

Abstract
The MHC class I chain-related MICA molecule is a stress-induced, highly polymorphic, epithelia-specific, membrane-bound glycoprotein interacting with the activating NK cell receptor NKG2D and/or gut-enriched Vδ1-bearing γδ T cells. We have previously reported the presence of a MICA transmembrane-encoded short-tandem repeat harboring a peculiar allele, A5.1, characterized by a frame shift mutation leading to a premature intradomain stop codon, thus denying the molecule of its 42-aa cytoplasmic tail. Given that this is the most common population-wide MICA allele found, we set out to analyze the functional consequences of cytoplasmic tail deletion. Here, we show native expression of MICA at the basolateral surface of human intestinal epithelium, the site of putative interaction with intraepithelial T and NK lymphocytes. We then demonstrate, in polarized epithelial cells, that although the full-length MICA protein is sorted to the basolateral membrane, the cytoplasmic tail-deleted construct as well as the naturally occurring A5.1 allele are aberrantly transported to the apical surface. Site-directed mutagenesis identified the cytoplasmic tail-encoded leucine-valine dihydrophobic tandem as the basolateral sorting signal. Hence, the physiological location of MICA within epithelial cells is governed by its cytoplasmic tail, implying impairment in A5.1 homozygous individuals, perhaps relevant to the immunological surveillance exerted by NK and T lymphocytes on epithelial malignancies.
Conventional MHC-I glycoproteins are widely expressed membrane-bound heterodimers consisting of a highly polymorphic MHC-encoded heavy chain noncovalently complexed with the β2-microglobulin (β2m) light chain (1). They present cytosol-derived short peptide antigens to the αβ T cell receptor of CD8+ cytotoxic T cells, thereby initiating the cell-mediated immune response (2). In humans there are three such genes, HLA-A, HLA-B, and HLA-C, also referred to as classical MHC-I. Similar to this first group is another trio, namely HLA-E, HLA-F, and HLA-G, dubbed nonclassical given their narrow pattern of tissue expression and especially their failure to participate in the canonical αβ T cell receptor interaction (3). Instead, they are recognized by a heterogeneous family of immunoreceptors including γδ T cell receptor, NK, T, and myeloid Ig and/or lectin-like inhibitory and activatory receptors (4). Despite this profound functional dichotomy, the HLA-A–G belong to the same structural lineage, as they collectively share over 70% amino acid identity. In this context, the identification of a second lineage of class I genes within the MHC per se was unexpected and intriguing. These MHC class I chain-related MIC genes are remote members of the extended MHC-I superfamily, with seven members scattered throughout the entire 1.8-Mb MHC class I region (5, 6). Within this family, the closely linked MICA and MICB loci are juxtaposed to the HLA-B locus on the centromeric end of the complex and encode single-chain (non β2m-linked), stress-induced, mucosa-specific class I-like glycoproteins that have been shown to interact with the gut-enriched Vδ1-bearing γδ T cells and/or T and NK cells coexpressing the C-type lectin NKG2D activating receptor (7–9). The induction of MIC upon microbial/tumoral challenge defines a stress beacon potentially relevant for triggering the innate arm of the immune system (10, 11).
The clear distinction between classical and nonclassical loci is, however, first and foremost genetic (12). Whereas classical genes are highly polymorphic, nonclassical loci are almost invariable. Surprising, therefore, was the identification of a large allelic repertoire in the MICA and MICB loci. Sequence analysis of MICA in not more than a few hundred chromosomes unveiled the existence of 54 alleles, among which 47 encode distinct putative glycoproteins (6, 13–15). A much less thorough analysis of MICB diversity has uncovered 16 alleles (6, 16, 17). While unraveling this rich allelic repertoire, we came across a polymorphic short-tandem repeat sequence within the MICA transmembrane segment encoding various numbers of alanine residues with repetitions of the (GCT)n codon, where n = 4, 5, 6, 9, or 10, with an additional allele, 5.1, characterized by a guanine insertion after the second GCT triplet (18). The latter is the origin of a frame shift mutation leading to the occurrence of a premature stop codon within the transmembrane segment, which chops off the 42-aa-long MICA cytoplasmic tail. Interestingly, several population-wide allelotyping analyses have reported that this MICA008/5.1 allele (008 refers to the extracellular sequence) is indeed the most frequent MICA allele in several populations of distinct ethnical backgrounds, e.g., over half of Caucasians carry this allele (14, 19, 20).
