

Abstract
The expression of the β-like globin genes is intricately regulated by a series of both general and tissue-restricted transcription factors. The hemapoietic lineage-specific transcription factor GATA-1 is important for erythroid differentiation and has been implicated in regulating the expression of the erythroid-specific genes including the genes of the β-globin locus. In the human erythroleukemic K562 cell line, only one DNA region has been identified previously as a putative site of GATA-1 interaction by in vivo footprinting studies. We mapped GATA-1 binding throughout the β-globin locus by using chIp-chip analysis of K562 cells. We found that GATA-1 binds in a region encompassing the HS2 core element, as was previously identified, and an additional region of GATA-1 binding upstream of the γG gene. This approach will be of general utility for mapping transcription factor binding sites within the β-globin locus and throughout the genome.
Cell differentiation and response pathways often culminate in gene expression programs mediated by transcription factor binding and/or activity at specific sites within the genome. Protein–DNA interactions are responsible for the erythroid-specific expression of the β-like globin genes and for the temporal regulation of the expression of these genes. The human 75-kb β-globin locus is composed of five globin genes (ɛ, γG, γA, δ, and β), one pseudogene (ψη), and a locus control region (LCR) located ≈6- to 30-kb upstream of the ɛ-globin gene (refs. 1–3). Transcription factor binding within the LCR at DNase I hypersensitive sites (HS) and at the promoter regions of the globin genes mediate the tissue-specific and stage-specific expression of the β-like globin genes (for review, see ref. 4). Both tissue-restricted transcription factors such as GATA-1, the erythroid Kruppel-like factor, NF-E2, ubiquitous factors, and chromatin modifiers control this specificity (for review, see ref. 5).
GATA-1, the founding member of the GATA family of zinc finger proteins, is a hematopoietic cell-specific transcription factor that recognizes the consensus sequence (A/T)GATA(A/G) (6). Nearly all erythroid cell-specific genes, including the α- and β-globin genes, contain functionally important GATA-1 binding sites within their regulatory regions (7). GATA-1 is essential for erythroid cell development, as erythroid precursors in mice deficient for GATA-1 fail to survive and mature, and the embryos die of anemia (8–10). GATA-1 also can induce terminal erythroid maturation when expressed in a G1E cell line, which lacks GATA-1 (11).
Some GATA-1 binding sites have been mapped within the β-globin locus in human and human-murine hybrid erythroleukemic cell lines by using in vivo footprinting. Studies in human K562 cells, which express the ɛ- and γ- globin genes, revealed footprints over GATA-1 motifs within the HS2 region only, although in HS3 region, the γG- and β-promoters and γA- and β-3′enhancers also were examined (12, 13). In the human-murine hybrid murine erythro-leukemic cells, DMS footprints over GATA-1 recognition elements were identified in regions HS1, HS3, and HS4 of the LCR, as well as the γG promoter and the β-3′-enhancer (14). In another human-murine cell line Hu11, GATA-1 sites were protected in the HS3 region (15). Although these footprinting studies precisely identify sites of protein-DNA interaction, they do not clearly identify the protein responsible for the footprint. Recently, chromatin immunoprecipitation assays of limited regions within the β-globin locus suggested GATA-1 recruitment to the γ-promoter, the HS3, as well as the HS2 element in human K562 cells (16). However, an extensive survey of the β-globin locus for direct, in vivo GATA-1 binding sites has not been performed.
We and others have recently developed a method in yeast to map the direct, in vivo binding sites of transcription factors on a genome-wide scale (17, 18). This approach involves chromatin immunoprecipitation (chIp) of protein-DNA complexes and microarray hybridization of labeled, immunopurified DNA (19). The approach is termed chIp-chip. There are many inherent challenges in applying this technique to human cells, including the large size of the genome, the complexity of gene regulation and chromatin structure, and the high proportion of repetitive elements. To optimize this technique and determine how to overcome these challenges, we used the well studied β-globin locus as a model system to develop this approach in mammalian cells. ChIp-chip was used to map the binding sites of GATA-1 in human erythroleukemic K562 cells within the 75-kb region of the β-globin locus, resulting in the identification of previously unidentified GATA-1 binding sites.
Materials and Methods
Cells.
K562 cells were grown in RPMI medium 1640 (with 300 mg/liter glutamine) supplemented with 10% (vol/vol) FBS, 1× antibiotics and antimycotic (100 units/ml ampicillin/100 units/ml streptomycin/0.25 μg/ml amphotericin). HeLa cells were grown in DMEM containing 10% (vol/vol) FCS.
Preparation of Protein Extracts.
To prepare nuclear extracts, cells were incubated in a hypotonic buffer solution (10 mM Hepes buffer, pH 7.9/1.5 mM MgCl2/10 mM KCl/0.5 mM DTT) at 4°C for 10 min. The swollen cells were harvested by centrifugation, resuspended in two cell pellet volumes of hypotonic solution, and lysed with a Dounce homogenizer. Nuclei were collected by centrifugation at 25,000 × g for 15 min. The nuclear pellet was used to prepare nuclear extracts and chromatin as described below.
