

Abstract
UV radiation is the most important environmental skin aggressor, causing cancer and other problems. This paper reports the use of oligonucleotide microarray technology to determine changes in gene expression in human keratinocytes after UVB treatment. Examination of the effects of different doses at different times after irradiation gave a global picture of the keratinocyte response to this type of insult. Five hundred thirty-nine regulated transcripts were found and organized into nine different clusters depending on behavior patterns. Classification of these genes into 23 functional categories revealed that several biological processes are globally affected by UVB. In addition to confirming a majority up-regulation of the transcripts related to the UV-specific inflammatory and stress responses, significant increases were seen in the expression of genes involved in basal transcription, splicing, and translation as well as in the proteasome-mediated degradation category. On the other hand, those transcripts belonging to the metabolism and adhesion categories were strongly downregulated. These results demonstrate the complexity of the transcriptional profile of the UVB response, describe several cellular processes previously not known to be affected by UV irradiation, and serve as a basis for the global characterization of UV-regulated genes and pathways.
The epidermis is a physiological barrier that protects the organism against pathogens and chemical or physical damage. UV radiation is the most important physical carcinogen in the environment, and the skin is its main target. In this tissue, UV induces photochemical changes that may lead to acute effects such as erythema or sunburn (1), or chronic effects that include premature skin aging (2, 3) and skin tumors (4, 5). In recent years, the importance of UV radiation has increased because of the thinning of the ozone layer (6) and increasing skin cancer rates (7). UV light affects the skin in different ways depending on its wavelength. UVA (320–400 nm) effects are primarily oxidative in nature. UVC (200–290 nm) hardly ever reaches the surface of the earth. UVB (290–320 nm) is the most important and is considered the causative agent of many of the effects attributed to UV (8), giving rise to mutations in DNA and modifying the pattern of gene expression. This transcriptional regulation is part of the cellular reaction to UV-induced stress and operates as a defense mechanism. The known UV response comprises, for instance, the rapid activation of genes with reparative, protective, or apoptotic functions and the communication of stress to other cells via secreted signaling molecules (9).
Although a number of UV-regulated genes and processes have been described (see refs. 9 and 10 for review), many of the molecular events involved in the UV response remain unknown. However, many studies on UV lack physiological relevance because they were performed by using UVC and established epithelial cell lines or fibroblasts. Habitually exposed to UV radiation, keratinocytes are more resistant to this insult than are fibroblasts (11), which probably means they have developed a specialized response.
To understand the mechanisms of UV response, we sought to obtain the transcriptional profile of primary keratinocytes after UVB irradiation. Cultured human epidermal keratinocytes were treated with three UV doses, and samples were collected at different intervals. Oligonucleotide microarrays containing over 6,000 genes (HuGeneFL, Affymetrix, Santa Clara, CA) were used to quantitatively assess changes in gene expression. The subset of regulated transcripts was classified into self-organizing maps according to their different behaviors. Biological functions were assigned to these genes, which were then correlated with expression patterns. These approaches provide new insights into the genes and biological processes involved in the UV response and demonstrate the complexity of the UVB transcriptional profile in keratinocytes, allowing global characterization of UV-regulated genes and pathways.
Materials and Methods
Cell Culture.
Primary human keratinocytes from foreskin were cultured on a fibroblast feeder layer (50-Gy-irradiated 3T3 J2 cells) as described by Rheinwald and Green (12). Cells were grown to 80% confluence in 100-cm2 plates in 3:1 DMEM/Ham's-F12 medium (GIBCO/BRL Life Technologies) supplemented with 10% FBS (BioWhittaker)/5 mg/ml of insulin (Sigma)/0.1 nM cholera toxin (Sigma)/10 ng/ml of epidermal growth factor (Serono, Geneva)/0.5 mg/ml of hydrocortisone (Sigma)/1% antibiotics (GIBCO/BRL Life Technologies).
Irradiation.
Fresh culture medium was supplied 24 h before treatment. For irradiation, the medium was removed and the keratinocytes treated with 10, 20, or 40 mJ/cm2 UVB. The collected medium was then replaced and the cells maintained in culture for 4 or 24 h. A bank of six filtered UVB Sankyo 615T8E 15-W lamps, which emit 80% of the energy in the UVB range as measured by a UVX radiometer, were used as the radiation source.
RNA Extraction and Preparation for Hybridization.
Total RNA was isolated by using TRIzol (GIBCO/BRL Life Technologies). RNA was amplified and labeled with biotin as described by Wodicka et al. (13).
Array Hybridization and Scanning.
Samples for oligonucleotide microarray hybridization were prepared as described by Lockhart et al. (14), except that hybridization was performed in 1× Mes buffer (0.1 M Mes, pH 6.7/1 M NaCl/0.01% Triton X-100), and the chips were washed in 0.1× Mes buffer. The target concentration was 15 μg of adjusted fragmented cRNA in 300 μm of hybridization solution. Samples were hybridized to Affymetrix GeneChip HuGeneFL arrays. Images were scanned at 3-μm resolution by using a GeneArray Scanner made for Affymetrix by Hewlett–Packard.