Cytoplasmic tail sequences are known to carry organelle-specific retention sequences or subcellular sorting motifs relevant for optimal functional behavior of a number of proteins (21, 22). Given the apparent localization of MICA in the intestinal epithelium, which serves as a paradigm for cellular polarity, the existence of such putative sequences within the cytoplasmic tail of the molecule seemed plausible. The intestinal epithelium is composed of a sheet of polarized epithelial cells, collectively the largest bodily interface with the environment (23). The plasma membrane of these cells is separated by tight junctions into apical (or luminal) and basolateral domains with distinct functional characteristics. The latter define the prime contact surface with the large number of the intraepithelial T and NK lymphocytes (IELs) shielding the entire gut surface against microbial or tumor invasion attempts (24). In the human small intestine and colon for instance, γδ T cells make up to 5–15% and 40% of IELs, respectively (25). However, our knowledge of the intracellular trafficking of MICA is nil. Intrigued by the identification of the peculiar MICA008/5.1 allele and its relatively high prevalence in the population, we set to analyze the functional consequences of loosing the cytoplasmic tail, known in some cases to harbor sorting motifs. To do so, we constructed various native as well as cytoplasmic tail-deleted MICA-green fluorescent protein (GFP) expression constructs and visualized the subcellular localization of the molecule in polarized Madin–Darby canine kidney (MDCK) epithelial cells by confocal microscopy. Furthermore, we analyzed the expression of native MICA in its natural setting, human intestinal epithelium by using newly generated anti-MICA antibodies. Finally, by site-directed mutagenesis, we define the targeting motif responsible for this subcellular localization.
Materials and Methods
Expression Vectors.
Human MICA cDNA clones with TM allele A5 were subcloned into the NheI/SmaI sites of the pEGFPN1 vector (pEGFP-MICA) (CLONTECH). The mutant MICA plasmid lacking the C-terminal 47 (pEGFP-MICAdel) residues was generated by ligating the N-terminal NheI--PvuII fragment from human MICA cDNA with TM allele A5 to the NheI/SmaI sites of the pEGFPN1 vector. Both constructs therefore express the reporter protein in fusion to the C terminus of MICA. Native MICA (TM alleles A5 and A5.1) expression constructs were generated in the pCAGGS vector (pCAGGS-MICA and pCAGGS-MICA-A5.1) (26). Site-directed mutagenesis was done by using the QuickChange site-directed mutagenesis kit (Stratagene). The orientation and the nucleotide sequence of each construct was verified by DNA sequencing.
Cell Cultures and Transfections.
MDCK cells were stably transfected with Lipofection reagent (DOTAP, Roche Molecular Biochemicals) by using 5 μg of pEGFP-MICA or pEGFPN1 according to the manufacturer's protocol and selected with 500 μg/ml of G418 (GIBCO/BRL) and screened for GFP fluorescence. Transient expression of the same cells with pEGFP-MICAdel, pCAGGS-MICA, and pCAGGS-MICA-A5.1 as well as amino acid substituted mutants were performed by using calcium phosphate (ProFection Mammalian Transfection Systems, Promega) following the manufacturer's instructions. Mouse myeloma (NS1) was transfected with 1 μg of pKJ2 (neomycin resistance gene) (27) and 5 μg of pCAGGS-MICA by using DOTAP lipofection reagent (Roche Molecular Biochemicals). Stable transfectants were selected in the presence of 500 μg/ml G418 (GIBCO/BRL) and cloned by limiting dilution.
Preparation of Anti-MICA Mouse Serum.
The G418-resistant NS1 clones were screened for the expression of the transferred MICA gene by reverse transcriptase–PCR and Northern blot analysis. BALB/c mice (CLEA Japan, Osaka2) were injected i.p. five times at weekly intervals with paraformaldehyde-fixed NS1 cells expressing MICA mRNA. Sera were collected 5 weeks after the first immunization.
Cell-Surface Domain Selective Biotinylation Assay.
The method described by Rosenthal and coworkers was slightly modified (28). Transwell polycarbonate filter-grown MDCK cells stably or transiently expressing GFP-tagged MICA molecules were labeled apically or basolaterally with EZ-Link Sulfo-NHS-LC-Biotin (Pierce). Biotinylated cell-surface proteins were labeled with anti-GFP rabbit (Molecular Probes) and anti-MICA mouse sera and then immunoprecipitated. Pellets were solubilized in Laemmli buffer, separated on SDS/PAGE (10% acrylamide), and blotted onto nitrocellulose membrane filters. Biotinylated proteins were detected by using horseradish peroxidase (HRP)-conjugated streptavidin (Amersham Pharmacia) and visualized with DAB (0.2 mg/ml 3,3′-diaminobenzidine, tetrahydrochloride/0.05 M Tris·HCl, pH 7.6/0.005% H2O2).
SDS/PAGE and Immunoblotting.
Cell lysates were separated on 10% SDS/PAGE and transferred to nitrocellulose filters. Filters were incubated with either anti-MICA mouse serum or anti-GFP rabbit serum followed by anti-mouse or anti-rabbit HRP-conjugated secondary antibody (Amersham Pharmacia). Blots were developed by DAB or enhanced chemiluminescence (Amersham Pharmacia) by using Hyperfilm ECL (Amersham Pharmacia).
Confocal Microscopy.
