

Abstract
Human chromosomes 21 and 22 (mainly the q-arms) were the first complete parts of the human genome released. Our analysis of genes, pseudogenes (Ψg), and Alu repeats across these chromosomes include the following findings: The number of gene structures containing untranslated exons exceeds 25%; the terminal exon tends to be the largest among exons, whereas, the initial intron tends to be the largest among introns; single-exon gene length is approximately the mean gene exon number times the mean internal exon length; processed Ψg lengths are on average approximately the same as single-exon gene length; and the G+C content and length of genes are uncorrelated. The counts and distribution of genes, Ψg, and Alu sequences and G+C variation are evaluated with respect to clusters and overdispersions. Other assessments concern comparisons of intergenic lengths, properties of Ψg sequences, and correlations between Alu and Ψg sequences.
Two “drafts” of the human genome have now been released: a public version (Human Genome Project) and the Celera version (1, 2). The first completely sequenced parts of the human genome included the euchromatic portions (q-arms) of chromosomes 21 and 22 (Chr21 and Chr22, respectively). A total of 34.55 Mb (about 97%) of Chr22q was sequenced in 12 contigs, and 33.6 Mb of Chr21q was sequenced in four contigs (3, 4). Neither p-arm of Chr21 and Chr22, mainly heterochromatin, was completely sequenced. The gene annotation available for Chr22 (as of March 6, 2001) is of two kinds: (i) complete gene structures specifying all exons and introns plus 5′ and 3′ untranslated regions (UTRs), and (ii) coding sequence structures (CDSs) restricted to exon regions translated into proteins and intervening introns. No CDS annotation is available for Chr21.
In this article we examine, among other things, the distribution of genes, pseudogenes (Ψg), repeats (mainly Alu elements), and G+C frequency (Fgc) variation. Comparisons, contrasts, and analysis of Chr21 and Chr22 will center on the following assessments: (i) correlations and associations of genes, Ψg, Alu counts, and Fgc variables; (ii) gene 5′ and 3′ intergenic lengths (see later text for precise definitions); (iii) numbers, lengths, and distribution of single-exon (intronless) genes; (iv) the distribution of genes with different exon numbers; (v) comparisons of intergenic lengths for consecutive pairs of genes with (−,−) orientations, (+,+) orientations, (−,+) divergent orientations, and (+,−) convergent orientations; (vi) the relative distribution of Alu and Ψg sequences in intergenic regions vs. introns; (vii) conspicuous genes (e.g., ribosomal protein genes) among Ψg sequences; (viii) the distribution of Ψg sequences associated with processed or small genes versus multiexon genes; (ix) the statistics of exons that are transcribed but not translated; and (x) to what extent genes, Ψg, and Alu sequences are clustered or overdispersed in Chr21 and Chr22.
There are at least three data annotations covering Chr21 and Chr22. The original Riken gene catalog of Chr21 (4), the Sanger Centre database of Chr22 (3), the University of California Santa Cruz (Golden Path) collection for Chr21 and Chr22, and refseq, maintained by the National Center for Biotechnology Information, derived, and extended from Golden Path. The sequence assemblies are virtually the same for each source. The known human genes, with recognized names, are in excellent (but not perfect) agreement across the data sets. However, there are many differences in annotation with respect to ORFs, predicted genes, matching spliced expressed sequence tags, and alternative splicings (5, 6). Our analysis concentrates on the Riken and Sanger Centre data but it appears to be consistent overall with the other data sets.
Chromosomal Counts of Genes, Ψg, and Alus
The Riken annotation of Chr21 (33.6 Mb) reports 214 complete gene structures, 53 Ψg, and 12,168 Alu elements (as of Jan. 16, 2001). On Chr22q (34.5 Mb), the Sanger annotation reports 552 genes, 145 Ψg, and 21,993 Alu elements have been identified. Thus, for the same approximate euchromatin extent, Chr22 has more than twice as many gene structures as Chr21, almost twice as many Alu sequences, and 3-fold more Ψg, consistent with the greater overall Fgc of Chr22 (48%) compared with Chr21 (42%) (3, 4). Chromosomes with more genes have more accessible genomic DNA with respect to Ψg and Alu sequences, partly because of more transcriptional activity, so a key determinant in these counts is the greater gene density and greater G+C content in Chr22 versus Chr21. Along these lines, among human chromosomes Chr19 has the highest G+C content (overall 49%), the highest gene density, the highest CpG dinucleotide bias, and more CpG islands, and next in these contexts is Chr22 (1, 7). In Chr21, the aggregate length of intergenic regions is 24,851 kb and the aggregate intron length is 8,241 kb, a ratio of about 3:1. For Chr22 the corresponding ratio is 20,611 kb to 11,758 kb, about 2:1. These data are based on the gene structure annotation and exclude the Ig gene segments.