Analysis.
All samples were prepared and hybridized in duplicate. genechip analysis suite software (Affymetrix) was used to analyze image data. Values representing genes not expressed at a given experimental point are unreliable and are referred to as “absent call” entries. To gain a more complete picture, those transcripts that showed at least one “present call” in one experimental point in both duplicates were included. When required, the expression levels provided by the “absent call” entries were modified to up- or down-regulation values no higher than 4-fold. The results of the whole experiment are published as supporting information on the PNAS web site (www.pnas.org). To classify the temporal profiles of gene expression, cluster analysis was performed by using the gene cluster 1.1 program [http://www.genome.wi.mit.edu/MPR/GeneCluster (15)]. Genes that, after irradiation, showed differences in their expression levels of at least a factor of 2.0 in at least one experimental point in both duplicates were selected for clustering. Biological functions were assigned by using the genecards database [http://bioinfo.weizmann.ac.il/cards (16)].
Cloning of Probes for Northern Blots.
Primers were designed by using GCG's Primer tool and NetPrimer (http://www.premierbiosoft.com/netprimer). Primer sequences can be provided on request. Total RNA was prepared from cultured keratinocytes by using TRIzol (GIBCO/BRL Life Technologies). Single-stranded cDNA was synthesized by using SuperScriptII RNase H Reverse Transcriptase (GIBCO/BRL Life Technologies). Probes were amplified by standard PCR by using Taq polymerase (Promega). PCR fragments were cloned into the pGEM-T Easy Vector (Promega) and the identity of the PCR fragments verified by sequencing.
Northern Blotting.
Twenty micrograms of each RNA sample were electrophoresed through 1.1% formaldehyde-agarose gels and transferred to Hybond-N nylon membranes (Amersham Pharmacia Biotech). Hybridization was performed in 50% formamide/1% SDS/5 × SSC/10 mM Tris, pH 7.5/10% dextran sulfate/100 mg/ml of salmon sperm. Probes were 32P-dCTP-labeled with Rediprime II (Amersham Pharmacia Biotech) and Northern blots analyzed and quantified by phosphorimaging by using quantity one 4.1.1 software (Bio-Rad).
Results and Discussion
Experimental System.
Primary human epidermal keratinocytes were grown to subconfluence using culture conditions that maintain many of the characteristics these cells show in vivo (12). The UVB irradiation doses applied (10, 20, and 40 mJ/cm2) represent low, middle, and high exposures to UV considering that, in our experimental system, few changes in expression are seen at 10 mJ/cm2, and nondesired high apoptotic rates are observed only at doses over 40 mJ/cm2 (data not shown). Irradiation with 40 mJ/cm2 caused undetectable apoptosis at 24 h and less than 4% of apoptotic cells at 48 h, as estimated by cell viability assays and analysis of DNA content by flow cytometry (data not shown). These conditions approach the situation in skin during moderate to intense exposure to the sun. Samples were collected at 4 and 24 h after irradiation, time points for early- and late-regulated genes. The final analysis therefore comprises the effects of both low and high doses in the short and long term.
Microarray experiment results were represented in log/log scatter plots to check the consistency of the data. Fig. 1 shows the expression pattern of untreated keratinocytes, on the horizontal axis, against its duplicate (Fig. 1A), a control sample from a different keratinocyte culture (Fig. 1B), and a sample of keratinocytes collected 24 h after irradiation with 40 mJ/cm2 (Fig. 1C), on the vertical axis. The borderline for discriminating differential expression was set at ±2-fold. The comparison of the two duplicate samples (Fig. 1A), with an almost perfectly matched scatter plot, demonstrates the reproducibility of the experimental approach. The representation of untreated vs. irradiated cells (Fig. 1C) displays a remarkable number of regulated genes that fall outside the borderlines. The comparison of untreated samples from different cultures (Fig. 1B) shows a reduced number of data points outside the ±2-fold lines, which probably indicates the existence of variability associated with the culture. These analyses illustrate the reproducibility of the method and demonstrate the suitability of this approach for studying the UVB keratinocyte response.
Figure 1.

Analysis of the experimental variability of the method. The data are represented as log/log scatter plots of the expression values provided by genechip software. For each graph, only those transcripts present in both samples are considered. The upper and lower boundaries represent a 2-fold change in expression. (A) Control 1 vs. Control 1′. Technical variability is provided by the comparison of the expression levels of two different chip analyses performed with the same sample of untreated keratinocytes. The vast majority of the points fall within the ±2-fold limit. (B) Control 1 vs. Control 2. Biological variability is provided by the comparison of two different control samples. A slight increase in the number of data points outside the borders indicates changes in the patterns of gene expression attributed to culture-specific characteristics. (C) Control 1 vs. 40 mJ/cm2 UVB, 24 h. Changes in gene expression because of the UVB response are represented. Up- and down-regulated genes are represented by the points located outside the ±2-fold lines, although most of the transcripts fall within these limits.