Nuclear extracts were prepared by lysing with RIPA buffer containing 10 mM Tris⋅Cl, pH 8, 140 mM NaCl, 0.025% sodium azide, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 1 mM PMSF and protease inhibitors (Sigma), and by incubating on ice for 30 min. Nuclear membranes were disrupted further by passage through a 21-gauge needle and by incubating on ice for another 30 min after refreshing PMSF. Extracts were clarified by centrifugation at 15,000 × g at 4°C for 15 min.
Chromatin Immunoprecipitation.
Cells (1–3 × 108) (see Table 1, which is published as supporting information on the PNAS web site, www.pnas.org) were fixed with formaldehyde at a final concentration of 1% for 15–20 min at 18°C. Crosslinks were quenched with glycine at a final concentration of 125 mM. Nuclear extracts were prepared as described above. Before clarification, chromatin was sheared with a Branson 250 Sonifier. At a power setting of 1.5 and a 100% duty cycle, the extracts were sonicated for three 10-s pulses, with two minutes on ice in between pulses. After clarification, extracts were precleared with 100 μl bed volume of protein A/G Sepharose beads (Pierce) for 1 h at 4°C on a rotating wheel. GATA-1 protein-DNA complexes were immunoprecipitated with either anti-GATA-1(C20) (Santa Cruz Biotechnology), anti-GATA-1(N6) (Santa Cruz Biotechnology), or anti-GATA-1 (Geneka Biotechnology, Montreal) at a final concentration of 1:500, incubated on ice for 3 h and then incubated with a 100-μl bed volume protein A/G beads on a rotating wheel at 4°C for 1 h. The beads were washed twice with 1 ml of RIPA buffer, once with 1 ml RIPA buffer at 500 mM NaCl concentration, once with 1 ml LiCl/detergent solution (10 mM Tris⋅Cl, pH 8/500 mM NaCl/0.025% sodium azide/1% Triton X-100/0.1% SDS/1% sodium deoxycholate), and once with 1 ml of 1× TBS (20 mM Tris⋅Cl, pH 7.6/150 mM NaCl). Beads were collected by centrifugation at 1,000 × g for 2 min at 4°C after each wash. Immunocomplexes were eluted from the beads with 100 ml of 1% SDS/1× TE (10 mM Tris⋅Cl, pH 7.6/1 mM EDTA, pH 8); the SDS was diluted with 150 ml of 0.67% SDS/1× TE. Crosslinks were reversed by heating at 65°C overnight. DNA was purified as described (20).
Immunoblots.
Protein extracts were separated in a 10% denaturing polyacrylamide gel and analyzed by immunoblot analysis using a 1:1,000 dilution of GATA-1 primary antibody and a 1:5,000 dilution of a horseradish-peroxidase conjugated secondary antibody. Antibodies used as negative controls, goat c-jun (Santa Cruz Biotechnology) and rabbit Nrf1 (Santa Cruz Biotechnology), also were used at a dilution of 1:1,000 for immunoblotting. Immunocomplexes were visualized with the enhanced chemiluminescence (ECL, Amersham Pharmacia) system.
PCR Assays.
Immunoprecipitated DNA was analyzed for specific enrichment by PCR assay.
Primers for the core HS1, HS2, and HS3 regions, the ɛ, γG, Ψη, δ, and β-promoters, and the γA and β-3′ enhancers and other regions assayed are listed in Table 2, which is published as supporting information on the PNAS web site. Immunoprecipitated DNA (1 ng) was amplified by PCR using 2× Taq Mastermix (Qiagen, Chatsworth, CA) and the following thermalcycling conditions: 94°C for 2 min, 30 cycles of 20 s at 94°C, 30 s at 50°C, and 1 min at 72°C, followed by a 10-min extension time at 72°C. Input DNA from sonicated K562 lysate (10 ng, 100 ng, and 1 μg) was amplified in parallel. Twenty to fifty percent of the products were separated by PAGE and visualized by ethidium bromide staining. Band intensity was determined by CHELIMAGER software (Alpha Innotech, San Leandro, CA).
Construction of the β-Globin Microarray.
The 75-kb sequence of the β-globin locus (http://www.ncbi.nlm.nih.gov, accession number v01317) was divided into 74 segments approximately 1 kb in length; primers (Research Genetics, Huntsville, AL) were designed to amplify each fragment. Table 2 lists the sequence of each primer and the position within the locus of the 5′ nucleotide; some fragments were further subdivided into 200- to 300-bp pieces, and primers were designed. Fragments were amplified from a BAC containing the β-globin locus in a 96-well format with Qiagen Taq Mastermix. The following thermalcycling conditions were used: 94°C for 5 min, 40 cycles of 10 s at 92°C, 30 s at 55°C, and 2 min at 72°C, followed by an extension time of 7 min at 72°C. Products were analyzed by agarose gel electrophoresis and ethidium bromide staining and were subsequently precipitated with sodium acetate/ethanol and centrifugation at 1,500 × g for 1 h at 4°C. Precipitated DNA was resuspended in water. Resuspended products (4 μl) were mixed with 4 μl of DMSO in a 384-well plate. The β-globin sequences then were arrayed onto slides along with control elements. Each PCR product was spotted 4–48 times on each array. The arrays were postprocessed according to manufacturer's instructions.