Chr22 contains 118 λ-Ig gene segments (variable V segments). Five consecutive Ψg of Ig κ-V region about locations 1329337–1359121 of Chr22q are included. Excluding these Ig gene segments, in Chr22 the mean number of exons per gene is 7.1 (median 5.5). The mode is 98 genes attained for single-exon genes. Chr21 has mean exon number 8.5 (median 6) and the mode occurs for genes of three exons, with 39 such genes (see Fig. 1).
Figure 1.

The first three graphs indicate the number of genes in Chr21 and Chr22 with different numbers of exons. The last graph shows the number of genes with counts for 3′ and 5′ UTEs in Chr22 (there is no corresponding data set for Chr21).
Numbers of Genes Containing Untranslated Exons (UTEs)
A total of 453 of the complete gene structures have their coding region specified in the CDS data set, 333 genes (73.5%) have no 5′ UTEs, 84 have a single 5′ UTE, 21 have two, seven have three, four have four, three have five, and one has eight.¶ A total of 403 (89%) genes have no 3′ UTEs, 36 have one, eight have two, three have three, two have five, and one has eight. These statistics are impressive for the proportion of genes (at least 25%) that possess UTEs. It is not known what kinds of controls these UTEs portend. Some possibilities are: UTEs probably play a role in regulating export of mRNA from the nucleus and 5′ UTEs with connecting introns participate in translation initiation; and 3′ UTEs also may assist in mRNA stability and with polyadenylation linkers. 5′ UTEs putatively contribute in regulating alternative splicing and translation efficiency (8). It has been established in Drosophila that the 3′ UTR plays a functional role in cytoplasmic localizations of mRNA transcripts (9, 10). There are also examples of sequential processing activities governed by 5′ alternative promoters [e.g., ultrabithorax (11)]. In human, the protein coding sectors of G protein-coupled receptors are predominantly intronless but at least l8% of the underlying genes contain 5′ UTEs (12, 13). Sosinsky et al. (13) proffer an excellent discussion of olfactory G protein-coupled receptors with intronless coding region that possess introns in their 5′ UTRs. These genes seem to involve retropositions at least in its early evolutionary stages and alternative splicing events using separate acceptor or donor splice sites of the same exon.
Do genes with greater numbers of exons and extended protein coding sequences tend to have more flanking UTEs? A correlation calculation yields no significant correlation between gene exon numbers and UTE counts and lengths.
What kinds of genes contain many UTEs (5′ and/or 3′)? Table 1 lists some examples of genes of Chr22 with five or more UTEs at both the 5′ and 3′ ends.
Table 1.
Genes of Chr22 with five or more UTEs
| Locus | No. of exons | No. of 5′ UTEs | 5′ UTL, bp | No. of 3′ UTEs | 3′ UTL, bp | Description |
|---|---|---|---|---|---|---|
| Genes with 5 or more 5′ UTEs | ||||||
| AC006285.5 | 9 | 5 | 714 | 0 | 3088 | Homo sapiens MIL1 protein mRNA |
| GGT1 | 17 | 5 | 668 | 0 | 1713 | Gamma-glutamyltransferase 1 |
| HMG2L1 | 12 | 5 | 565 | 0 | 2155 | High mobility group protein 2-like 1 |
| LZTR1 | 21 | 8 | 860 | 0 | 1713 | Leucine-zipper-like transcriptional regulator 1 |
| Genes with 5 or more 3′ UTEs | ||||||
| DJ319F24.C22.1 | 11 | 0 | 0 | 5 | 981 | Matches expressed sequence tag sequences |
| DJ671014.C22.2 | 13 | 2 | 275 | 5 | 661 | Homo sapiens gamma-parvin mRNA |
| DJ402G11.C22.6 | 16 | 1 | 247 | 8 | 1423 | Matches expressed sequence tag cluster |
UTL, untranslated exon length.
A gene in possession of one or more 5′ UTEs does not necessarily involve 3′ UTEs. A direct calculation shows that the flanking UTR exon counts are basically uncorrelated: correlation (5′ UTE, 3′ UTE) = 0.006; correlation (5′ untranslated exon length, 3′ untranslated exon length) = 0.10.
Correlations of Genes, Ψg, Alu Counts, and Fgc Variables
We traversed Chr21 and Chr22 and compared the counts of genes, Ψg, Alu sequences, and the average Fgc in 25-kb, 50-kb, and 100-kb sliding windows with 5-kb displacements. The correlations between these variables are displayed in Table 2. The correlations are largely consistent with the familiar facts that in eukaryotes the density of genes increases with Fgc (e.g., ref. 14), and Alu sequences are predominantly G+C rich (15). Interestingly, the correlations increase with window size, probably as a consequence of the statistical law of large numbers. Explicitly, in Chr21, correlation (gene, Fgc: window size, W = 25 kb) = 0.32, correlation (gene, Fgc: W = 50) = 0.43, correlation (gene, Fgc: W = 100) = 0.54. A corresponding pattern prevails in Chr22.
Table 2.