To identify UVB-regulated genes, the expression levels of each irradiated sample were compared with those of control keratinocytes. Transcripts that showed at least a 2-fold change and a “present call” in at least one experimental point in both duplicates were selected. On the basis of these criteria, 539 regulated transcripts were identified (see supporting information, which is published on the PNAS web site), accounting for approximately 10% of the genes represented in the HuGeneFL Array.
Transcriptional Profiling of the UV Response in Keratinocytes.
The genecluster 1.1 (http://www.genome.wi.mit.edu/MPR/GeneCluster) program, based on self-organizing map algorithms, was used to organize the regulated genes into different expression patterns. Nine clusters were obtained, representing specific patterns of regulation (Fig. 2). Roughly, clusters 0–4 correspond to five different kinetics of gene activation, whereas clusters 5–8 show kinetics of repression. Cluster 0 (117 transcripts) includes genes with a specific late response to high doses; they are induced only at 40 mJ/cm2 and 24 h. An example of this group is Bag-1, a potent inhibitor of apoptosis and Fas-induced cell death (17). Cluster 1 (79 transcripts) comprises genes that respond to UV radiation at 24 h in a dose-dependent manner; a good example is SprpII, a constituent of the cornified envelope that appears during epidermal differentiation, previously shown to be induced by UV (18). Cluster 2 (57 transcripts) includes genes slightly repressed at 4 h and induced at 24 h in a dose-independent manner, for example, cyclin G1, a DNA damage-induced transcript (19). Cluster 3 (24 transcripts) is the only one that shows a generalized pattern of induction maintained from 4 to 24 h in a dose-dependent manner. Some well-known UV-responding genes, such as IL-8, IEX1, and COX2, belong to this cluster. In cluster 4, 20 interesting transcripts are present that do not change at 4 h but are specifically induced at 24 h with a dose of 20 mJ/cm2.
Figure 2.

Cluster analysis of gene expression after UV irradiation. Five hundred thirty-nine genes that changed their expression in at least one experimental point after UV irradiation were grouped into nine clusters with different kinetics. The number of genes in each cluster is indicated. Each cluster is represented by the centroid (average pattern) for genes in the cluster. For each graph, the x axis represents time after irradiation (0, 4, and 24 h) and the y axis the normalized factor of induction or repression. Doses: ◊, 10 mJ/cm2; □, 20 mJ/cm2; Δ, 40 mJ/cm2.
Clusters 5–8 represent kinetics of repression by UV. Cluster 5 (49 transcripts) includes genes (e.g., integrin β4) down-regulated at 4 h in a dose-independent manner, which return to basal levels at 24 h, except for 40 mJ/cm2. In cluster 6 (42 transcripts), genes suffer up-regulation at 4 h and down-regulation at 24 h when 40 mJ/cm2 are provided; examples of this group are junD and gadd45. Genes in cluster 7 (74 transcripts) are repressed at 24 h in a dose-dependent manner (e.g., PEPCK). Cluster 8 (77 transcripts) represents genes whose expression is decreased at 4 h but, in contrast to cluster 5, continue to be down-regulated at 24 h in a dose-dependent way. Thrombospondin 2 is a good example of this group.
Of the set of 539 regulated genes, only 249 transcripts changed their expression 4 h after UV irradiation: 183 were repressed and 66 induced. However, the 539 were regulated at 24 h, and the number of induced and repressed genes was similar at this time (297 and 242, respectively). These data suggest that the early UV response (4 h) leads mainly to transcriptional arrest with the induction of a small number of genes (those included in cluster 3), whereas the late response (24 h) is more complex, involving a higher number of genes up- and down-regulated in similar proportions.
Northern blots were performed to confirm microarray results. Some examples are shown in Fig. 3, which presents genes from six of the nine clusters. The results for Bag-1, cyclin G1, IL-8, PEPCK, thrombospondin 2, and sprp2 followed the patterns obtained with the microarrays, whereas APR showed slight deviations at 24 h. Northern blots confirm the experimental approach but also indicate that the degree of change in gene expression calculated by the genechip software is clearly underestimated, in agreement with other reports (20).
Figure 3.