Transwell permeable filter-grown pEGFP-MICA and/or pEGFP-MICAdel-transfected MDCK cells were stained with Hoechst 33258 (Sigma) to visualize the nuclei. For the detection of MICA-A5.1 molecules, pCAGGS-MICA-A5.1-transfected polarized MDCK cells were apically surface labeled with biotin, incubated first with anti-MICA mouse serum, and then with FITC-conjugated sheep anti-mouse Ig and Texas Red-conjugated streptavidin (both from Amersham Pharmacia) and counterstained with Hoechst 33258. Filters were excised and transferred with the cell side up to a slide. Images were acquired by using a Zeiss Axioskop microscope equipped with a laser-scanning confocal unit (LSM-410, Carl Zeiss, Jena, Germany). GFP and/or FITC fluorescence and Texas Red fluorescence were excited by using the 488-nm Argon and 543-nm Helium Neon laser line and collected by using a dichroic beam splitter (NT 80/20/543, Zeiss) with emission filters, BP510–525 (Zeiss) and LP570 (Zeiss), respectively. Fluorescence associated with Hoechst 33258 was simultaneously excited by using the 351/364-nm Argon laser line and collected by using a dichroic beam splitter (NT 80/20/543, Zeiss) with an emission filter, LP397 (Zeiss). Three-dimensional reconstructions used the LSM software version 3.98 (Zeiss). Acquired images were imported into the Adobe PHOTOSHOP 5.0 for processing.
Immunofluorescence and Immunoelectron Microscopy.
Tissue samples from surgical specimens were fixed overnight at 4°C with 4% paraformaldehyde/0.1 M sodium phosphate buffer and embedded in Tissue-Tek OCT compound (Sakura Fine Technical). Six-micromolar Cryostat sections were mounted on saline-coated slides and air dried, incubated with anti-MICA mouse serum and rabbit anti-alkaline phosphatase antibody, and revealed by using FITC-conjugated sheep anti-mouse Ig and Texas Red-conjugated donkey anti-rabbit Ig (Amersham Pharmacia). Sections with coverslips were examined by an Olympus fluorescence microscope. For immunoelectron microscopy, cryostat sections were labeled with anti-MICA mouse serum, followed by anti-mouse HRP-conjugated secondary antibody and stained with DAB solution. The preparations were postfixed in 2% osmic acid at room temperature for 1 h, dehydrated in a graded ethanol series, and embedded in Epon 812, according to the standard inverted gelatin capsule method. Ultrathin sections were prepared on an LKB ultramicrotome and observed with a JEOL 1200 EX electron microscope without counterstaining. As a negative control, the primary antibody was replaced by nonimmune mouse serum.
Results and Discussion
MICA is the key member of a novel family of MHC-encoded class I-related loci involved in mucosal immunity (6). The expression of MICA and possibly MICB in the human gut foretold a putative strategic role for these molecules as a first line of defense in signaling stressed (infected, transformed) epithelial cells to neighboring NK and γδ lymphocytes, the so called intraepithelial lymphocytes. This is corroborated by their augmented cell-surface expression during viral and bacterial infections as well as in tumors. Given the close contact of IELs with the basal and basolateral surfaces of enterocytes, it was of interest to investigate whether MICA was selectively routed to this location and, if so, to identify the nature and location of the sorting motif in the molecule. Given precedents having defined sorting signals in the cytoplasmic tails of other polarized molecules, we concentrated our attention on the MICA cytoplasmic tail and took advantage of the existence of a natural mutant that lacks this tail. Further impetus for our attention to this allele was its high frequency within several populations surveyed to date. To experimentally mimic the in vivo situation in the human gut, we carried out our studies in the MDCK system, widely used as a prototype for epithelial polarity (22, 29).
Expression and Intracellular Localization of GFP-Tagged MICA Molecules in MDCK Epithelial Cells.
We first established an MDCK epithelial cell line stably expressing a GFP-tagged MICA (pEGFP-MICA) protein and traced GFP fluorescence to investigate the intracellular localization of MICA. Untransfected MDCK cells used as a negative control revealed no fluorescence signals, and cells transfected with the GFP expression plasmid (pEGFPN1) used as positive control revealed fluorescence evenly distributed throughout the intracellular compartments (Fig. 1A Left and Center). However, intense fluorescence of the MICA-GFP fusion proteins was clearly found at the plasma membrane (Fig. 1A Right). Fig. 1B shows immunoblot analysis of the same MDCK cells by using anti-MICA and anti-GFP sera. Immunoreactive proteins were recognized as a single 95-kDa band in MDCK cells stably expressing MICA-GFP fusion molecules, by using either anti-MICA mouse serum (Left) or anti-GFP rabbit serum (Right). Anti-MICA mouse serum raised in this report reacted specifically with a protein of 65 kDa in pCAGGS-MICA-transfected NS1 cells but did not detect any bands in untransfected NS1 cells, pCAGGS-MICB NS1 cells, or pEGFP-MICB MDCK cells (data not shown). Finally and as shown in the Fig. 1B Right, GFP expression alone yielded the expected 30-kDa single band as evidenced by using anti-GFP rabbit serum.
Figure 1.
