPC differentiation can respond to both TRβ and TRα1 at least in a knockout context. However, because limited changes have been observed in TRβ−/− but not in TRα0/0 animals, TRβ seems to play a predominant role in OPC differentiation. This would be consistent with a significant expression of TRβ in these cells (9, 17). However, our data do not rule out the possibility that OPC start to express TRβ shortly after an initial step in their differentiation pathway. This controversial matter can be addressed only by in vitro experiments.
Retina Degeneration as an Indirect Consequence of OPC Persistence.
The abnormal differentiation of oligodendrocytes seems to be a primary defect responsible for a late retina degeneration in a fraction of the TRα0/0β−/− knockout mice. This is supported by the fact that cell death is restricted to RGC, the only retina cells to contact optic nerve oligodendrocytes. Furthermore, RGC express only TRα (3), and their death in TRα0/0β−/− but not TRα0/0 knockout mice is therefore very unlikely to be a cell-autonomous process. However, as OPC persistence is observed in TRβ−/− mice in the absence of retina degeneration, the possibility remains that the TRα mutation renders RGC more sensitive to a defect in axon myelination. The limited penetrance of this phenotypic trait suggests that the occurrence or timing of retina degeneration during TRα0/0β−/− knockout mice aging is sensitive to unknown environmental or genetic factors.
Whereas the persistence of OPC in human adult brain is found in several pathological situations, including multiple sclerosis as a consequence of demyelination or various injuries (24), it has not been reported, to our knowledge, in the optic nerve. Retina degeneration limited to RGC is also rarely observed in human and is associated with optic atrophy (36). It might have been unnoticed in the only reported case of TRβ deletion in human who suffered from very low visual acuity and color blindness (37). In conclusion, it seems that T3 is playing a subtle, but crucial, role in OPC differentiation, which has visible consequences on optic nerve and retina upon aging.
Acknowledgments
This work was supported by the Association pour la Recherche contre le Cancer, the Comité Départemental de la Loire de la Ligue Nationale contre le Cancer, and the Human Frontier Scientific Program (RGO347/1999.M). D.B. was supported by a fellowship of the Société de Secours pour les Amis des Sciences.
Abbreviations
- OPC
oligodendrocyte precursor cells
- T3
thyroid hormone or 3,5,3′-triiodo-l-thyronine
- TR
thyroid hormone receptor
- RGC
retinal ganglion cells
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Oppenheimer J H, Schwartz H L. Endocr Rev. 1997;18:462–475. doi: 10.1210/edrv.18.4.0309. [DOI] [PubMed] [Google Scholar]
- 2.Bernal J, Nunez J. Eur J Endocrinol. 1995;133:390–398. doi: 10.1530/eje.0.1330390. [DOI] [PubMed] [Google Scholar]
- 3.Ng L, Hurley J B, Dierks B, Srinivas M, Salto C, Vennstrom B, Reh T A, Forrest D. Nat Genet. 2001;27:94–98. doi: 10.1038/83829. [DOI] [PubMed] [Google Scholar]
- 4.Forrest D, Erway L C, Ng L, Altschuler R, Curran T. Nat Genet. 1996;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- 5.Hashimoto K, Curty F H, Borges P P, Lee C E, Abel E D, Elmquist J K, Cohen R N, Wondisford F E. Proc Natl Acad Sci USA. 2001;98:3998–4003. doi: 10.1073/pnas.051454698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter T A, Kazlauskaite R, Pankratz D G, Wynshaw-Boris A, Refetoff S, et al. Proc Natl Acad Sci USA. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itoh Y, Esaki T, Kaneshige M, Suzuki H, Cook M, Sokoloff L, Cheng S Y, Nunez J. Proc Natl Acad Sci USA. 2001;98:9913–9918. doi: 10.1073/pnas.171319498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barres B A, Lazar M A, Raff M C. Development (Cambridge, UK) 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- 9.Durand B, Raff M. BioEssays. 2000;22:64–71. doi: 10.1002/(SICI)1521-1878(200001)22:1<64::AID-BIES11>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 10.Ahlgren S C, Wallace H, Bishop J, Neophytou C, Raff M C. Mol Cell Neurosci. 1997;9:420–432. doi: 10.1006/mcne.1997.0631. [DOI] [PubMed] [Google Scholar]