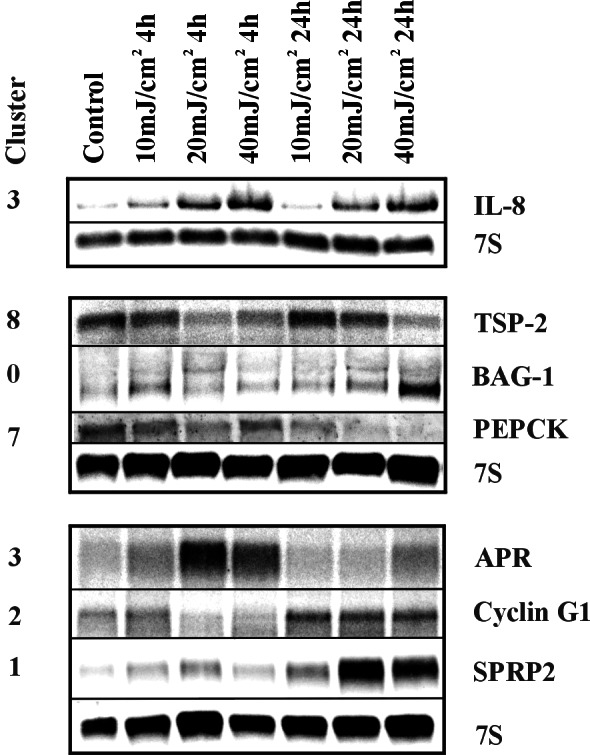
Confirmation of microarray results by Northern blot analysis. RNAs were prepared from keratinocytes treated with the indicated doses either 4 or 24 h after exposure to UVB. Representative Northern blots for genes with altered expression after UVB irradiation are shown. 7S RNA was used as a loading control. The molecular weights of the hybridizing bands agreed with the reported sizes of the corresponding transcripts.
Groups of Genes Regulated by UVB According to Their Biological Role.
The different behaviors revealed by clustering analysis reflect the complexity of the UV response and should help elucidate the intricate processes that take place in UV-irradiated cells. To this end, the regulated transcripts were classified according to biological function. Because of the relatively small size of the sample, it was not convenient to use a very detailed classification of gene function [such as that at reported for yeast by the MIPS (http://mips.gsf.de/proj/yeast/catalogues/funcat/)]. Therefore, we established a list of 23 different categories (or functional groups) to sort the 539 transcripts according to their biological roles as defined by the genecards database (16) (Table 1). Some genes may have several functions, so we have chosen the function for which each gene is best known. In some cases, transcripts in the same category may have opposite roles (for instance, activators and inhibitors).
Table 1.
Functional classification of UV-regulated genes
| Function | c0 | c1 | c2 | c3 | c4 | c5 | c6 | c7 | c8 | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| Adhesion | 2 | 3 | 3 | 1 | 2 | 6 | 7 | 13 | 37 | |
| Angiogenesis | 2 | 3 | 5 | |||||||
| Apoptosis and survival | 1 | 4 | 1 | 2 | 1 | 1 | 3 | 13 | ||
| Cell cycle | 1 | 2 | 3 | 1 | 1 | 3 | 2 | 13 | ||
| Cell growth | 2 | 1 | 2 | 2 | 7 | |||||
| Development | 3 | 1 | 2 | 1 | 3 | 10 | ||||
| Homeostasis | 4 | 3 | 1 | 8 | ||||||
| Inflammatory response | 6 | 3 | 1 | 5 | 1 | 2 | 18 | |||
| Intracellular signaling | 22 | 10 | 9 | 5 | 3 | 13 | 6 | 8 | 11 | 87 |
| Lysosomal enzymes | 2 | 1 | 1 | 1 | 2 | 1 | 8 | |||
| Metabolism | 4 | 2 | 4 | 0 | 2 | 10 | 19 | 10 | 51 | |
| Proteases | 2 | 2 | 1 | 2 | 1 | 4 | 12 | |||
| Protease inhibitors | 2 | 2 | 1 | 2 | 7 | |||||
| Proteasome | 6 | 8 | 4 | 1 | 1 | 1 | 21 | |||
| Replication | 1 | 2 | 1 | 1 | 2 | 2 | 9 | |||
| Respiratory chain | 2 | 1 | 1 | 1 | 5 | |||||
| Splicing | 3 | 2 | 3 | 3 | 1 | 1 | 13 | |||
| Stress response | 7 | 5 | 2 | 2 | 1 | 2 | 4 | 2 | 25 | |
| Structural proteins | 5 | 12 | 5 | 3 | 1 | 2 | 4 | 5 | 5 | 42 |
| Transcription | 21 | 3 | 8 | 2 | 1 | 13 | 4 | 4 | 4 | 60 |
| Translation | 6 | 4 | 1 | 1 | 1 | 1 | 2 | 1 | 17 | |
| Transport | 8 | 7 | 2 | 2 | 4 | 1 | 4 | 5 | 33 | |
| Others | 9 | 5 | 6 | 1 | 1 | 5 | 2 | 3 | 6 | 38 |
Columns c0–c8 indicate the number of genes of each cluster in the functional group. Functions in bold type are globally up- or down-regulated.
