

Abstract
MICA are distant homologs of MHC class I molecules expressed in the normal intestinal epithelium. They are ligands of the NKG2D activating receptor expressed on most γδ T cells, CD8+ αβ T cells, and natural killer cells and therefore play a critical role in innate immune responses. We investigated MICA cell-surface expression on infection of epithelial cell lines by enteric bacteria and show here that MICA expression can be markedly increased by bacteria of the diffusely adherent Escherichia coli diarrheagenic group. This effect is mediated by the specific interaction between bacterial adhesin AfaE and its cellular receptor, CD55, or decay-accelerating factor. It is extremely rapid after AfaE binding, consistent with a stress-induced signal. MICA induction on epithelial cells triggered IFN-γ release by the NKG2D expressing natural killer cell line NKL. This host–bacteria interaction pathway could play a role in the pathogenesis of inflammatory bowel disease, a condition that implicates a bacterial trigger in genetically susceptible individuals. This was supported by the increased MICA expression at the surface of epithelial cells in colonic biopsies from Crohn's disease-affected patients compared with controls.
Environmental pathogens stimulate host immune responses because of recognition of antigenic peptides bound to MHC molecules by T lymphocytes mainly expressing αβ T cell receptors. In addition to the antigen-specific acquired immunity, innate cellular immune responses may involve γδ TCR-expressing T lymphocytes, natural killer (NK), or NK T cells, and the recognition of ligands distinct from classical MHC class I or class II molecules.
MHC class I-related MICA and MICB are polymorphic molecules encoded within the MHC locus centromeric to HLA-B (1). MICA and MICB are closely related with an 84% amino acid identity (2). MICA molecules are heavily glycosylated because of eight potential glycosylation sites accounting for a 65–75-kDa molecular mass range in the absence of N-glycanase treatment (3). MICA crystallographic structure revealed crucial differences with classical MHC class I folding (4) explaining the lack of MICA peptide binding, β2 microglobulin association, and CD8 binding. NKG2D, a lectin-like receptor widely distributed on γδ T cells, CD8+ αβ T cells, and NK cells, is a functional ligand for MICA (5) to which it binds as a homodimer (6, 7). This interaction mediates effector cell activation through the DAP10 signaling adaptor (8). The importance of MICA expression in human diseases is further illustrated by their recognition on epithelial tumors by infiltrating γδ T cells (8–10). Two other properties distinguish MICA molecules: first, they have a limited normal tissue distribution in the gastrointestinal epithelium; second, they are stress-inducible (3). It has been recently demonstrated that MICA molecules were induced on cytomegalovirus-infected fibroblast or endothelial cells and could positively modulate antigen-specific CD28− CD8αβ T cell responses (11). Infection of dendritic cells and epithelial cells by Mycobacterium tuberculosis could also increase MICA expression and enhance the Vγ2δ2 T cell recognition via MICA–NKG2D interaction (12). However, there is no study of MICA expression in response to enteric bacterial infection (Shigella spp., Salmonella spp., Escherichia coli), an especially important issue regarding the normal pattern of MICA tissue distribution.
We provide here direct evidence that strains of E. coli of the diffusely adherent E. coli (DAEC) pathogenic group trigger a rapid MICA expression increase at the surface of Caco-2 intestinal epithelial or HeLa cell lines. Adhesion of DAEC to epithelial cells is mediated by adhesins belonging to a family that includes the AfaE afimbrial adhesins and the fimbrial F1845 one (13, 14). The specific cellular receptor for these adhesins is CD55, also known as the decay-accelerating factor (DAF), a glycophosphatidylinositol-anchored protein expressed notably on epithelial cells (15). We show that the increase in MICA expression is triggered by the CD55–AfaE interaction and induces the release of IFN-γ by the NKG2D-expressing NK cell line NKL. Therefore, we describe a new function for MICA as a molecule involved in the innate immune antibacterial response as well as a new function for CD55 besides its role of complement regulatory protein. These data could be relevant to the pathogenesis of autoimmune conditions triggered by a bacterial infection, for instance Crohn's disease (CD), where DAEC have been implicated (16, 17).
Methods
Bacterial Infection of Epithelial Cell Lines.