Probe Labeling and Hybridization.
Labeling of chromatin-immunoprecipitated DNA involved three amplification steps. The first step used a random octamer with a fixed sequence linker (5′-GTTTCCCAGTCACGATCNNNNNNNN-3′) and two cycles of T7 sequenase (United States Biochemical) polymerization. The thermalcycling conditions were as follows: 2 min at 94°C, 2 min at 8°C, ramped for 8 min to 37°C and 8 min at 37°C. This reaction mix was diluted in 1× TE and purified by Qiagen purification system. One half of the products were amplified by PCR using the fixed sequence primer (5′-GTTTCCCAGTCACGATC-3′) and 2× Qiagen Taq Mastermix in a 100-μl volume. The thermalcycling conditions were as follows: 25 cycles of 30 s at 92°C, 30 s at 40°C, 30 s at 50°C, and 1 min at 72°C. The products were purified, and 50% were used for the amplification and labeling step with the same primer and thermalcycling conditions, but with a 4:1 ratio of amino-allyl-conjugated dUTP (Sigma) to unconjugated dTTP; all other nucleotides were at a final concentration of 0.25 mM Qiagen Taq polymerase in appropriate buffer conditions. Products were purified and concentrated with Microcon-30 filters (Millipore); amino-allyl conjugates were crosslinked with monofunctional NHS-ester Cy3 or Cy5 dye (Amersham Pharmacia) for 1 hr. The crosslinking reactions were quenched with glycine, and the Cy3-conjugated DNA and Cy5-conjugated background DNA were combined and purified with the Qiagen Minelute kit. Purified probe was combined with hybridization buffer [5× SSC/25% formamide/0.1% SDS/20 μg human Cot1 DNA (GIBCO/BRL)]. The hybridization mix was boiled for 5 min and centrifuged for 2 min before applying to a prehybridized β-globin microarray. Microarrays were prehybridized with 5 × SSC/25% formamide/0.1% SDS/1% BSA for 45 min at 42°C. Slides were hybridized for 12–16 h at 42°C and washed according to manufacturer's conditions.
Data Analysis.
Microarrays were scanned with an Axon 4000A scanner (Axon Instruments, Foster City, CA), and images were analyzed with GENEPIX PRO3.0 software. The data were filtered to remove spots with aberrant morphology or those with intensities below the threshold of detection; then, the median ratios were normalized to background. The median ratio for each sequence element was determined, and the ratios for replicate experiments were scaled to the top-ranking elements for the anti-GATA-1 chIp experiments. For the mock chIp experiments, median ratios were scaled to the median value of all fragments because there were no consistent top-ranking elements. Error bars (see Fig. 2) represent the 75th percentile value of median ratios determined for each fragment.
Figure 2.

(a) GATA-1 chIp-chip analysis from K562 cells across the β-globin locus shows anti-GATA-1 enrichment in regions 009BG and 032BG. The bars for each region on the microarray are in the order they appear within the β-globin sequence and are in the approximate position shown relative to the schematic of the locus drawn in c. Each bar represents the median value for eight experiments, with the error bars to the 75th percentile value of all median ratios determined for that fragment. (b) ChIp-chip analysis of mock immunoprecipitated DNA from K562 cells shows no peaks in enrichment. Each bar represents the median value for three experiments with error bars to the 75th percentile value. (c) The number of GATA-1 consensus sites within each segment of the β-globin locus are presented in the order that they appear within the locus. A schematic of the β-globin locus also is shown to indicate the relative position of each of the array segments to relevant features along the locus.
Results
GATA-1 Chromatin Immunoprecipitations.
We identified β-globin regions bound by GATA-1 in K562 cells by chIp-chip. This approach involves first formaldehyde cross-linking to fix protein–DNA interactions followed by cell lysis and sonication to shear chromatin to 500-bp fragments. Protein–DNA complexes then are immunoprecipitated with an antibody against the transcription factor of interest. After reversal of the crosslinks, immunopurified DNA is extracted, labeled, and hybridized to a microarray of human β-globin locus sequences. Three different GATA-1 antibodies were used to map GATA-1 binding sites within the β-globin locus; each was raised against a different epitope. Anti-GATA-1(C20) (Santa Cruz Biotechnology) is a goat polyclonal antibody prepared against a carboxyl-terminal peptide; human-reactive anti-GATA-1(N6) (Santa Cruz Biotechnology) is a rat monoclonal antibody raised against a recombinant mouse GATA-1 protein; and anti-GATA-1(amino acids 66–78) is a rabbit polyclonal prepared against amino acid residues 66–78 of human GATA-1. In K562 cell lysates, all three antibodies recognize a protein that migrates at 45 kDa (Fig. 1a). Weaker reacting bands also are observed; the size of these bands differs with the different GATA-1 antibodies. A 45-kDa peptide was not seen when control antibodies, goat polyclonal c-jun antibody, which recognizes a protein of approximately 50 kDa, and the rabbit polyclonal Nrf1 antibody, which recognizes a protein of 35 kDa, were used to probe blots of K562 extracts.