Correlations were sought between the genes in functional groups and their expression patterns (activation/repression). In many categories, correlations were not evident, perhaps in part because of the complexity of the categories, as shown by the transcription and intracellular signaling groups. However, there were eight functional categories (marked in bold type in Table 1) in which more than two-thirds of the components behaved in a similar manner, indicating the existence of biological processes globally induced or repressed by UV. A generalized down-regulation, shared by at least 75% of the components, was observed in the cell-adhesion and metabolism categories. On the other hand, global induction was seen in the stress response, structural proteins, inflammatory response, mRNA splicing, proteasome-related proteins, and translation categories.
Genes Involved in Stress Response, Inflammation, and Apoptosis.
Genotoxic stress, such as that caused by UV irradiation, is known to increase the transcription of certain genes. Among our data, transcripts involved in the oxidative stress response, such as manganese superoxide dismutase [cluster 1 (c1)] and some heat-shock proteins (c0 and c1), are up-regulated after UVB treatment, as described by others (reviewed in ref. 21). Up-regulation of genes related to DNA damage, such as IEX-1 (c3), XPC-complementing proteins (c0 and c4), and Gadd45 (c6) was also apparent. It is also well known that UV light produces an inflammatory reaction in the skin in which inflammation-related genes are induced (reviewed in ref. 22). In agreement, our data show that 83% of the altered genes in the inflammatory response category are up-regulated. These include cytokines such as IL-8 (c3), melanoma growth stimulatory activity (c0), and the metabolic enzyme COX2 (c3), previously shown to be induced by UV (23, 24). The pathway of IL-1 is particularly activated, with increases in the transcription of several forms of the cytokine and receptor, as well as TRAF6 (c0), a signal transducer in the NFκB pathway, which activates IκB kinase in response to the proinflammatory cytokines IL-1 (25) and the IL-1 adapter protein MyD88 (c3) (26). Other molecules, such as major histocompatibility complex class I E (c0), are also up-regulated.
Depending on the dose, UVB radiation can drive cells to apoptosis. As previously mentioned, these experiments were performed by using physiological doses of between 10 and 40 mJ/cm2—and even at the latter, apoptotic levels were not significant. When genes involved in apoptosis and survival are analyzed, a complex pattern appears. In control keratinocytes, apoptosis/survival transcripts in the array are called absent or are expressed only at very low levels. After UV irradiation, the expression of some of the most important genes related to keratinocyte apoptosis [tumor necrosis factor (TNF)α, FasL, and bak, for instance] remained unaltered (note that genes like Bax, bad, and bid are not represented in the chip). However, several other transcripts, some previously undescribed in keratinocytes, underwent changes in expression. At the short interval (4 h), two proapoptotic genes were induced in a dose-dependent manner: IPL (c3), also known as TSSC3, whose murine homologue TDAG51 is implicated in FasL-mediated apoptosis (27), and APR (c3), the ortholog of the murine p53-dependent proapoptotic gene Noxa (28). In addition, three of the four genes repressed at 4 h are antiapoptotic [semaphorin E (c8), the FAS-associated protein tyrosine phosphatase 1 (c8), and MCL1 (c5)], whereas only one [TRAIL (c8)] is proapoptotic. At 24 h after irradiation, however, there was an induction of both proapoptotic [BNIP3 (c1), TIAR (c2), and TNFR (c4)] and antiapoptotic [PEA15 (c1), NAIP (c1), TNFAIP3 (c1), and BAG-1 (c0)] transcripts. Although the functions of many of these genes are still insufficiently clear to develop a model, it seems that after UV irradiation, there could be an initial shift toward apoptosis (five of the six gene changes at 4 h favor apoptosis), which stabilizes at 24 h when the expression of proapoptotic and antiapoptotic genes is balanced. Because no significant apoptosis was observed in the cultures, the “proapoptotic” response at 4 h could represent preparation of the cells to undergo apoptosis in the case of severe cell damage.
Structural Proteins.
Forty-two genes coding for structural proteins changed their expression in response to UV, and most of them were induced. Transcripts in this category can be subdivided into three groups: cytoskeletal, nuclear-related, and keratinocyte-specific genes. The group of transcripts coding for cytoskeletal proteins showed no common behavior. However, 9 of the 13 genes coding for nuclear structural proteins were up-regulated, including several histones [H2A (c3), H2B (c3), and H3 (c0) isoforms]. Interestingly, the induction of genes involved in transcriptional repression by histone deacetylation was also observed, as were those of histone deacetylase HDAC1/RPD3 (c1) and its associated proteins SAP18 (c0), involved in transcriptional repression via the tethering of the sin3 complex to core histone proteins (29), and the GLI-Kruppel-related protein YY1 (c2) (these last two factors are included in the “transcription” category). It has been proposed that the complex HDAC1/sin3 is recruited to the promoters of specific genes by factors like YY1, where it deacetylates histones in a targeted way, leading to local changes in chromatin structure and specific inactivation of gene expression (30, 31). SMRT (c5), a corepressor involved in this mechanism of inhibition for retinoid and thyroid-hormone receptors genes (32, 33), is repressed at 4 h and recovered at 24 h. Although currently it is not known which genes are silenced by these factors (except in the case of SMRT), it is conceivable that this mechanism could be responsible for at least part of the transcriptional inhibition caused by UV.