Two human epithelial cell lines were used, the cervical carcinoma cell line HeLa (homozygous for MICA*008; ref. 9) and Caco-2 (MICA*033) derived from a human colorectal adenocarcinoma. HeLa cells were grown in DMEM with 4.5 g of Glc/liter/2 mM L-Gln/100 units/ml penicillin/100 μg/ml streptomycin/10% FCS in 5% CO2 at 37°C. Caco-2 cells were grown in the same medium with 20% FCS in 10% CO2 and used before being differentiated. Wild-type bacterial strains were Salmonella typhimurium (CNRSS 96-479), E. coli A30 (18), and M90T (19), an invasive serotype 5 of Shigella flexneri that carries the 220-kb virulence plasmid pWR100. Its mutant derivative BS176 is an avirulent strain cured of pWR100. Transformed S. flexneri strains derived from M90T and BS176 (named, respectively, SC301 and SC300) contain the plasmid pIL22 including the cloned afa-1 operon (20). E. coli A30 and S. typhimurium were grown in Luria broth at 37°C, and S. flexneri strains in trypticase soy broth were supplemented with 50 μg/ml ampicillin for SC300 and SC301. Overnight cultures of bacteria were diluted in Luria broth or in tryptic soy broth (1:100) and grown to exponential phase for 2 h at 37°C. Epithelial cells were washed with PBS and infected with washed bacteria resuspended in FCS-free and antibiotic-free DMEM. Infections were done unless otherwise mentioned with 100 bacteria/cell for SC301 and SC300, 200 bacteria/cell for A30, S. typhimurium, and up to 500 bacteria/cell for M90T and BS176. Cells were incubated 1 h at 37°C for S. flexneri and 2 h for E. coli and S. typhimurium. After several washes, cells were incubated in FCS-free DMEM with gentamicin (100 μg/ml) for 30 min. In addition, M90T and BS176 were centrifuged for 10 min at 2,000 rpm to promote adherence of these bacteria to cells. Bacterial adherence on cells was visualized by Giemsa staining. Uninfected cells were treated identically to infected ones at every step.
AfaE- and AfaD-Coated Beads.
AfaE-III and AfaD products encoded by the afa-3 gene cluster were obtained by purification of histidine-tagged recombinant fusion proteins as described (13). These proteins were then coated on 1-μm-diameter carboxylated microspheres as recommended by the supplier (Molecular Probes). BSA-coated beads were used as a control. The concentration of beads was ≈1.8 × 1010 beads/ml, and the amount of protein bound to each microsphere was estimated at 15 fg. After two washes in PBS, 40 μl of beads in FCS-free DMEM were added for 2 h at 37°C to epithelial cells seeded at 0.7 × 106 in 35-mm dishes the day before experiment.
Antibodies and Flow Cytometry Analysis.
A polyclonal serum against the α2 domain of the MICA molecule was generated by immunization of rabbits with a synthetic peptide of the 140–160 translated sequence of MICA as in ref. 21 (Syntem, Montpellier, France). In all experiments, this serum was tested in parallel with the preimmune rabbit serum as a negative control. The monomorphic HLA-A, -B, -C mAb W6/32 was used for classical class I staining (22) and visualized with FITC-conjugated goat anti-mouse Ig (Sigma). For cell-surface staining of MICA molecules, epithelial cells were incubated with 1:100 diluted rabbit sera in PBS, 0.02% sodium azide at 4°C for 30 min, washed, and then stained with FITC-labeled goat anti-rabbit Ig (Sigma). After fixation (1% paraformaldehyde in PBS), they were analyzed (104 events) on a FACScan (Becton Dickinson). In blocking experiments, mAbs recognizing the CD55 short consensus repeat domain 1 (SCR1) (IA10, PharMingen), SCR2 (BRIC 110, International Blood Group Reference Laboratory, Bristol, Great Britain), SCR3 (BRIC 216, Serotec; and IH4, kindly provided by D. M. Lublin, Washington University, St. Louis), or SCR4 (8D11, kindly provided by D. M. Lublin) were added 1 h before incubation with AfaE-coated beads at 50 μg/ml purified mAbs (BRIC 216 and IA10) or at a 1:10 dilution of culture supernatant (BRIC 110, IH4, 8D11).