Correlations among counts of genes, Ψg, Alu sequences, and Fgc
| Window size | Chr21
|
Chr22
|
|||||
|---|---|---|---|---|---|---|---|
| Ψg | Alu | Fgc | Ψg | Alu | Fgc | ||
| 25 k | g | 0.02 | 0.15 | 0.32 | 0.04 | 0.13 | 0.26 |
| Ψg | 0.09 | −0.02 | 0.01 | −0.10 | |||
| Alu | 0.31 | 0.03 | |||||
| 50 kb | g | 0.05 | 0.23 | 0.43 | 0.09 | 0.22 | 0.29 |
| Ψg | 0.13 | −0.03 | 0.01 | −0.14 | |||
| Alu | 0.34 | 0.08 | |||||
| 100 kb | g | 0.05 | 0.33 | 0.54 | 0.16 | 0.30 | 0.33 |
| Ψg | 0.16 | −0.04 | 0.01 | −0.18 | |||
| Alu | 0.37 | 0.13 | |||||
Apparently, because gene and Alu counts correlate positively with G+C levels, they correlate positively with each other. However, a manifest contrast between Chr21 and Chr22 is that Alu counts and Fgc values are positively correlated in Chr21 but uncorrelated in Chr22. Possible reasons are: There could be different target sites or sources for the Alu distributions in the two chromosomes or the Alu samples may differ sharply in their age composition and base composition. In both chromosomes, we also observe that Ψg locations are uncorrelated with gene locations. This finding could signify that Ψg sequences are generated randomly throughout the human genome and randomly inserted into the genome mostly by reverse transcription.
Comparison of Intergenic Lengths
For Chr21, we concentrated on intergenic regions that do not cross the three unsequenced gaps, also removing overlapping gene groups and excluding intergenic regions exceeding 1 Mb as outliers. A corresponding scheme was applied to study the intergenic regions of the largest five contigs in Chr22 (these contain 491 genes).
The 5′ extension of a gene is defined as the intergenic region extending from the 5′ end of the gene proceeding upstream to the next gene, which can be in either orientation (see Table 3). The 3′ extension refers to the intergenic region extending from the 3′ end of the gene proceeding downstream to the next gene. There are 190 consecutive pairs of genes in Chr21, which we divide into four groups (Table 4). There are 51 intergenic lengths for (−,−) gene pairs, where both genes share a negative orientation relative to the reported sequence. The median intergenic length is 35,568 bp. The group with (−,+) orientation comprises 48 pairs of genes, also called divergent pairs. In such an orientation, the promoter sequences of the two genes are roughly adjacent. The median intergenic length here is 73,116 bp. For (+,−) gene pairs (convergent pairs), there are 47 gene pairs with a common downstream intergenic separation of median length 22,077 bp. There are a total of 44 pairs of (+,+) genes with median intergenic length 28,950 bp. The median intergenic lengths, 35,568 bp, of (−,−) and 28,905 bp of (+,+) gene pairs differ by about 6,500 bp, consistent within statistical fluctuation. The fact that divergent gene pairs show the greatest intergenic separation makes sense because there are more regulatory sequences in the common intergenic region upstream of both genes including promoter and enhancer sequences of both genes. The convergent gene pairs generally have small intergenic separations. For Chr22, the corresponding results parallel those of Chr21.
Table 3.
5′ and 3′ extension lengths for genes of different exon counts
| Genes | Chr21 median,
bp
|
Chr22 median, bp
|
||||||
|---|---|---|---|---|---|---|---|---|
| Gene count | 5′ Extension | Gene count | 3′ Extension | Gene count | 5′ Extension | Gene count | 3′ Extension | |
| Single exon genes | 14 | 77,249 | 14 | 40,140 | 69 | 20,174 | 66 | 15,191 |
| Genes of 2 exons | 17 | 124,854 | 15 | 53,163 | 53 | 17,829 | 53 | 16,818 |
| Genes of 3 exons | 34 | 85,143 | 33 | 33,183 | 42 | 23,172 | 42 | 13,459 |
| Genes of 4 exons | 12 | 59,389 | 12 | 40,616 | 44 | 12,415 | 41 | 13,155 |
| Genes of 5 exons and more | 111 | 29,940 | 110 | 23,332 | 242 | 18,358 | 243 | 8,851 |
Table 4.
Comparisons of intergenic lengths
| Chr21 median, bp | Chr22 median, bp | |||
|---|---|---|---|---|
| 5′ Extension | | | |→ | 46,979 | 18,397 |
| 3′ Extension | →| | | | 28,260 | 10,783 |
| Intergenic region of (−,−) gene pairs | ←| | |← | 35,568 | 17,998 |
| Intergenic regions of (−,+) gene pairs | ←| | |→ | 73,116 | 19,623 |
| Intergenic regions of (+,−) gene pairs | →| | |← | 22,077 | 5,814 |
| Intergenic regions of (+,+) gene pairs | →| | |→ | 28,905 | 14,291 |
In Chr21, the intergenic lengths do not include the unsequenced gaps and overlapping gene groups. In Chr22, the intergenic lengths encompass the largest five contigs and exclude overlapping gene groups. Intergenic lengths of (−,−) are the intergenic lengths between two successive genes on the (−) strand. The other categories of (−,+), (+,−), and (+,+) are determined in the corresponding manner.