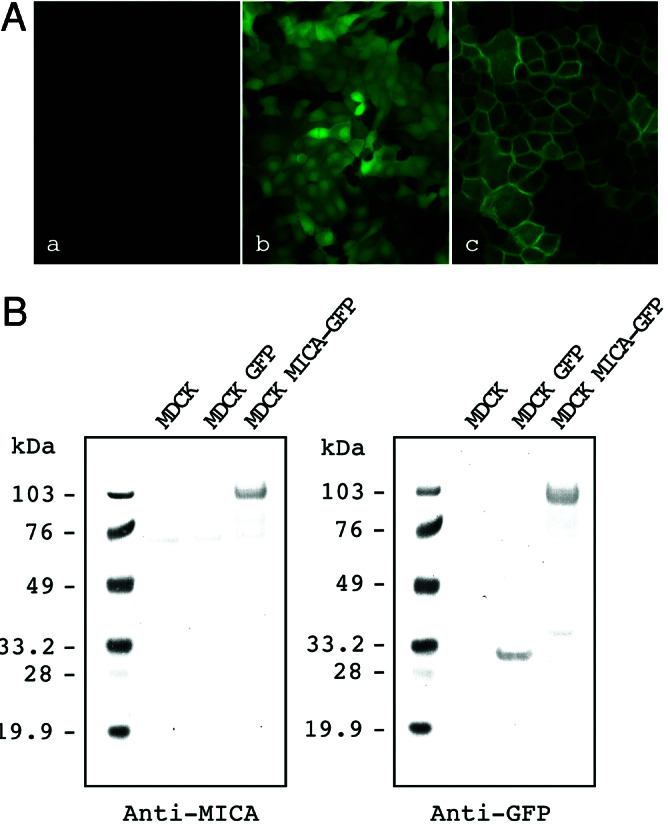
Expression of GFP-tagged MICA molecules in MDCK cells. (A) Green fluorescence signals were detected by using fluorescence microscopy in MDCK cells that were nontransfected (Left), stably transfected with the control GFP (Center), or MICA-GFP expression plasmids (Right). (B) Immunoblot analysis of GFP-tagged MICA molecules in MDCK cells: nontransfected MDCK cells (Control), MDCK cell lines stably expressing GFP (GFP), and MICA-GFP. Transferred membrane was stained with anti-MICA mouse serum (Left) or anti-GFP rabbit serum (Right). Protein size markers (kDa) are shown on the left.
Subcellular routing of MICA was analyzed by laser-scanning confocal microscopy in monolayers of MDCK cells stably expressing GFP-tagged MICA molecules. En face images reveal a honeycomb pattern of GFP signals (Fig. 2A Upper Right), whereas the vertical scan confirms an almost exclusive basolateral localization of the GFP-tagged MICA (Fig. 2 A Lower Right) (with apparently more protein at the lateral with respect to the basal surface).
Figure 2.
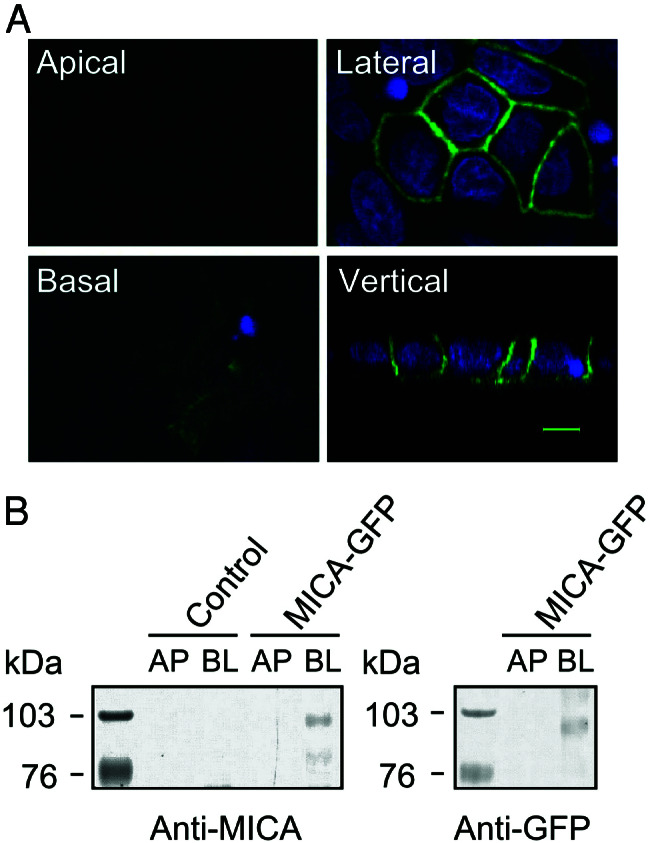
Intracellular localization of GFP-tagged MICA molecules in polarized MDCK cells. (A) MDCK cells stably expressing GFP-tagged MICA molecules. MICA-GFP fluorescence is colored by green, and nuclei are colored by blue. Fluorescence signals were analyzed by laser-scanning confocal microscopy. Confocal fluorescence micrographs (x–y plane) were acquired at the apical (Upper Left), lateral (Upper Right), and basal region (Lower Left). Confocal fluorescence micrographs (x–z plane) showing the distribution of MICA-GFP along the apical-basal axis (vertical). The scale bar is 5 μm. (B) Domain-selective cell-surface biotinylation assay of MDCK cells. Stably transfected MDCK cells expressing GFP-tagged MICA molecules were biotinylated from either the apical (AP) or basolateral (BL) domain. Biotinylated proteins were recovered by immunoprecipitation with anti-MICA mouse serum (Left) or anti-GFP rabbit serum (Right) and detected by SDS/PAGE-immunoblot analysis by using HRP-conjugated streptavidin. Control lanes on the left panel correspond to untransfected cells. Protein size markers (kDa) are shown on the left.