Cell-Surface Labeling and Immunoprecipitation of MICA.
After cell-surface biotinylation with Sulfo-NHS-LC-biotin (Pierce), MICA molecules were immunoprecipitated with the immune serum, and an aliquot was subjected to N-glycanase treatment before analysis on an SDS/polyacrylamide gel as described (5).
COS-7 Cell Transient Transfections.
The MICA specificity of the rabbit antiserum was assessed in COS-7 cell transient transfections. Full-length MICA cDNA was obtained from HeLa cells (allele MICA*008) by using PCR conditions, as described in ref. 23, and was cloned in the pTargeT mammalian expression vector (Promega). COS-7 cells were transfected by electroporation with the pTargeT–MICA cDNA construct at 250 V and 960 μF in culture medium containing 1.25% DMSO; 48 h after transfection, MICA molecules transiently expressed in COS-7 cells were detected by flow cytometry by using the immune serum.
IFN-γ Release by NK Cells in Presence of MICA-Expressing Caco-2 Targets.
To evaluate NKL cell line recognition of MICA, Caco-2 cells infected or not with bacteria during 2 h were harvested and fixed in 1% PBS–paraformaldehyde for 15 min. After two washes, Caco-2 (105 cells/well) were incubated overnight with NKL (105 cells/well) in round-bottom 96-well plates in 200 μl of DMEM supplemented with 10% heat-inactivated human serum, 2 mM L-Gln, 100 units/ml penicillin, 100 μg/ml streptomycin. NKL, grown in RPMI 1640 medium with 10% heat-inactivated human serum and 100 IU/ml IL-2, were deprived of IL-2 for 18 h before contact with target cells. In CD55 blocking experiments, targets were incubated before addition of bacteria for 30 min at room temperature with the CD55-specific mAb, BRIC 216 (10 μg/ml) or an isotypic control. In MICA-blocking experiments, diluted immune or preimmune sera (1:50) were added after bacterial infection. To assess whether the phosphatidylinositol 3-kinase signaling pathway was involved in NKL activation, NKL cells were pretreated with 25 mM wortmannin 1 h before incubation with fixed Caco-2 cells. Supernatants from each well were assessed for NKL cell reactivity in duplicate, by measuring specific secretion of IFN-γ using commercially available ELISA kits (Diaclone, Besançon, France).
Immunohistochemical Localization of MICA in Surgically Resected Colonic Specimens.
Samples of colonic mucosa were obtained in macroscopically normal areas of colon surgically removed for cancer (n = 6) or damaged areas of colon surgically removed for CD without recent steroid therapy (n = 18). Samples were immediately snap-frozen, and 5-μm-thick cryocut sections were performed. An indirect immunoperoxidase method was used with MICA-specific and the preimmune rabbit antisera at a dilution of 1:100. Slides were then treated with anti-rabbit biotinylated antibodies and visualized with diaminobenzidine according to automated procedures indicated by the manufacturer (Ventana Medical Systems, Tucson, AZ). Analysis of qualitative staining was performed independently by two observers and focused on the type of cell stained, the pattern of staining, and its distribution.
Results
E. coli Strains Expressing afa Operons Increase MICA Cell-Surface Expression on HeLa and Caco-2 Intestinal Epithelial Cell Lines.
MICA molecules were detected with a polyclonal rabbit antiserum against a synthetic peptide of the MICA second domain amino acid conserved sequence, which recognizes native MICA in flow cytometry and immunoprecipitation assays (21). The specificity of this antiserum was confirmed by cell-surface immunoprecipitation from HeLa cell lysates and by cell-surface staining of the COS-7 cell line transiently transfected with the HeLa MICA allele 008 (Fig. 1 a and b).
Figure 1.

MICA specificity of the rabbit antiserum tested by immunoprecipitation (a) and flow cytometry detection of transfected COS-7 (b). (a) Cell-surface proteins were labeled with biotin, and MICA molecules were immunoprecipitated from the lysates of 10 × 106 C1R cells, a human lymphoblastoid cell line used as a negative control (C), and of 10 × 106 HeLa cells treated (+) or nontreated (−) with N-glycanase. HeLa is homozygous for the MICA allele 008, whose size is 38 kDa because of a truncation in the cytoplasmic tail of the molecule (9). (b) Full-length MICA cDNA from HeLa cells were transfected by electroporation in COS-7 cells. Preimmune rabbit antiserum (dotted line) and the MICA-specific immune serum (solid line) were tested with untransfected (Left) and MICA*008-transfected COS-7 cells.