Table 4 suggests that 5′ regulatory regions are more extensive than 3′ regulatory regions. How is this affected by the extent of each gene and by the number of exons?
Table 3 highlights longer lengths in 5′ regions (with the single exception of genes of four exons in Chr22, perhaps because of few gene numbers).
Comparison of Lengths of Different Exon and Intron Types
Three types of exons—initial, internal, and terminal—are usually discriminated. The initial exons, which may play a role in transcription initiation, tend to be longer than internal exons (Tables 5 and 6). Internal exon lengths average about 150 bp and are reasonably constant for genes with at least five exons. The terminal exon length is relatively large and variable because such exons often contain 3′ UTR sequences.
Table 5.
Exon and intron lengths in gene structures
| Chr21
|
Chr22
|
|||
|---|---|---|---|---|
| Mean, bp | Median, bp | Mean, bp | Median, bp | |
| Single exon gene length | 1,209 | 674 | 1,322 | 947 |
| Initial exon length | 197 | 135 | 231 | 139 |
| Internal exon length | 158 | 129 | 142 | 120 |
| Terminal exon length | 784 | 365 | 1,009 | 653 |
| Initial intron | 13,311 | 3,844 | 8,928 | 2,592 |
| Internal intron | 4,423 | 1,845 | 3,510 | 1,312 |
| Terminal intron | 6,160 | 2,187 | 2,282 | 1,057 |
There are 180 genes in Chr21 and 389 genes in Chr22 with three or more exons. There are 141 genes in Chr21 and 341 genes in Chr22 with three or more introns.
Table 6.
Chr22 coding region exon and intron lengths
| Exon length, bp | Mean | Median | Intron length, bp | Mean | Median |
|---|---|---|---|---|---|
| Initial exon | 162 | 101 | Initial intron | 7,876 | 2,706 |
| Internal exon | 138 | 121 | Internal intron | 3,071 | 1,271 |
| Terminal exon | 206 | 132 | Terminal intron | 2,402 | 1,050 |
In Chr22, there are 354 genes with three or more translated exons. There are 310 genes with three or more introns. Data are not available for Chr21.
The exon length tends to be greatest for single-exon genes in both chromosomes. Internal exon and intron lengths are generally the smallest in Chr21 (Table 5). In multiple-exon genes, the terminal exon length is generally longer than internal exon lengths. This is not true for intron lengths. In Chr22, the terminal intron length is generally shorter than the internal intron length and the largest intron is principally the initial one (Table 6). This applies to both the complete gene structure annotations and also CDS data consonant with the impression that the first intron often carry some controls on transcription initiation and gene processing.
Is there a correlation between gene length and G+C content? On the basis of isochore studies it is observed that high G+C regions are more dense with genes. However from analysis of long genes in conjunction with expressed sequence tag data, it was suggested that long genes (i.e., genes with many exons) prefer DNA regions of reduced Fgc (16). We examined this hypothesis relative to the q-arms of Chr21 and Chr22. For the variables of exon number in gene structures we found for all genes correlation (exon no., G+C) = 0.021 (in Chr21) and −0.019 (in Chr22). For all genes with at least three exons, we ascertained correlation (exon no., mean internal exon length) = 0.082 (in Chr21) and −0.151 (in Chr22); and for all genes with at least four exons, we have correlation (exon no., mean internal intron length) = −0.073 (in Chr21) and −0.014 (in Chr22). These determinations effectively indicate that long genes are uncorrelated with respect to Fgc and with respect to internal exon and intron lengths.
Distinctive Features of Single-Exon Genes
Chr21 contains 15 single-exon (intronless) genes from a total of 214 genes (7%), with one located in an intron of another gene. Chr22 has 98 single-exon genes excluding the λ-Ig V gene segments. There are 13 single-exon genes located in intron regions of Chr22. Thus, in Chr22 the percent of single-exon genes, 98/552 = 17.8%, is significantly greater than the 7% in Chr21. Single-exon lengths are more than 2-fold longer than most exon lengths of multiexon genes (Tables 5 and 6).
In Chr21 and Chr22, the 5′ and 3′ extensions for single-exon genes generally exceed those of multiexon genes, and the 5′ extension length of a gene exceeds the 3′ intergenic length independent of exon numbers (Table 3). For example, the median of 5′ and 3′ extension lengths of the single-exon genes are 77,249 bp and 40,140 bp, respectively, in Chr21 and 20,174 bp and 15,191 bp in Chr22. Apparently, single-exon genes need more space to function properly. An evolutionary scenario may propose that most single-exon genes derive from a single intronless progenitor of recent evolutionary history with insufficient time to allow for gain of introns (“introns late” theory). This scenario putatively allows a rapid diversification in invertebrates, whereas vertebrates have acquired introns at a slower rate. A more likely possibility is that single-exon genes can be formed from fusions of exons (presumably by means of reverse transcription, transposition, or recombination). In this context, many single-exon genes need to be processed rapidly to achieve appropriate expression and for this reason avoid introns. An enticing observation is that in both chromosomes the mean single-exon gene length is close to the mean gene exon number times the mean internal exon length (Chr21: 1,209 ≈ 8.5*158; Chr22: 1,322 ≈ 7*142).