To confirm the steady-state distribution of GFP-tagged MICA, a domain-selective cell-surface biotinylation assay in which transfected MDCK monolayers were selectively biotinylated from either surface was carried out. In agreement with confocal results, the majority of GFP-tagged MICA molecules were found to be expressed basolaterally with an apparent molecular mass of 95 kDa (Fig. 2B). The tightness of the epithelial monolayers used in these experiments was verified by quantification of biotin leakage to the opposite side of the filters (30), which was found to be less than 0.2% over 3 days (data not shown).
MICA Is Expressed at the Basolateral Surface of the Human Intestinal Epithelial Cells.
As described above, we showed that human GFP-tagged MICA is located at the basolateral plasma membrane of stably transfected canine polarized epithelial cells. Because it is not known whether MICA molecules are similarly polarized to the basolateral plasma membrane of human enterocytes, we visualized the intracellular localization of MICA in human intestine by using immunofluorescence and immunohistochemistry. Double labeling of cryosections of the human intestinal epithelium with anti-MICA and anti-intestinal alkaline phosphatase sera depicts a nonoverlapping fluorescent pattern establishing distinctive cellular distribution for these molecules. Whereas anti-intestinal alkaline phosphatase is well known to specifically label the apical membrane, the anti-MICA staining establishes a basal as well as lateral expression pattern (Fig. 3A). Similarly, immunoperoxidase staining by using the anti-MICA mouse serum revealed MICA expression at the basolateral plasma membrane of intestinal epithelium (Fig. 3B). This was further confirmed, at high resolution, by immunoelectron microscopy, which revealed an identical subcellular localization for MICA (Fig. 3C). A similar staining pattern was observed in human epithelial cells isolated from different individuals (data not shown).
Figure 3.
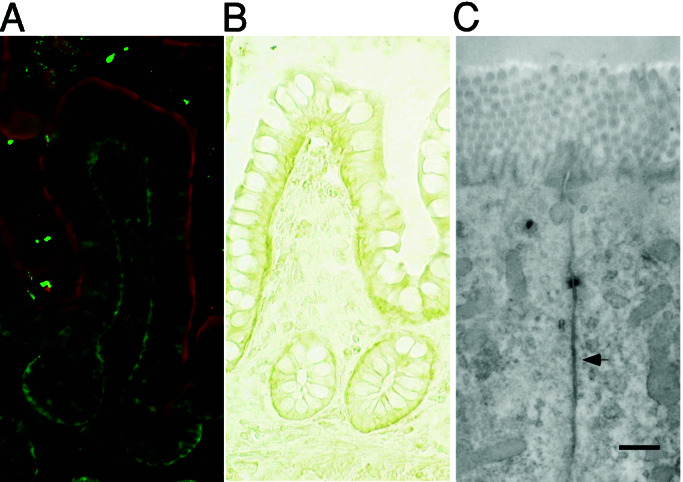
Immunofluorescence and immunoelectron micrographs of human intestinal epithelium sections stained with anti-MICA mouse serum. (A) Human intestinal epithelium was double-stained with anti-MICA mouse serum and FITC-conjugated anti-mouse Ig (green) and anti-intestinal alkaline phosphatase rabbit serum and Texas Red-conjugated anti-rabbit Ig (red). (B) Immunohistochemical detection of MICA in human intestinal epithelium by staining with anti-MICA mouse serum and HRP-conjugated anti-mouse Ig. (C) The ultrastructural localization of MICA molecules in human intestinal epithelial cells was analyzed by immunoelectron microscope employing anti-MICA mouse serum and HRP-conjugated secondary antibody. (Scale bar = 500 nm.)
Dihydrophobic Amino Acids Leu-Val at Codons 344–345 of the MICA Cytoplasmic Tail Are Required for Basolateral Sorting.
Efficient basolateral sorting of some transmembrane proteins has been reported to depend on a sorting signal within their cytoplasmic domains (22). To test whether the cytoplasmic tail of MICA contains such a signal, C-terminally truncated MICA cDNA lacking the entire cytoplasmic domain plus 5 aa of the transmembrane domain (47 aa in total) was fused to a GFP reporter gene and transiently expressed in MDCK cells. The hybrid glycoprotein had an expected molecular mass of 91 kDa (roughly 5 kDa smaller than the 95 kDa for full-length MICA-GFP fusion protein) as revealed by both anti-MICA (Fig. 4C) and anti-GFP sera (data not shown). Confocal laser-scanning microscopy clearly indicated that C-terminally truncated MICA-GFP molecule was predominantly localized to the apical side of the cell (whether this apical staining represents cell-surface expression on the plasma membrane and/or reflects the localization of the mutant protein in as yet to be identified intracellular storage/transit compartment remains an open question), as opposed to basolateral targeting for native MICA (Fig. 4 A and B). This result suggests that the C-terminal 47-aa residues of MICA contain the necessary information required for this basolateral targeting.
Figure 4.