- 11.Tokumoto Y M, Durand B, Raff M C. Dev Biol. 1999;213:327–339. doi: 10.1006/dbio.1999.9397. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Pena A. J Neurobiol. 1999;40:497–512. doi: 10.1002/(sici)1097-4695(19990915)40:4<497::aid-neu7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 13.Baas D, Bourbeau D, Sarlieve L L, Ittel M E, Dussault J H, Puymirat J. Glia. 1997;19:324–332. doi: 10.1002/(sici)1098-1136(199704)19:4<324::aid-glia5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 14.Baas D, Fressinaud C, Ittel M E, Reeber A, Dalencon D, Puymirat J, Sarlieve L L. Neurosci Lett. 1994;176:47–51. doi: 10.1016/0304-3940(94)90868-0. [DOI] [PubMed] [Google Scholar]
- 15.Carre J L, Demerens C, Rodriguez-Pena A, Floch H H, Vincendon G, Sarlieve L L. J Neurosci Res. 1998;54:584–594. doi: 10.1002/(SICI)1097-4547(19981201)54:5<584::AID-JNR3>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 16.Billon N, Tokumoto Y, Forrest D, Raff M. Dev Biol. 2001;235:110–120. doi: 10.1006/dbio.2001.0293. [DOI] [PubMed] [Google Scholar]
- 17.Gao F B, Apperly J, Raff M. Dev Biol. 1998;197:54–66. doi: 10.1006/dbio.1998.8877. [DOI] [PubMed] [Google Scholar]
- 18.Gauthier K, Plateroti M, Harvey C B, Williams G R, Weiss R E, Refetoff S, Willott J F, Sundin V, Roux J P, Malaval L, et al. Mol Cell Biol. 2001;21:4748–4760. doi: 10.1128/MCB.21.14.4748-4760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauthier K, Chassande O, Plateroti M, Roux J P, Legrand C, Pain B, Rousset B, Weiss R, Trouillas J, Samarut J. EMBO J. 1999;18:623–631. doi: 10.1093/emboj/18.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraichard A, Chassande O, Plateroti M, Roux J P, Trouillas J, Dehay C, Legrand C, Gauthier K, Kedinger M, Malaval L, et al. EMBO J. 1997;16:4412–4420. doi: 10.1093/emboj/16.14.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plateroti M, Gauthier K, Domon-Dell C, Freund J N, Samarut J, Chassande O. Mol Cell Biol. 2001;21:4761–4772. doi: 10.1128/MCB.21.14.4761-4772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gothe S, Wang Z, Ng L, Kindblom J M, Barros A C, Ohlsson C, Vennstrom B, Forrest D. Genes Dev. 1999;13:1329–1341. doi: 10.1101/gad.13.10.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine J M, Reynolds R, Fawcett J W. Trends Neurosci. 2001;24:39–47. doi: 10.1016/s0166-2236(00)01691-x. [DOI] [PubMed] [Google Scholar]
- 24.Dawson M R, Levine J M, Reynolds R. J Neurosci Res. 2000;61:471–479. doi: 10.1002/1097-4547(20000901)61:5<471::AID-JNR1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 25.Baas D, Barnstable C J. Glia. 1998;23:169–179. [PubMed] [Google Scholar]
- 26.Forrest D, Hanebuth E, Smeyne R J, Everds N, Stewart C L, Wehner J M, Curran T. EMBO J. 1996;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- 27.Schachner M, Bartsch U. Glia. 2000;29:154–165. doi: 10.1002/(sici)1098-1136(20000115)29:2<154::aid-glia9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 28.Biffiger K, Bartsch S, Montag D, Aguzzi A, Schachner M, Bartsch U. J Neurosci. 2000;20:7430–7437. doi: 10.1523/JNEUROSCI.20-19-07430.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flamant F, Poguet A L, Plateroti M, Chassande O, Gauthier K, Streichenberger N, Mansouri A, Samarut J. Mol Endocrinol. 2002;16:24–32. doi: 10.1210/mend.16.1.0766. [DOI] [PubMed] [Google Scholar]
- 30.Tang D G, Tokumoto Y M, Raff M C. J Cell Biol. 2000;148:971–984. doi: 10.1083/jcb.148.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang D G, Tokumoto Y M, Apperly J A, Lloyd A C, Raff M C. Science. 2001;291:868–871. doi: 10.1126/science.1056780. [DOI] [PubMed] [Google Scholar]
- 32.Barres B A, Raff M C. Neuron. 1994;12:935–942. doi: 10.1016/0896-6273(94)90305-0. [DOI] [PubMed] [Google Scholar]
- 33.Gao F B, Durand B, Raff M. Curr Biol. 1997;7:152–155. doi: 10.1016/s0960-9822(06)00060-1. [DOI] [PubMed] [Google Scholar]
- 34.Gao F B, Raff M. J Cell Biol. 1997;138:1367–1377. doi: 10.1083/jcb.138.6.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez P A, Tang D G, Cheng L, Prochiantz A, Mudge A W, Raff M C. Neuron. 2000;28:81–90. doi: 10.1016/s0896-6273(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 36.Votruba M, Moore A T, Bhattacharya S S. J Med Genet. 1998;35:793–800. doi: 10.1136/jmg.35.10.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newell F W, Diddle K R. Klin Monatsbl Augenheilkd. 1977;171:731–734. [PubMed] [Google Scholar]