Distribution and Properties of Ψg Sequences
Ψg are nonfunctioning copies of genes that may result either from reverse transcription by means of a mRNA transcript (processed) or from gene duplication and subsequent disablement (17). A recent study of Ψg from Chr21 and Chr22 was set forth by Harrison et al. (18). Ψg sequences tend to be biased toward highly expressed genes. For example, many highly expressed ribosomal protein genes generate Ψg in eukaryotes. Clusters of ribosomal protein Ψg occur more frequently at the carboxyl end of Chr21 and Chr22, these regions also being somewhat higher in Fgc. Other frequent sources of Ψg include cytochrome subunits and membrane proteins (Table 7).
Table 7.
Pseudogene types with at least two occurrences
| Chr21 Ψg types of at least two occurrences indicating starting positions |
| Ribosomal protein components, 17 occurrences 4930891, 6631155, 7467697, 12311851, 14370507, 15947825, 22421026, 22673718, 22965074, 22999209, 23081472, 23117970, 23252636, 26075948, 26119393, 30480511, 33617159 |
| Cytochrome components (cytochrome p450 and cytochrome c subunits) two occurrences: 883308, 2527156 |
| Chr22 Ψg types of at least two occurrences indicating starting positions |
| Ribosomal protein components, 26 occurrences 1617744, 2429706, 3091507, 3645358, 10389068, 10853902, 14003472, 14035049, 14552659, 15436724, 15457833, 15714964, 19683973, 21032645, 23923189, 24403570, 26581122, 26896944, 27579196, 27776958, 29114107, 31431878, 31782364, 33006302, 33547793, 34546362 |
| GGT related (gamma-glutamyltransferase), 7 occurrences: 2622592, 2626735, 5131371, 6567692, 7583805, 8214982, 8599941 |
| Human membrane protein, 7 occurrences: 2700170, 2850276, 4618174, 5054329, 5210968, 8219216, 8624564 |
| Cytochrome c oxidase proteins, 3 occurrences; cytochrome p450 2 occurrences: 19093074, 20019122, 22937160; 25944997, 25954605 |
| Immunoglobulin kappa variable region pG, 5 occurrences: 1329337, 1339353, 1346561, 1351060, 1358639 |
| Homeotic Drosophila homolog, 4 occurrences: 6267591, 6294253, 6348328, 6374843 |
| Mitochondrial precursor, 3 occurrences: 694062, 18487456, 22539612 |
| Transcriptional repressor, 3 occurrences: 2687511, 5069545, 5196078 |
| Human keratin type 1 cytoskeletal 18 (cytokeratin 18), 3 occurrences: 3110597, 4413964, 28405473 |
| Human NADH-ubiquinone oxidoreductase chain 1, 2 occurrences: 7919065, 19742955 |
| Phorbolin 1, 2 occurrences: 22766055, 22887693 |
| Similar to mouse tubulin alpha-3 alpha-7 chain, 2 occurrences: 4930817, 4993379 |
| Actin like-protein, 2 occurrences: 911820, 8653947 |
| IGLC immunoglobulin lambda light chain C region: 16098291, 16255043 |
In Chr21, 49 Ψg are presumably processed into one exon each, whereas four have at least two exons; in Chr22, 123 Ψg are processed, whereas 22 involve two or more partially processed exons (eight consist of two exons, two of three exons, two of four exons, three of five exons, one of seven exons, two of eight exons, one of nine exons, two of 10 exons, and one of 15 exons). Table 7 displays all Ψg types that occur at least twice (see also ref. 18).
There are Ψg shared by both chromosomes. In this respect, the ribosomal protein gene Ψg are conspicuous. Thus, the 60S L23a has two copies in Chr21 and one copy in Chr22. One L10 Ψg is identified in Chr21 and one in Chr22. Table 8 presents some data on Ψg types that occur in both chromosomes.
Table 8.
Common Ψg types in Chr21 and Chr22
| Common Ψg types in both Chr21 and Chr22 | Locations in Chr21 | Locations in Chr22 |
|---|---|---|
| 60S ribosomal protein L23 | 15947825, 22965074, 33617159 | 34546362 |
| 60S ribosomal protein L10 | 14370507 | 31782364 |
| 60S ribosomal protein L34 | 22421026 | 3091507 |
| 40S ribosomal protein S3 | 7467697 | 14035049, 10389068 |
| Human keratin type I cytoskeletal 18 (cytokeratin 18) | 7462183 | 3110597, 4413946, 28405473 |
| Cytochrome c pseudogenes | 2527156 | 19093074, 20019122, 22937160 |
| Cytochrome P450 subfamily IID | 883308 | 25944997, 25954605 |
Comparisons of Alu and Ψg Sequences
Alu sequences are found predominantly near the 5′ UTR of genes rather than the 3′ UTR. This makes sense because Alus are G+C rich and CpG islands tend to be located near the 5′ end of genes (19). Actually, the gene structure annotation of Chr22 estimates 540 extant CpG islands of which 248 overlap the 5′ end of genes (4). It is thought that for Alu sequences to survive under transposition, they fare best by targeting CpG islands. In this environment, Alus gain CpG dinucleotides (20).