Figure 1.
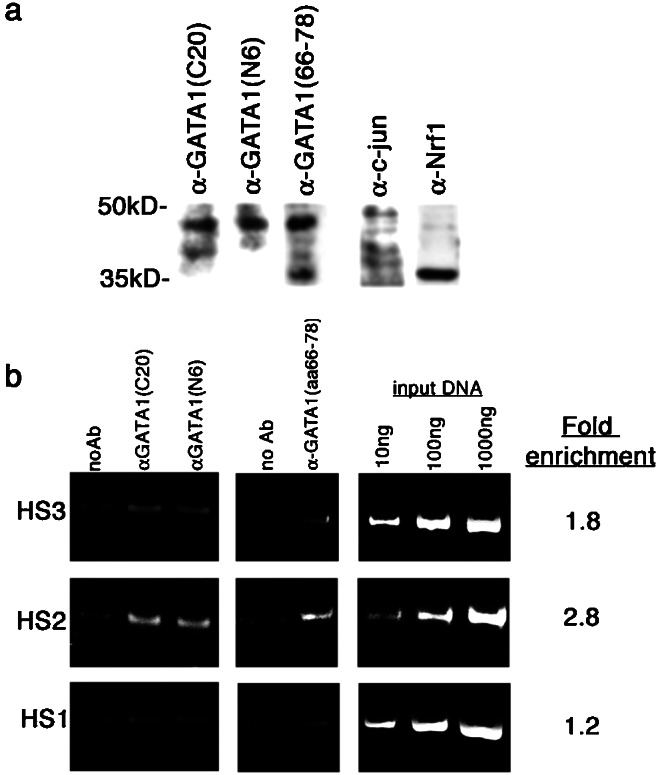
(a) GATA-1 antibodies, anti-GATA-1(C20), anti-GATA-1(N6), and anti-GATA-1(amino acids 67–78) recognize a protein that migrates at approximately 45 kDa in human K562 nuclear extracts by immunoblot analysis, whereas sera raised against c-jun and Nrf1 recognize proteins of 50 kDa and 35 kDa, respectively, in similar extracts. (b) PCR assays of the core HS3, HS2, and HS1 regions with chromatin that was mock immunoprecipitated (noAb lanes) or immunoprecipitated with one of three GATA-1 antibodies [GATA-1(C20), GATA-1(N6), and GATA-1(amino acids 67–78) lanes]. As positive controls, 10 ng, 100 ng, and 1 μg of whole lysate DNA was amplified in parallel.
Ikuta et al. (12) and Reddy et al. (13) had previously shown footprinting of a GATA-1 consensus site within the HS2 region, but not within the HS3 region in K562 cells. Conflicting evidence was presented recently by GATA-1 chIp-PCR assays in K562 cells showing anti-GATA-1 interaction with the HS3 region, in addition to the HS2 element (16). Footprinting analysis in other cell lines also presents conflicting results of GATA-1 sites protected within the HS1, HS3, and HS4 regions, but not within the HS2 region (14, 15). Here, we first examined the HS1, HS2, and HS3 core elements for GATA-1 binding by chromatin immunoprecipitation and PCR assays. The GATA-1 transcription factor was immunoprecipitated from fixed K562 cells by using different GATA-1 antibodies. GATA-1-associated DNA was assayed for specific enrichment of the hypersensitive regions by PCR.
We observe consistent enrichment of only the HS2 core element in GATA-1 immunoprecipitates as compared with mock immunoprecipitates (Fig. 1b). The mean fold-enrichment (intensity of product band for GATA-1 immunoprecipitates over the intensity of the product band for the mock immunoprecipitates) for all three GATA-1 antibodies is shown. All three antibodies yield consistent enrichment ratios, and only the HS2 region was enriched 2- to 4-fold for each of the antibodies. The HS1 element shows no GATA-1 enrichment over the mock immunoprecipitates for any of the GATA-1 antibodies with fold-enrichment ratios of 1.3, 1.1 and 1.2 for anti-GATA-1(C20), anti-GATA-1(N6), and anti-GATA-1(amino acids 66–78), respectively. The HS3 region shows a slight enrichment of 2.2, 1.7 and 1.4-fold in anti-GATA-1(amino acids 66–78) immunoprecipitates, anti-GATA-1(C20) and anti-GATA-1(N6), respectively.