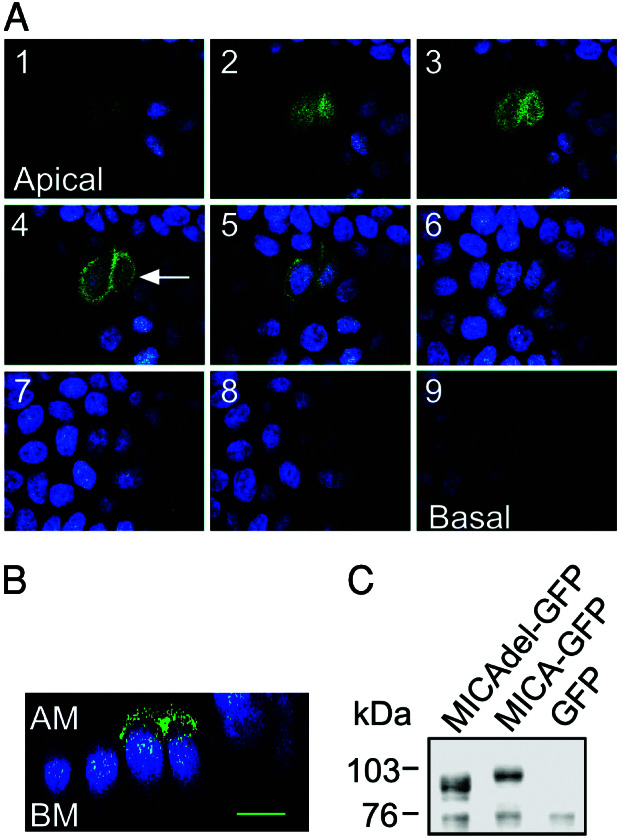
Intracellular localization of GFP-tagged MICA mutant molecules lacking the cytoplasmic domain in polarized MDCK cells. (A) MDCK cells were transiently transfected with the pEGFP-MICAdel plasmid (which lacks the 47 residues C terminus of the protein). Confocal fluorescence micrographs (x–y plane), beginning at the apical membrane (section 1, Upper Left) and ending at the basal membrane (section 9, Lower Right) were acquired in 1.0-μm increments. Arrow in optical section 4 indicates the plane of vertical section. (B) Confocal fluorescence micrograph (x–z plane) shows the distribution of GFP-tagged MICA mutant molecules lacking the cytoplasmic domain along the apical–basal axis. GFP-tagged mutant MICA-derived fluorescence is colored by green and nuclei by blue. AM, apical membrane; BM, basal membrane. The scale bar is 10 μm. (C) Immunoblot analysis of transiently expressed GFP-tagged mutant MICA molecules in MDCK cells. MDCK cells, transiently transfected with the pEGFP-MICA (MICA-GFP) or with pEGFP-MICAdel (MICAdel-GFP) plasmids, were immunoblotted with anti-MICA mouse serum. A control consisted of MDCK cells stably expressing GFP molecules alone. Sizes of marker proteins used in kDa are shown on the left.
Cytoplasmic tail dihydrophobic sequences have been shown to be responsible for subcellular sorting of CD44 in MDCK cells (31). There are three such dihydrophobic pairs in the cytoplasmic tail of MICA (numbering according to that in A5 allele): Leu-Val (344–345), Val-Leu (349–350), and Leu-Met (371–372). These pairs were sequentially changed to Ala-Ala by site-directed mutagenesis of the pEGFP-MICA plasmid and transfected back into MDCK cells (Fig. 5A). Although mutagenesis of the last two pairs did not affect the subcellular localization of the recombinant molecule, that of the first pair, Leu-Val (344–345), shifted the MICA localization from basolateral to apical, defining hence this discrete sequence as the MICA basolateral targeting sequence (Fig. 5B).
Figure 5.

Determination of the basolateral sorting motif within the MICA cytoplasmic domain by alanine substitution analysis. (A) The MICA cytoplasmic tail sequence. Boxes highlight dihydrophobic amino acids that were substituted by Ala-Ala via site-directed mutagenesis. (B) MDCK cells were transiently transfected with the alanine-substituted pEGFP-MICA plasmids. Confocal fluorescence micrographs were acquired in 1.0-μm increments. Confocal fluorescence images showing the distribution of alanine-substituted GFP-tagged MICA molecules along the apical-basal axis. GFP-tagged MICA-derived fluorescence is colored by green and nuclei by blue. AM, apical membrane; BM, basal membrane. (Scale bar = 10 μm.)
The A5.1 Frame Shift Mutation in the MICA Transmembrane Domain Creates a Presumably Defective Allele Resulting in a Loss of Basolateral Targeting.