How are Alu and Ψg distributed in intergenic regions versus introns, and how many Alu and Ψg sequences overlap with gene exons? Explicitly, in Chr21 there are 14 (of 12,168) Alu sequences that overlap exons, of which only four overlap internal exons. Also, there are 20 Alu sequences within or containing exon sequences and only four of these contact internal exons. The corresponding Alu count in Chr22 is 30 (of 21,993) that overlap exon sequences, of which 28 overlap boundary exons (cf. ref. 21). Also, there are 54 Alu sequences totally contained within or enveloping exon sequences and 46 Alu sequences in contact with boundary (mostly untranslated) exons. In Chr22, the same analysis was applied to the protein CDSs. The results reveal only two Alu sequences, both overlapping boundary exons. Also, one short internal exon (136 bp) is completely contained within an Alu sequence. There are no Ψg sequences overlapping exon sequences in Chr21. In Chr22, there is a single Ψg that overlaps with an internal exon sequence and two Ψg are contained within boundary exon sequences. The Alu densities (counts/kb) in Chr21 for intergenic and intron regions are 0.33 and 0.47, respectively. In Chr22, the density numbers are 0.62 and 0.77, respectively, and in both Chr21 and Chr22 the Alu density is higher in introns than in intergenic regions. However, Ψg sequences prefer intergenic regions. Size of the sequence may be a decisive factor. The Ψg density values (counts/kb) are as follows: Chr21, 0.0018 (intergenic) and 0.0011 (intronic); Chr22, 0.0053 (intergenic) and 0.0028 (intronic). The foregoing data are organized in Tables 9 and 10.
Table 9.
Chr21 distribution of Ψg and Alu sequences in intergenic regions and introns
| Total | Intergenic | Intron | Overlapping with exons | |
|---|---|---|---|---|
| Ψg counts | 53 | 44 | 9 | 0 |
| Alu counts | 12,168 | 8,250 | 3,884 | 34 |
| Length of region, kb | 33,092 | 24,851 | 8,241 | |
| Marker density, count per kb | ||||
| Ψg | 0.0016 | 0.0018 | 0.0011 | |
| Alu | 0.3677 | 0.3321 | 0.4713 | |
There are 34 Alu sequences overlapping with exons. Eight of 34 overlap with internal exons and 26 overlap with boundary exons.
Table 10.
Chr22 distribution of Ψg and Alu sequences in intergenic regions and introns
| Total | Intergenic | Intron | Overlapping with exons | |
|---|---|---|---|---|
| Ψg counts | 145 | 109 | 33 | 3 (0) |
| Alu counts | 21,993 | 12,841 | 9,068 | 84 (3) |
| Length of region, kb | 32,369 | 20,611 | 11,758 | |
| Marker density, count per kb | ||||
| Ψg | 0.0045 | 0.0053 | 0.0028 | |
| Alu | 0.6794 | 0.6230 | 0.7712 | |
There are 84 Alu sequences overlapping with exons. Eleven of 84 overlap with internal exons and 73 overlap with boundary exons. There are only three Alu sequences overlapping with translated exons. Three Ψg overlap with terminal exons but no Ψg overlap with translated exons. The locations of the three overlapping pairs of gene and pseudogene are as follows: gene (novel gene): 26358694∼26379157, Ψg: 26378836∼26386771; gene (Homo sapiens cDNA): 15058600∼15105383, Ψg: (similar to H. sapiens angiotensin II receptor gene): 15103686∼15103899; and gene (tissue inhibitor of metalloproteinase 3, related to Sorsby fundus dystrophy): 16700083∼16761409, Ψg: 16758391–16758808.
What are the lengths of the different Ψg sequences? Of the 49 processed Ψg in Chr21, the mean length is 1,250 bp (940-bp median). The four Chr21 multiexon Ψg lengths consist of three two-exon constructs and one of three exons. Explicitly they have exon-(intron)-exon lengths of 278-(75)-461 bp; 122-(309)-570 bp; 185-(17)-110 bp; and a three-exon Ψg with lengths of 92-(68)-152-(1273)-104 bp. The small sizes of both exons and introns among the multiexon Ψg putatively reflect corrupted gene structures. It seems evident that most Ψg arise from processed multiexon genes. The mean length parallels that of single-exon genes. Chr22 contains 123 processed Ψg with an average length of 1,082 bp (median 744) roughly the same as in Chr21. The 22 multiexon Ψg of Chr22 have mean exon length of 182 bp (median 153), again strikingly small compared with the single-exon Ψg types. The mean exon number per multiexon Ψg is about five. The three longest Ψg have lengths of 19,168 bp, 16,318 bp, and 11,585 bp, and nine others have lengths in the range of 4 to 10 kb.