There are also keratinocyte-specific proteins in this group. A generalized induction of transcripts involved in the formation of the cornified envelope was found. These up-regulated genes include keratinocyte transglutaminase (c1), its substrates sprpII (c1) and involucrin (c4), as well as genes from other categories also involved in this process, such as PAI-2 (c1), annexin (c2) (34), and cystatin M (c1) (35). Only the up-regulation of sprpII has been previously reported (17). Although the genes described above code for proteins characteristic of suprabasal, differentiated skin keratinocytes and were also expressed in control cells (in agreement with the observation of sporadic foci of differentiation), the cultures showed a strong prevalence of keratin K5 and K14 mRNAs (the most abundant transcripts according to the microarray data and characteristic of proliferative keratinocytes) over keratins K1 and K10 (typical of differentiated cells). Other components of the late steps of differentiation (for instance, filagrin and loricrin) were not detected (data not shown). UV did not affect other keratins or keratinocyte-specific cytoskeletal genes except for the induction of K18 (c1), present at very low levels in nonirradiated cultures (not shown).
Proteasome Degradation.
Genes involved in the proteasome-ubiquitin degradation pathway form another important group. Nineteen of the 21 transcripts in this category were up-regulated only at 24 h. This generalized behavior indicates that irradiated cells increase the turnover of many proteins, which could be due either to the replacement of damaged molecules or to the need to significantly change the proteome of the cell. Only two genes were down-regulated in this category, one of which [cyclin-selective ubiquitin carrier protein (c7)] is involved in the destruction of mitotic cyclins in the exit from mitosis into G1. This transcript was down-regulated at 24 h, and its repression may be required for cell cycle arrest, because a dominant negative form of this protein prevents entry into the cell cycle (36).
Transcription, mRNA Splicing, and Translation.
Transcription is a complex category that includes many genes and has no evident bias toward activation or repression. However, there was a generalized up-regulation of basal transcriptional machinery, which included different subunits of RNA Pol II such as TFIIA (c0), TFIIB (c0), TAFII32 (c0), and TAFII55 (c0), as well as up-regulation of genes involved in mRNA elongation. Ten of the 11 transcripts of this type were induced, most belonging to cluster 0 (induction at 24 h, 40 mJ/cm2). In addition, many transcriptional activators [e.g., HIF-1α (c2), AP-2 (c2), p65 (c0)] and repressors [e.g., YY1 (c2), SAP18 (c0), ATF3 (c3)] were increased with different kinetics. Some of these transcriptional regulators are highly relevant in keratinocyte biology, and their regulation by UVB may affect different cellular functions, e.g., AP-2 (c2) a transcription factor proposed to be involved in epidermal specificity (37). AP-2-binding sites are often found in epidermal promoters, particularly in cytoskeletal genes (ref. 37 and refs. therein; refs. 38, 39). However, AP-2-responsive genes are not up-regulated in our experiments, in agreement with the described requirement for cooperation of AP-2 with other transcription factors (37).
mRNA splicing-related transcripts underwent generalized induction (11 of 13), as did those involved in protein translation (13 of 17). Nine of these latter genes are related to the initiation phase of translation, and eight were up-regulated. Moreover, the only down-regulated gene in this group was the INF-inducible RNA-dependent protein kinase PKR (c5), an inhibitor of protein synthesis. In addition, four genes related to posttranslational modifications were also up-regulated. The generalized increase of transcripts involved in translation further confirms that the experimental conditions do not induce significant levels of apoptosis, because this process rapidly inhibits the rate of protein synthesis at the level of polypeptide chain initiation (40).
Metabolism.
More than 75% of the genes in this category were down-regulated, especially those related to glucidic, lipidic, and amino acidic metabolism. This was the case with key enzymes such as hexokinase 1 (c8), phosphoenolpyruvate carboxykinase (c7), and isocitrate and succinate dehydrogenases (c7 and c8, respectively), which are involved in glucose metabolism; fatty acid and long chain acyl-CoA synthetases (c8 and c7, respectively), which are involved in fatty acid metabolism; and several amino acid synthetases. However, some important transcripts in this group were up-regulated, such as glycogen synthase kinase 3α (c0) and glycogenin (c1), which take part in glycogen metabolism. This complex pattern of gene regulation might reflect the alteration of energetic requirements and metabolic pathways that cells undergo to cope with the effects of UVB irradiation, in particular an inhibition of glycolysis and an increase in gluconeogenesis.