DNA from sonicated nuclear extracts (input control DNA) at 10-, 100-, and 1,000-fold higher concentrations than the immunoprecipitated DNA were PCR amplified in parallel. The intensity of the HS2 band for anti-GATA-1-immunoprecipitated DNA is greater than the intensity of the PCR product obtained when a 10-fold higher concentration of control DNA is used as template (Fig. 1b, 10-ng lane). This observation suggests a specific enrichment of HS2 sequences in GATA-1 immunoprecipitates compared with the total pool of genomic DNA. The intensity of the HS1 and HS3 PCR products in anti-GATA-1 immunoprecipitates is less than that observed for the 10-ng input control, indicating a lack of significant enrichment.
Binding Profile for GATA-1 Across the β-Globin Locus Using ChIp-chip.
To determine the distribution of GATA-1 binding sites across the entire 75-kb β-globin locus, chIp-chip analysis was performed in which the sequence was divided into 74 contiguous fragments (fragments 001BG-074BG) approximately 1 kb in length. Each fragment was amplified with sequence-specific primers (Table 2) and printed onto microarrays 4–48 times. These β-globin arrays were probed with Cy3-labeled (red) DNA from anti-GATA-1 immunoprecipitates and Cy5-labeled (green) background DNA. The Cy3/Cy5 median intensity ratios were determined for each sequence element. These experiments were repeated eight times with different GATA-1 antibodies, different reference DNA, as well as a different number of cells (Table 1, experiments 1–8). We obtained similar results whether we used a pool of total genomic DNA as a reference or DNA that was immunoprecipitated with an antibody of the same seratype. The results are summarized in Fig. 2a.
Two β-globin locus sequence regions were consistently enriched greater than 2-fold with this analysis. One of these regions, 009BG, encompasses the HS2 region of the LCR and the other, 032BG, a region located 822–1,784-bp upstream of the start codon of the γG gene. Enrichments are not observed in the three mock chromatin immunoprecipitation experiments summarized in Fig. 2b (Table 1, experiments 9–11), which were performed with no antibody, but similar results are observed when rabbit preimmune serum is used for immunoprecipitation (Table 1, experiment 12; data not shown). GATA-1 chIp-chip experiments with HeLa cells, which do not express the globin genes, also lack enrichment of regions 009BG and 032BG (Table 1, experiment 13; data not shown). We also looked at the binding profiles for other transcription factors known to bind within the locus, such as Nrf1 and CBP (CREB-binding protein), by using the same approach. Different binding profiles were obtained for these proteins.
For any of these chIp-chip experiments, sporadic, nonreproducible enrichments occur, particularly within the L1 regions of the locus, which contain repetitive DNA. These regions are indicated in Fig. 2 by large error bars for these segments. It is possible that GATA-1 binds in or near an L1 line element repeat somewhere within the genome; the sequence homology between this fragment and the L1 elements on the β-globin array result in sporadic enrichments.
In addition to uninduced cells, chIp-chip experiments (Table 1, experiment 7–8) were performed from hemin-induced K562 cells. Previous studies have suggested, but not directly demonstrated, that the up-regulation of γ-globin expression upon hemin treatment is a posttranscriptional effect. Consistent with this interpretation, the GATA-1 binding profile for these treated cells was the same as that for the uninduced cells (data not shown; combined with other experiments). Therefore, the change in γ-globin gene expression observed upon hemin-induction is not caused by differential GATA-1 binding.
Distribution of GATA-1 Consensus Binding Sites.
We surveyed the locus for GATA-1 consensus sites to see whether there is a relationship between the density of sites and enrichment in GATA-1 immunoprecipitates. Fig. 2c shows the number of consensus recognition elements in each 1-kb segment of the β-globin locus. There seems to be no correlation between the number of sites and the observed enrichment. Regions 009BG and 032BG contain 5 and 8 sites, respectively. However, segments with 10–12 consensus sites, including regions 005BG, 022BG, 025BG, 028BG, and 038BG show no significant enrichments, with Cy3/Cy5 median ratios that range from 1.0–1.5. Sites of GATA-1 binding do not correspond to regions with a high density of GATA-1 recognition elements.
Fine Mapping of GATA-1 Binding Within Enriched β-Globin Fragments.
PCR assays were performed in an effort to confirm the anti-GATA-1 chIp-chip results and to determine more precisely the location of GATA-1 transcription factor binding within the enriched fragments. Regions 009BG and 032BG were subdivided into 250- to 300-bp fragments, and primers were designed (Table 2). Primers also were designed for smaller fragments of region 033BG, immediately upstream of the γG gene, bearing GATA-1 consensus binding sites (Table 2). Enrichment of these smaller fragments in anti-GATA-1 immunoprecipitates as compared with mock immunoprecipitates was assessed by PCR assay. The results of these assays on region 009BG containing HS2 are shown in Fig. 3a. The middle panel shows a 2.9-fold enrichment of the center 276-bp fragment of region 009BG, which contains the “core” HS2 sequence. The regions flanking the core element are not enriched. The entire sequence of the core enriched region is shown in Fig. 3b. Only one consensus GATA-1 site is present in this region (indicated by the black box). Previously, this site has been shown to be footprinted in K562 cells (12, 13).