Distribution of Genes and Ψg Along the Chromosomes
Chr22 contains 26 Ψg in a 1.5-Mb region proximal to the
centromere (18). This is unusually high. Genomic heterogeneity occurs
broadly and on different scales. In probing the organization of a
genome, the general problem arises of how to characterize anomalies in
the spacings of markers in a long sequence of nucleotides or amino
acids. These include properties of clustering/clumping (too many
neighboring short spacings), overdispersion (too many long gaps between
markers), and excessive evenness (too few short spacings and/or too
few long gaps). Questions concerning the spacings in a marker array can
be approached by consideration of the cumulative lengths of
r consecutive distances along the marker array where
R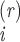 is the distance
(number of letters) between marker i and marker
i+r designated r-scan lengths (e.g.,
ref. 22). The spans of the longest and shortest r-scans are
useful statistics for detecting significant clumping, significant
overdispersion, or excessive regularity in the spacings of the marker.
The use of sums of r consecutive fragment lengths, rather
than single (r = 1) fragment lengths, can provide
sensitivity and better tolerate measurement errors.
is the distance
(number of letters) between marker i and marker
i+r designated r-scan lengths (e.g.,
ref. 22). The spans of the longest and shortest r-scans are
useful statistics for detecting significant clumping, significant
overdispersion, or excessive regularity in the spacings of the marker.
The use of sums of r consecutive fragment lengths, rather
than single (r = 1) fragment lengths, can provide
sensitivity and better tolerate measurement errors.
We apply the r-scan test for r = 5 under 0.95 significance to analyze the distributions of genes in Chr21 and Chr22. Clusters are identified from significantly small five-scan intervals, and the C+G contents are calculated by masking out those intervals. A similar scheme is applied to determine regions of significant overdispersion. Clusters occur in relatively high G+C regions and overdispersed regions occur in comparatively low G+C regions. Specifically, in Chr21, there are three clusters and one overdispersed region (Table 11).
Table 11.
Distribution of genes
| Location | Size, Mb | Gene no. | Fgc | |
|---|---|---|---|---|
| Chr21 | ||||
| Cluster | 20,351,850–20,590,675 | 0.24 | 8 | 0.4262 |
| 31,156,109–31,472,069 | 0.32 | 7 | 0.5549 | |
| 33,062,659–33,372,115 | 0.31 | 7 | 0.5160 | |
| Overdispersion | 1,997,831–12,535,699 | 10 | 21 | 0.3721 |
| Chr22 | ||||
| Cluster | 4,622,749–4,818,802 | 0.2 | 6 | 0.4837 |
| Overdispersion | 10,527,428–12,563,741 | 2 | 10 | 0.4430 |
| 16,334,074–19,246,348 | 2.9 | 10 | 0.4336 |
We also applied the r-scan test to the set of ribosomal protein Ψg in both Chr21 and Chr22. We found that the ribosomal Ψg are distributed quite randomly in Chr22. However, the distribution is not so random in Chr21. There is a region of 1 Mb (the expanse of 22,421,026–23,436,159 with an average G+C level of 0.44), which contains seven ribosomal protein Ψg (17 in the whole chromosome). For the Ψg distribution, in Chr21, there is a cluster in the 0.8-Mb interval (region of 22,673,718–23,436,157 with an average G+C level of 0.44) containing 11 Ψg; in Chr22, there is a cluster of seven Ψg in a 0.1-Mb stretch (region of 283,333–371,454 with an average G+C level of 0.42) and another seven Ψg, including five successive Ig κ variable Ψg, clustered between positions 1282766 and 1359121 with an average G+C of 0.41. An interesting observation from the three Ψg clusters is that the orientations of these Ψg are significantly nonrandom. For example, the 11 Ψg in Chr21 are all on the positive (reported) strand except for the first Ψg. In Chr22, the seven Ψg of the first cluster are also all on the positive strand and the seven Ψg in the second cluster are all on the minus strand except for the first Ψg.
Concluding Comments
The median size and distribution of processed Ψg are about the same as the length of single-exon genes. Also, the median range of single-exon genes is remarkably similar to the average internal exon length times the average number of exons per gene. These properties support the hypothesis that most single-exon genes derive from processed multiexon genes in dynamic regions. An analysis of Chr22 reveals that at least 25% of gene structures possess 5′ and 3′ UTEs. Many of these UTEs may have an important role in alternative splicing, as is the case with G protein-coupled receptor membrane proteins (13). The larger length for the 5′ extension region suggests that 5′ regulatory regions are more extensive than 3′ regulatory regions. The intergenic length of convergent orientation is also longer than the intergenic length of divergent orientations. Ψg appear to derive predominantly from highly expressed genes, especially ribosomal protein genes and cytochrome c proteins. The largest exons and introns are foremostly the first or last exon or intron. The counts of genes are significantly correlated with G+C chromosomal content. As expected, in the presence of increased transcription activity, there are more genes, Alu sequences, and Ψg numbers (cf. ref. 23).