A triplet repeat short-tandem repeat polymorphism was previously identified in the transmembrane region of MICA, and six distinct alleles of this microsatellite were reported (18, 32). These are defined by (GCT = Alanine)4,5,6,9,10 and one allele, A5.1, containing an additional one-base insertion (after the second GCT). The latter creates a frame shift mutation resulting in a premature termination codon within the transmembrane region itself, thus denying the molecule the sorting signal identified within the cytoplasmic domain. However, this particular allele has been frequently observed in all population studies carried out thus far, with the high gene frequencies up to 55% in Caucasians (14, 19, 20). To confirm that this prevalent allele loses the ability to be targeted to the basolateral surface, MDCK cells were transiently transfected with MICA-A5.1 expression vector (pCAGGS-MICA-A5.1) and intracellular routing of the molecule traced. Transfected MDCK monolayers, in which the apical membrane was labeled with biotin, were fixed, permeabilized, and incubated with anti-MICA mouse serum on both the apical and basolateral sides. These cells were then stained with a sheep anti-mouse FITC-conjugated secondary antibody and Texas Red-conjugated streptavidin. Confocal laser-scanning microscopy analysis revealed that MICA-A5.1 molecules were predominantly localized to the apical plasma membrane region in fully polarized MDCK cells cultured on permeable filter supports. This finding was confirmed by double-labeling experiments by using polarized MDCK cells. After domain-selective cell-surface biotinylation to label the apical membrane, FITC signals marking the localization of MICA-A5.1 molecules were found to be colocalized with Texas Red signals derived from the biotin-labeled apical surface membrane (Fig. 6 A and B). On the other hand, FITC fluorescent signals in MDCK cells transfected with the pCAGGS-MICA plasmid expressing the MICA A5 allele were predominantly localized to the basolateral plasma membrane (Fig. 6C). Thus, these data establish that MICA-A5.1 moves to an apparently incorrect destination, i.e., apical plasma membrane in contrast to the basolateral localization of the A5 and presumably all other alleles.
Figure 6.

Intracellular localization of MICA-A5.1 molecules in polarized MDCK cells. (A) MDCK cells were transiently transfected with the pCAGGS-MICA-A5.1 plasmid. Monolayers were labeled for the apical membrane with biotin and then washed, fixed, and stained with anti-MICA mouse serum and FITC-conjugated secondary antibody. The apical cell-surface membrane was visualized by Texas Red-conjugated streptavidin. Fluorescence of MICA-A5.1 is colored by green and the apical plasma membrane by red. The fluorescence signals were analyzed by laser-scanning confocal microscopy. Confocal fluorescence micrographs (x–y plane) were acquired at the apical (Left), lateral (Center), and basal (Right) regions. (B) Confocal fluorescence micrographs (x–z plane) showing the distribution of MICA-A5.1 along the apical-basal axis. (C) MDCK cells were transiently transfected with pCAGGS-MICA and stained with anti-MICA mouse serum and FITC-conjugated secondary antibody. Nuclei stained with Hoechst 33258 are colored in blue. AM, apical membrane; BM, basal membrane. (Scale bar = 10 μm.)
In summary, we report the identification of a dihydrophobic Leu-Val tandem sequence in the cytoplasmic tail of MICA responsible for targeting the protein to the basolateral plasma membrane of the gut epithelial cells, the prime site of contact with the effector NK and T IELs. Thus, the incorrect membrane localization of MICA-A5.1 is plausibly associated with functional deficiency, and A5.1 homozygous individuals are possibly defective in immune surveillance exerted by intestinal IELs against various epithelial malignancies as recently corroborated by the demonstration of an analogous function for the NKG2D-interacting RAE1/H60 loci, murine functional homologs of MIC genes (33–35).
Acknowledgments
We thank Dr. Johbu Ito and Mr. Akira Akatsuka for technical help in confocal microscopy, and Dr. Masataka Kuwana for providing the MICA-A5.1 cDNA clone. S.B. acknowledges financial support from the Institut National de la Santé et de la Recherche Médicale-Contrat de Recherche Stratégique and the Actions Concertées Incitatives Jeunes Chercheurs and Biologie du Développement et Physiologie Intégrative-Ministère de la Recherche.
Abbreviations
- IELs
intraepithelial T and NK lymphocytes
- MDCK
Madin–Darby canine kidney
- GFP
green fluorescent protein
- HRP
horseradish peroxidase
References
- 1.Madden D R. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- 2.Rock K L, Goldberg A L. Annu Rev Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 3.Maenaka K, Jones E Y. Curr Opin Struct Biol. 1999;9:745–753. doi: 10.1016/s0959-440x(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 4.Lanier L L. Cell. 1998;92:705–707. doi: 10.1016/s0092-8674(00)81398-7. [DOI] [PubMed] [Google Scholar]