Figure 3.
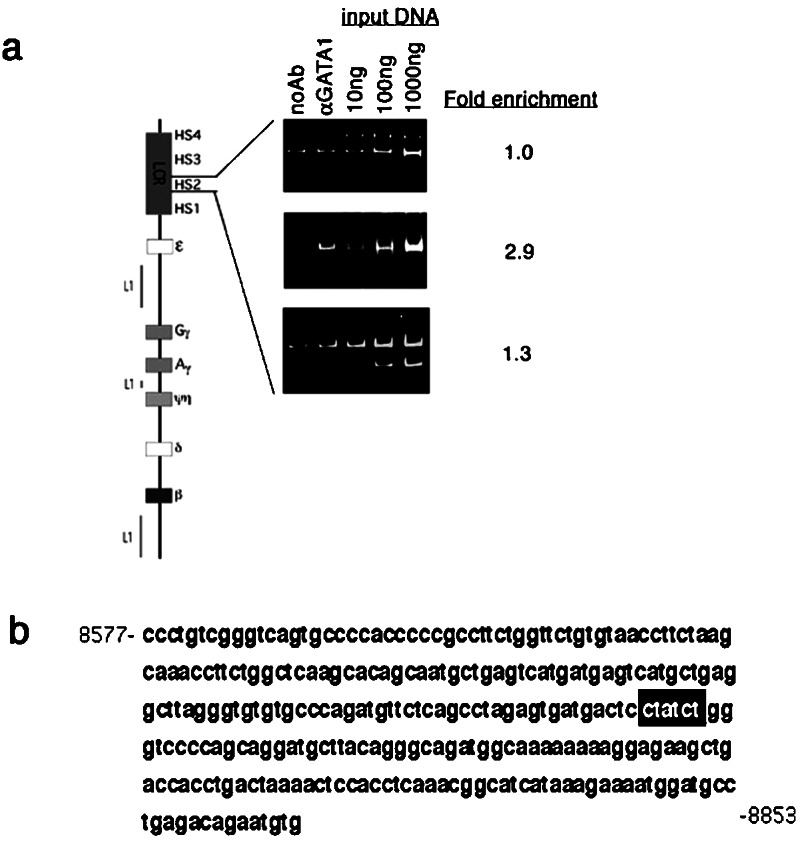
(a) GATA-1 chIp-PCR assays for subdivisions of region 009BG containing the HS2 element show specific enrichment of the core sequence (Middle) but not of the flanking sequences (Top and Bottom). The lanes are described in Fig. 1. (b) The sequence of the core HS2 region amplified is shown, and the consensus GATA-1 site is boxed in black.
The PCR assay results of sequences upstream of the γG gene are shown in Fig. 4a. Peak enrichments of 2.8- and 4.6-fold are observed for two of these small fragments, −1557 to −1289 and −1055 to −822 from the γG start codon, respectively. A weak enrichment of 2.0-fold is observed for the fragment just upstream of the −1557 to −1289 sequence. Enrichment is not observed for sequences more proximal to the γG gene. The sequences of the strongly enriched sites are shown in Fig. 4 b and c. The first sequence bears three GATA-1 recognition elements, including one near the 5′-end of the sequence (Fig. 4b). The flanking, weakly enriched fragment slightly overlaps with the 5′-end of this fragment, so GATA-1 binding at this recognition site may account for the enrichment observed in the −1557 to −1289 site and the weak enrichment observed in the neighboring fragment. The −1055 to −822 fragment contains two GATA-1 consensus sites (Fig. 4c). Three of the five GATA-1 sites within these two enriched regions have the sequence “tgataa.” There are 23 other “tgataa” sites with the same orientation within the β-globin locus, but they are evenly distributed along the locus (data not shown).
Figure 4.

(a) GATA-1 chIp-PCR assay of subdivisions of region 032BG and the promoter region of the γG genes shows two regions of enrichment in GATA-1 immuno-precipitates represented in the second and fourth panels. The lanes are described in Fig. 1. (b) The sequence of enriched region represented in the second panel above is shown with GATA-1 consensus motifs boxed in black. (c) The sequence of enriched region represented in the fourth panel above is shown.
PCR assays also were performed on anti-GATA-1 immunoprecipitates for other selected regions within the locus, including the ɛ-, δ-, β-promoters, the γA- and β-enhancers, and regions in and around the pseudo-β gene and between the HS2 and HS1 regions. Significant enrichments are not observed for the other regions (data not shown).
Discussion
In the past, only limited regions of the β-globin locus have been examined for transcription factor binding. The scope of in vivo footprinting and traditional chIp-PCR analysis is limited to regions predicted to play a role in gene regulation. ChIp-chip technology allows for a comprehensive survey of the entire locus. By using this approach, we find GATA-1 binding within two 1-kb segments along the β-globin locus in K562 cells. One of these sites contains the HS2 core element. The other is a previously undescribed region of GATA-1 binding upstream of the γG gene.