Acknowledgments
We are grateful to Drs. E. Zuckerkandel, A. M. Campbell, B. E. Blaisdell, U. Francke, and D. Petrov for helpful discussions regarding this manuscript. This work was supported in part by National Institutes of Health Grants 5R01GM10452-36 and 5R01HG00335-14.
Abbreviations
- Chr21
chromosome 21
- Chr22
chromosome 22
- UTR
untranslated region
- CDS
coding sequence structure
- Ψg
pseudogenes
- Fgc
G+C frequency
- UTE
untranslated exon
Footnotes
We assume that for genes where the coding sequence annotation agrees exactly with the complete gene structure annotation, no UTEs are present. The main results are unchanged even if this is not always correct; they would then represent lower bounds on the occurrence of UTEs.
References
- 1.International Human Genome Sequencing Consortium. Nature (London) 2001;409:860–921. [Google Scholar]
- 2.Venter J C, Adams M D, Myers E W, Li P W, Mural R J, Sutton G G, Smith H O, Yandell M, Evans C A, Holt R A, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Dunham I, Shimizu N, Roe B A, Chissoe S, Hunt A R, Collins J E, Bruskiewich R, Beare D M, Clamp M, Smink L J. Nature (London) 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- 4.Hattori M, Fujiyama A, Taylor T D, Watanabe H, Yada T, Park H S, Toyoda A, Ishii K, Totoki Y, Choi D K. Nature (London) 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 5.Reymond A, Friedli M, Henrichsen C N, Chapot F, Deutsch S, Ucla C, Rossier C, Lyle R, Guipponi M, Antonarakis S E. Genomics. 2001;78:46–54. doi: 10.1006/geno.2001.6640. [DOI] [PubMed] [Google Scholar]
- 6.Antonarakis S E. Curr Opin Genet Dev. 2001;11:241–246. doi: 10.1016/s0959-437x(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 7.Gentles A J, Karlin S. Genome Res. 2001;11:540–546. doi: 10.1101/gr.163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huo L, Scarpulla R C. Gene. 1999;11:213–224. doi: 10.1016/s0378-1119(99)00135-3. [DOI] [PubMed] [Google Scholar]
- 9.Macdonald P M, Kerr K. Mol Cell Biol. 1998;18:3788–3795. doi: 10.1128/mcb.18.7.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancebo R, Zhou X L, Shillinglaw W, Henzel W, Macdonald P M. Mol Cell Biol. 2001;21:3462–3471. doi: 10.1128/MCB.21.10.3462-3471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez A J. Annu Rev Genet. 1998;32:279–305. doi: 10.1146/annurev.genet.32.1.279. [DOI] [PubMed] [Google Scholar]
- 12.Gentles A J, Karlin S. Trends Genet. 1999;15:47–49. doi: 10.1016/s0168-9525(98)01648-5. [DOI] [PubMed] [Google Scholar]
- 13.Sosinsky A, Glusman G, Lancet D. Genomics. 2000;70:49–61. doi: 10.1006/geno.2000.6363. [DOI] [PubMed] [Google Scholar]
- 14.Donofrio G, Jabbari K, Musto H, Alvarez-Valin F, Cruveiller S, Bernardi G. Ann NY Acad Sci. 1999;870:81–94. doi: 10.1111/j.1749-6632.1999.tb08867.x. [DOI] [PubMed] [Google Scholar]
- 15.Jurka J. Curr Opin Struct Biol. 1998;8:333–337. doi: 10.1016/s0959-440x(98)80067-5. [DOI] [PubMed] [Google Scholar]
- 16.Duret L, Mouchiroud D, Gautier C. J Mol Evol. 1995;40:308–317. doi: 10.1007/BF00163235. [DOI] [PubMed] [Google Scholar]
- 17.Vanin E F. Annu Rev Genet. 1985;19:253–272. doi: 10.1146/annurev.ge.19.120185.001345. [DOI] [PubMed] [Google Scholar]
- 18.Harrison P M, Hegyi H, Bertone P, Echols N, Johnson T, Balasubramanian S, Luscombe N, Gerstein M. Genome Res. 2002;12:273–281. doi: 10.1101/gr.207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross S H, Bird A P. Curr Opin Genet Dev. 1995;5:309–314. doi: 10.1016/0959-437x(95)80044-1. [DOI] [PubMed] [Google Scholar]
- 20.Jurka J, Milosavljevic A. J Mol Evol. 1991;32:105–121. doi: 10.1007/BF02515383. [DOI] [PubMed] [Google Scholar]
- 21.Batzer M A, Arcot S S, Phinney J W, Alegria-Hartman M, Kass D H, Milligan S M, Kimpton C, Gill P, Hochmeister M, Ioannou P A. J Mol Evol. 1996;42:22–29. doi: 10.1007/BF00163207. [DOI] [PubMed] [Google Scholar]
- 22.Karlin S, Brendel V. Science. 1992;257:39–49. doi: 10.1126/science.1621093. [DOI] [PubMed] [Google Scholar]
- 23.Zhang M Q. Hum Mol Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]