Adhesion.
The adhesion category was also clearly affected by UVB radiation. Remarkably, 75% of changes were down-regulations. Analysis of the altered genes strongly suggests a decrease in the adhesive properties of irradiated keratinocytes. For instance, hemidesmosomal [e.g., integrins α6 (c7) and β4 (c5), BPAG1 (c8) and BPAG2 (c8)], desmosomal [e.g., desmoplakin I (c8) and desmocollin I (c8)], gap junctional [e.g., connexin 26 (c8)], nonhemidesmosomal integrins [e.g., α3 (c8), β1 (c7) and β5 (6)], and other cell-to-matrix attachment-related components were down-regulated. To our knowledge, this is the first study reporting global repression of these molecules after UVB irradiation.
In summary, these results describe and help explain the processes that occur during the UV response. The expression patterns obtained in the cluster analysis are suggestive of a wide and dose-independent transcriptional arrest during the early UV response (4 h), plus the induction of a concrete set of genes (c3) involved in the activation of well-known UV-response pathways (inflammation, stress response, etc.) At 24 h, the transcriptional arrest is overcome, and more complex transcriptional patterns are found. Besides confirming the induction of genes related to the UV-specific inflammatory and stress responses, the regulation of several processes mainly affected at 24 h postirradiation was revealed. Analysis of functional categories suggests that keratinocytes undergo an activation of cornified envelope synthesis (induction of keratinocyte-specific structural proteins), changes in their energetic requirements (repression of glycolysis and other catabolic pathways and induction of gluconeogenesis), and a generalized decrease in their adhesive properties. At 40 mJ/cm2, the data reflect a targeted elimination of proteins because of the need to either remove UV-damaged proteins or change the proteome of the cells, as well as an increase in general mechanisms of transcription and translation, probably to obtain new molecules involved in the UV response or in the replacement of UV-damaged proteins. These results unveil the complexity of the UVB response in keratinocytes and serve as a basis for undertaking a global characterization of UV-regulated genes and pathways.
Supplementary Material
Acknowledgments
We thank Dr. C. Johnson (Pfizer) for help with the microarray experiments and Drs. M. Del Río, P. Perez, and F. Larcher (CIEMAT) for assisting with keratinocyte cultures. The technical assistance provided by P. Hernandez is acknowledged. This project was supported by Grant SAF98–0047 from the Spanish Comision Interministerial de Ciencia y Tecnologia (Ministry for Science and Technology).
Abbreviation
- cn
cluster n
Note Added in Proof.
Recently, Li et al. (41) also reported the transcriptional program of human epidermal keratinocytes in response to UV.
References
- 1.Young A R. Photodermatology. 1987;4:127–134. [Google Scholar]
- 2.Gilchrest B A. In: Skin and Ageing Processes. Gilchrest B A, editor. Boca Raton, FL: CRC; 1989. pp. 97–116. [Google Scholar]
- 3.Fisher G J, Datta S C, Talwar H S, Wang Z Q, Varani J, Kang S, Voorhees J J. Nature (London) 1996;379:335–339. doi: 10.1038/379335a0. [DOI] [PubMed] [Google Scholar]
- 4.Lee J A, Frederick J E, Haywood E K, Stevens R G. Med J Aust. 1989;150:540. doi: 10.5694/j.1326-5377.1989.tb136672.x. [DOI] [PubMed] [Google Scholar]
- 5.Rogers G S, Gilchrest B A. Br J Dermatol. 1990;122 Suppl. 35:55–60. doi: 10.1111/j.1365-2133.1990.tb16126.x. [DOI] [PubMed] [Google Scholar]
- 6.Young A R. Br J Clin Pract Suppl. 1997;89:10–15. [PubMed] [Google Scholar]
- 7.Green A, Whiteman D, Frost C, Battistutta D. J Epidemiol. 1999;9:S7–S13. doi: 10.2188/jea.9.6sup_7. [DOI] [PubMed] [Google Scholar]
- 8.International Agency for Research on Cancer. Solar and Ultraviolet Radiation. Vol. 55. Lyon, France: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, IARC; 1992. [PMC free article] [PubMed] [Google Scholar]
- 9.Bender K, Blattner C, Knebel A, Iordanov M, Herrlich P, Rahmsdorf H J. J Photochem Photobiol B. 1997;37:1–17. doi: 10.1016/s1011-1344(96)07459-3. [DOI] [PubMed] [Google Scholar]
- 10.Tyrrell R M. Bioessays. 1996;18:139–148. doi: 10.1002/bies.950180210. [DOI] [PubMed] [Google Scholar]