- 5.Bahram S, Bresnahan M, Geraghty D E, Spies T. Proc Natl Acad Sci USA. 1994;91:6259–6263. doi: 10.1073/pnas.91.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bahram S. Adv Immunol. 2000;76:1–60. doi: 10.1016/s0065-2776(01)76018-x. [DOI] [PubMed] [Google Scholar]
- 7.Groh V, Steinle A, Bauer S, Spies T. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 8.Bauer S, Groh V, Wu J, Steinle A, Phillips J H, Lanier L L, Spies T. Science. 1999;285:727–729. [PubMed] [Google Scholar]
- 9.Wu J, Song Y, Bakker A B, Bauer S, Spies T, Lanier L L, Phillips J H. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 10.Groh V, Rhinehart R, Randolph-Habecker J, Topp M S, Riddell S R, Spies T. Nat Immunol. 2001;2:255–260. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 11.Das H, Groh V, Kuijl C, Sugita M, Morita C T, Spies T, Bukowski J F. Immunity. 2001;15:83–93. doi: 10.1016/s1074-7613(01)00168-6. [DOI] [PubMed] [Google Scholar]
- 12.Parham P, Ohta T. Science. 1996;272:67–74. doi: 10.1126/science.272.5258.67. [DOI] [PubMed] [Google Scholar]
- 13.Fodil N, Laloux L, Wanner V, Pellet P, Hauptmann G, Mizuki N, Inoko H, Spies T, Theodorou I, Bahram S. Immunogenetics. 1996;44:351–357. doi: 10.1007/BF02602779. [DOI] [PubMed] [Google Scholar]
- 14.Fodil N, Pellet P, Laloux L, Hauptmann G, Theodorou I, Bahram S. Immunogenetics. 1999;49:557–560. doi: 10.1007/s002510050536. [DOI] [PubMed] [Google Scholar]
- 15.Robinson J, Perez-Rodriguez M, Waller M J, Cuillerier B, Bahram S, Yao Z, Albert E D, Madrigal J A, Marsh S G. Immunogenetics. 2001;53:150–169. doi: 10.1007/s002510100303. [DOI] [PubMed] [Google Scholar]
- 16.Pellet P, Renaud M, Fodil N, Laloux L, Inoko H, Hauptmann G, Debre P, Bahram S, Theodorou I. Immunogenetics. 1997;46:434–436. doi: 10.1007/s002510050299. [DOI] [PubMed] [Google Scholar]
- 17.Ando H, Mizuki N, Ota M, Yamazaki M, Ohno S, Goto K, Miyata Y, Wakisaka K, Bahram S, Inoko H. Immunogenetics. 1997;46:499–508. doi: 10.1007/s002510050311. [DOI] [PubMed] [Google Scholar]
- 18.Mizuki N, Ota M, Kimura M, Ohno S, Ando H, Katsuyama Y, Yamazaki M, Watanabe K, Goto K, Nakamura S, et al. Proc Natl Acad Sci USA. 1997;94:1298–1303. doi: 10.1073/pnas.94.4.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersdorf E W, Shuler K B, Longton G M, Spies T, Hansen J A. Immunogenetics. 1999;49:605–612. doi: 10.1007/s002510050655. [DOI] [PubMed] [Google Scholar]
- 20.Fischer G, Arguello J R, Perez-Rodriguez M, McWhinnie A, Marsh S G, Travers P J, Madrigal J A. Immunogenetics. 2000;51:591–599. doi: 10.1007/s002510000179. [DOI] [PubMed] [Google Scholar]
- 21.Pelham H R. Philos Trans R Soc London B. 1999;354:1471–1478. doi: 10.1098/rstb.1999.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mostov K E, Verges M, Altschuler Y. Curr Opin Cell Biol. 2000;12:483–490. doi: 10.1016/s0955-0674(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 23.Shao L, Serrano D, Mayer L. Semin Immunol. 2001;13:163–176. doi: 10.1006/smim.2000.0311. [DOI] [PubMed] [Google Scholar]
- 24.Lefrancois L, Fuller B, Huleatt J W, Olson S, Puddington L. Springer Semin Immunopathol. 1997;18:463–475. doi: 10.1007/BF00824053. [DOI] [PubMed] [Google Scholar]
- 25.Kagnoff M F. Am J Physiol. 1998;274:G455–G458. doi: 10.1152/ajpgi.1998.274.3.G455. [DOI] [PubMed] [Google Scholar]
- 26.Niwa H, Yamamura K, Miyazaki J. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 27.Boer P H, Potten H, Adra C N, Jardine K, Mullhofer G, McBurney M W. Biochem Genet. 1990;28:299–308. doi: 10.1007/BF02401419. [DOI] [PubMed] [Google Scholar]
- 28.Schulein R, Lorenz D, Oksche A, Wiesner B, Hermosilla R, Ebert J, Rosenthal W. FEBS Lett. 1998;441:170–176. doi: 10.1016/s0014-5793(98)01519-1. [DOI] [PubMed] [Google Scholar]
- 29.Ikonen E, Simons K. Semin Cell Dev Biol. 1998;9:503–509. doi: 10.1006/scdb.1998.0258. [DOI] [PubMed] [Google Scholar]
- 30.Hershberg R M, Cho D H, Youakim A, Bradley M B, Lee J S, Framson P E, Nepom G T. J Clin Invest. 1998;102:792–803. doi: 10.1172/JCI3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheikh H, Isacke C M. J Biol Chem. 1996;271:12185–12190. doi: 10.1074/jbc.271.21.12185. [DOI] [PubMed] [Google Scholar]
- 32.Perez-Rodriguez M, Corell A, Arguello J R, Cox S T, McWhinnie A, Marsh S G E, Madrigal J A. Tissue Antigens. 2000;55:162–165. doi: 10.1034/j.1399-0039.2000.550209.x. [DOI] [PubMed] [Google Scholar]
- 33.Diefenbach A, Jensen E R, Jamieson A M, Raulet D H. Nature (London) 2001;413:165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerwenka A, Baron J L, Lanier L L. Proc Natl Acad Sci USA. 2001;98:11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Girardi M, Oppenheim D E, Steele C R, Lewis J M, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar R E, Hayday A C. Science. 2001;294:605–609. [PubMed] [Google Scholar]