We refined the location of GATA-1 binding within these 1-kb segments by chIp-PCR analysis. PCR assay results of the “core” HS2 and flanking regions, indicate that GATA-1 binds within the core HS2 element and not within the neighboring sequences. The DNA sequence of this core region contains only one consensus GATA-1 site. In vivo footprinting studies (12, 13) show protection at this GATA-1 recognition element within the HS2. The functional significance of GATA-1 binding at this site has yet to be determined. It has been shown that mutating this recognition site has no effect on γA expression in K562 hemin-induced and uninduced cells (21). However, this experiment was performed in the context of a recombinant adeno-associated virus, so it is possible that in its natural context, GATA-1 influences the expression of γG and/or γA from the HS2 region of the LCR. Also, it is possible that other GATA-1 binding sites contribute to the expression of γ-globin gene expression.
Consistent with this hypothesis, we found by PCR assay GATA-1 binding at least two additional regions upstream of the γG gene, within −1557 to −1289 and within −1055 to −822 before the start codon. These sites are several hundred bp removed from the canonical promoter sequence, which was not enriched in GATA-1 immunoprecipitates. It is interesting to note that three of the five potential GATA-1 binding sites in these two regions are the sequence “tgataa.” The recognition sequence in HS2 is “agatag,” so it would seem that not all GATA-1 in vivo binding sites are identical, but perhaps in a particular context or environment, GATA-1 prefers a specific binding motif. Similarly, it has been shown by in vivo footprinting in the human-mouse hybrid cell line Hu11 that three “agatag” GATA-1 sites are protected in the HS3 region of the LCR (15). Promoter/enhancer environment may dictate the affinity of GATA-1 for a particular sequence.
The effect of GATA-1 binding at these sites upstream of the γG gene on γ-globin expression in K562 cells is not known. However, because γG is the most highly expressed globin in K562 cells, the new binding sites will most likely be important for γG transcription. As the region of binding is removed from the TATA box at −30 bp from the γG gene start codon, GATA-1 may not directly interact with the basal transcriptional machinery. It is possible that these regions of GATA-1 binding link to the transcriptional machinery via another protein or directly by a contortion in the DNA. Several proteins, such as Friend of GATA (FOG) and CBP/p300, are known to interact with GATA-1 and may mediate the interaction with the RNA polymerase (22–24). Alternatively, GATA-1 binding may initiate a change in chromatin structure that primes the region for transcription. Individually mutating these potential GATA-1 binding sites or mutating them in combination would be important to understanding their contribution to γ-globin gene expression.
The results of our GATA-1 chIp-chip analysis are in concordance with in vivo footprinting studies in K562 cells (12, 13), which again show protection of a GATA-1 site in the HS2 core region, but not in the HS3 region, the γG- and β-promoters, or in the γA 3′- and β-3′-enhancers. However, results of GATA-1 chIp-PCR analysis in K562 cells by Duan et al. (16) conflict with our findings and the footprinting studies. They report GATA-1 enrichment of the HS3 region and the γ-promoter. However, they fail to show regions that are not enriched in anti-GATA-1 immunoprecipitates, and their enrichments are not above control input DNA. Perhaps these regions are enriched as a result of a higher concentration of DNA template, or perhaps chromatin shearing was not complete, and large DNA fragments from the β-globin locus created the observed enrichment at these other regions in GATA-1 immunoprecipitates.
The lack of detectable binding of GATA-1 in the proximal promoter region of the γ-globin genes is noteworthy. The K562 cell line expresses a relatively low level of globin, and it is conceivable that, at any one time, the γ-globin promoters in the majority of the cells are not active. Also, these cells presumably resemble early erythroid precursors that produce a smaller fraction of total mRNA as globin mRNA than do more mature cells. This finding may reflect different specific mechanisms of globin gene activation. Finally, K562 cells are derived form a leukemic cell, and globin regulation may be aberrant in these cells.
Footprinting analysis of specific, functionally important sites within the β-globin locus using human murine hybrid cells have identified putative GATA-1 binding sites in the HS4, HS3, and HS1 regions, the γG promoter, and the β 3′-enhancer (14, 15). We did not observe GATA-1 binding in any of these regions by chIp-chip analysis or chIp-PCR assay. It is possible that we were not able to detect enrichment of these regions because the GATA-1 epitope was inaccessible, but because we used three different GATA-1 antibodies recognizing three different epitopes, this explanation seems unlikely. A more probable explanation is that the different cell lines exhibit different GATA-1 binding. Variations in the relative concentrations and types of other transcription factors or chromatin-binding proteins, as well as DNA sequence, lead to alterations in GATA-1 binding in different cell lines.
The chIp-chip technique is of enormous utility for understanding regulation of the globin locus and can be applied to other transcription factors involved in globin expression, such as NF-E2, erythroid Kruppel-like factor, FOG, and CBP/p300. Binding analysis of these factors in different cell lines under different conditions would lend an understanding to how the β-globin locus is temporally regulated. The position of transcription factors and chromatin modifiers within the locus in relation to the actively transcribed globin genes can perhaps help distinguish between the looping and linking models of globin gene expression (for review, see ref. 1).