- 11.Otto A I, Riou L, Marionnet C, Mori T, Sarasin A, Magnaldo T. Cancer Res. 1999;59:1212–1218. [PubMed] [Google Scholar]
- 12.Rheinwald J G, Green H. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 13.Wodicka L, Dong H, Mittman M, Ho M H, Lockhart D J. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 14.Lockhart D J, Dong H, Byrne M C, Follettie M T, Gallo M V, Chee M S, Mittmann M, Wang C, Kobayashi M, Horton H, et al. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 15.Tamayo P, Slonim D, Mesirov J, Zhu Q, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: Encyclopedia for Genes, Proteins and Diseases. Rehovot, Israel: Weizmann Institute of Science, Bioinformatics Unit and Genome Center; 1997. [Google Scholar]
- 17.Takayama S, Sato T, Krajewski S, Kochel K, Irie S, Millan J A, Reed J C. Cell. 1995;80:279–284. doi: 10.1016/0092-8674(95)90410-7. [DOI] [PubMed] [Google Scholar]
- 18.Kartasova T, van de Putte P. Mol Cell Biol. 1988;8:2195–2203. doi: 10.1128/mcb.8.5.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bates S, Rowan S, Vousden K H. Oncogene. 1996;13:1103–1109. [PubMed] [Google Scholar]
- 20.Muller H, Bracken A P, Vernell R, Moroni M C, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner J D, Helin K. Genes Dev. 2001;15:267–285. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyrrell R M. EXS. 1996;77:255–271. [PubMed] [Google Scholar]
- 22.Kondo S. J Invest Dermatol Symp Proc. 1999;4:177–183. doi: 10.1038/sj.jidsp.5640205. [DOI] [PubMed] [Google Scholar]
- 23.Venner T J, Sauder D N, Feliciani C, Mckenzie R C. Exp Dermatol. 1995;4:138–145. doi: 10.1111/j.1600-0625.1995.tb00237.x. [DOI] [PubMed] [Google Scholar]
- 24.Isoherranen K, Punnonen K, Jansen C, Uotila P. Br J Dermatol. 1999;140:1017–1022. doi: 10.1046/j.1365-2133.1999.02897.x. [DOI] [PubMed] [Google Scholar]
- 25.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel D V. Nature (London) 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 26.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer J L, Di Marco F, French L, Tschopp J. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 27.Hu R J, Lee M P, Connors T D, Johnson L A, Burn T C, Su K, Landes G M, Feinberg A P. Genomics. 1997;46:9–17. doi: 10.1006/geno.1997.4981. [DOI] [PubMed] [Google Scholar]
- 28.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D. Cell. 1997;89:357–364. doi: 10.1016/s0092-8674(00)80216-0. [DOI] [PubMed] [Google Scholar]
- 30.Kadosh D, Struhl K. Genes Dev. 1998;12:797–805. doi: 10.1101/gad.12.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Struhl K. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 32.Chen J D, Evans R M. Nature (London) 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 33.Nagy L, Kao H-Y, Chakravarti D, Lin R J, Hassig C A, Ayer D E, Schreiber S L, Evans R M. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 34.Robinson N A, Lapic S, Welter J F, Eckert R L. J Biol Chem. 1997;272:12035–12046. doi: 10.1074/jbc.272.18.12035. [DOI] [PubMed] [Google Scholar]
- 35.Zeeuwen P L, Van Vlijmen-Willems I M, Jansen B J, Sotiropoulou G, Curfs J H, Meis J F, Janssen J J, Van Ruissen F, Schalkwijk J. J Invest Dermatol. 2001;116:693–701. doi: 10.1046/j.1523-1747.2001.01309.x. [DOI] [PubMed] [Google Scholar]
- 36.Townsley F M, Aristarkhov A, Beck S, Hershko A, Ruderman J V. Proc Natl Acad Sci USA. 1997;94:2362–2367. doi: 10.1073/pnas.94.6.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinha S, Degenstein L, Copenhaver C, Fuchs E. Mol Cell Biol. 2000;20:2543–2555. doi: 10.1128/mcb.20.7.2543-2555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magnaldo T, Vidal R G, Ohtsuki M, Freedberg I M, Blumenberg M. Gene Expression. 1993;3:307–315. [PMC free article] [PubMed] [Google Scholar]
- 39.Navarro J M, Casatorres J, Jorcano J L. J Biol Chem. 1995;270:21362–21367. doi: 10.1074/jbc.270.36.21362. [DOI] [PubMed] [Google Scholar]
- 40.Clemens M J, Bushell M, Jeffrey I W, Pain V M, Morley S J. Cell Death Differ. 2000;7:603–615. doi: 10.1038/sj.cdd.4400695. [DOI] [PubMed] [Google Scholar]
- 41.Li D T, Turi T G, Schuck A, Freedberg I M, Khitrov G, Blumenberg M. FASEB J. 2001;15:2533–2535. doi: 10.1096/fj.01-0172fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.