It would be of general interest to apply the chIp-chip approach on a genome-wide scale for mammalian systems. As chIp-chip technology continues to improve, it should be possible to use smaller numbers of cells, to obtain higher levels of relative enrichment of bound fragments, and to analyze even larger chromosomal regions.
Supplementary Material
Acknowledgments
We thank those who critically read this manuscript. The work in M.S.'s laboratory was funded by National Institutes of Health Grant CA77808. S.M.W.'s laboratory is supported by National Institutes of Health Grant CA42556. Bioinformatics assistance was provided by the Gerstein laboratory, which is supported by the Keck Foundation. N.M.L. is sponsored by the Anna Fuller Fund, and C.E.H. is funded by a Howard Hughes Predoctoral Fellowship.
Abbreviations
- LCR
locus control region
- HS
DNase I hypersensitive region
- chip
chromatin immunoprecipitation
References
- 1.Engel J D, Tanimoto K. Cell. 2000;100:499–502. doi: 10.1016/s0092-8674(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 2.Forrester W C, Thompson C, Elder J T, Groudine M. Proc Natl Acad Sci USA. 1986;83:1359–1363. doi: 10.1073/pnas.83.5.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuan D, Solomon W, Li Q, London I M. Proc Natl Acad Sci USA. 1985;82:6384–6388. doi: 10.1073/pnas.82.19.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orkin S H. Curr Opin Cell Biol. 1995;7:870–877. doi: 10.1016/0955-0674(95)80072-7. [DOI] [PubMed] [Google Scholar]
- 5.Weiss M J, Orkin S H. Exp Hematol (Charlottesville, VA) 1995;23:99–107. [PubMed] [Google Scholar]
- 6.Martin D I K, Orkin S H. Genes Dev. 1990;4:1886–1898. doi: 10.1101/gad.4.11.1886. [DOI] [PubMed] [Google Scholar]
- 7.Orkin S H. Blood. 1992;80:575–581. [PubMed] [Google Scholar]
- 8.Fujiwara Y, Browne C P, Cunniff K, Goff S C, Orkin S H. Proc Natl Acad Sci USA. 1996;93:12355–12358. doi: 10.1073/pnas.93.22.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss M J, Keller G, Orkin S H. Genes Dev. 1994;8:1184–1197. doi: 10.1101/gad.8.10.1184. [DOI] [PubMed] [Google Scholar]
- 10.Weiss M J, Orkin S H. Proc Natl Acad Sci USA. 1995;92:9623–9627. doi: 10.1073/pnas.92.21.9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss M J, Yu C, Orkin S H. Mol Cell Biol. 1997;17:1642–1651. doi: 10.1128/mcb.17.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikuta T, Kan Y W. Proc Natl Acad Sci USA. 1991;88:10188–10192. doi: 10.1073/pnas.88.22.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy P M S, Shen C-K J. Proc Natl Acad Sci USA. 1991;88:8676–8680. doi: 10.1073/pnas.88.19.8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikuta T, Papayannopoulou T, Stamatoyannopoulos G, Kan Y W. J Biol Chem. 1996;271:14082–14091. doi: 10.1074/jbc.271.24.14082. [DOI] [PubMed] [Google Scholar]
- 15.Strauss E C, Orkin S H. Proc Natl Acad Sci USA. 1992;89:5809–5813. doi: 10.1073/pnas.89.13.5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duan Z, Stamatoyannopoulos G, Li Q. Mol Cell Biol. 2001;21:3083–3095. doi: 10.1128/MCB.21.9.3083-3095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer V R, Horak C E, Scafe C S, Botstein D, Snyder M, Brown P O. Nature (London) 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 18.Ren B, Robert F, Wyrick J J, Aparicio O, Jennings E G, Itamar S, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 19.Horak C E, Snyder M. Methods Enzymol. 2002;350:469–483. doi: 10.1016/s0076-6879(02)50979-4. [DOI] [PubMed] [Google Scholar]
- 20.Orlando V, Paro R. Cell. 1993;75:1187–1198. doi: 10.1016/0092-8674(93)90328-n. [DOI] [PubMed] [Google Scholar]
- 21.Miller J L, Walsh C E, Ney P A, Samulski R J, Nienhuis A W. Blood. 1993;82:1900–1906. [PubMed] [Google Scholar]
- 22.Blobel G A, Nakajima T, Eckner R, Montminy M, Orkin S H. Proc Natl Acad Sci USA. 1998;95:2061–2066. doi: 10.1073/pnas.95.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hung H-L, Lau J, Kim A Y, Weiss M J, Blobel G A. Mol Cell Biol. 1999;19:3496–3505. doi: 10.1128/mcb.19.5.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsang A P, Visvader J E, Turner C A, Fujiwara Y, Yu C, Weiss M J, Crossley M, Orkin S H. Cell. 1997;90:109–119. doi: 10.1016/s0092-8674(00)80318-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.