Modification of MICA surface expression was tested on the Caco-2 intestinal epithelial cell line and on HeLa with various strains of enterobacteriaceae characterized by different host–pathogen interaction mechanisms. Wild-type strains of the enteroinvasive bacteria S. flexneri (M90T) and S. typhimurium did not increase MICA surface expression even after a 2-h or 4-h infection of S. flexneri or S. typhimurium on HeLa and Caco-2 cell lines (data not shown). Other enteric pathogens with a different mode of host interaction were tested. Diarrheagenic E. coli are heterogeneous and presently divided into at least six groups (24): enteroinvasive, enterotoxigenic, enteropathogenic, enterohemorrhagic, enteroaggregative, and DAEC. The main pathogenic factor for DAEC is bacterial adhesin-mediated attachment encoded by the afa family of operons that contains six genes (afaA to afaF) (18). AfaE is the structural adhesin-encoding gene and is polymorphic (25). AfaE-I and AfaE-III adhesins are produced by diarrhea-associated strains (14). Strains producing the AfaE-III and AfaE-I adhesins were tested. A30, an AfaE-III strain (18), induced the higher and most reproducible increase in MICA surface expression on HeLa (Fig. 2a) and on Caco-2 cells (data not shown). MICA specific mean fluorescence intensity increased 6.53 ± 2.57-fold in a logarithmic scale as a mean from 12 different experiments in A30 infected HeLa cells.
Figure 2.

E. coli-induced MICA cell-surface expression is mediated by the interaction between bacterial adhesin AfaE and its cellular receptor DAF/CD55. In each flow cytometry experiment, MICA-specific immune serum (solid line) and preimmune serum (dotted line) as a negative control were used in parallel. (a) HeLa epithelial cell lines were infected with a wild-type invasive strain of S. flexneri (M90T), a wild-type strain of E. coli expressing the afa-3 operon (A30), and invasive and noninvasive S. flexneri strains transformed with the plasmid pIL22 containing the afa-1 operon (SC301 and SC300, respectively). S. flexneri strains were in contact with cells for 1 h and E. coli strain for 2 h before addition of gentamicin for 30 min. Horizontal axis, fluorescence intensity (logarithmic scale); vertical axis, relative number of cells. Controls are uninfected cells treated in the same conditions. (b) Dose effect of bacteria/cell ratio on MICA surface expression level. HeLa cells were infected with SC300 at 5, 20, 100, and 500 bacteria/cell for 30 min, washed, and incubated for 30 min in gentamicin containing medium before analysis. The expression level of MHC class I molecules was detected with the monomorphic W6/32 mAb (shown in bold). (c) HeLa cells were incubated for 2 h with polystyrene beads coated with recombinant AfaE-III (AfaE), AfaD proteins from an afa-3-expressing E. coli strain (13), or BSA-coated beads (HeLa control). (d) Effect of CD55-specific mAbs on AfaE-induced MICA cell-surface expression increase. HeLa cells were incubated 1 h at 37°C with mAbs specific of CD55 domain SCR1 (IA10), SCR2 (BRIC 110), SCR3 (BRIC 216), or SCR4 (8D11) before addition of AfaE beads for 2 h.
We therefore assumed that the adherent phenotype of this pathogenic E. coli strain was associated with the observed increase in MICA cell-surface expression. The role of the afa operon was further tested with derivatives of S. flexneri strains producing AfaE-I adhesin encoded by plasmid pIL22 (20). SC301 and SC300 are, respectively, the pIL22-expressing equivalents of S. flexneri wild-type M90T and of BS176, the noninvasive mutant of M90T (19). Whereas BS176 and M90T had no effect, both SC300 and SC301 reproducibly induced a high level of MICA on HeLa and Caco-2, clearly indicating that the increase of MICA expression was an effect of bacterial products encoded by the afa operon (Fig. 2a). The noninvasive Shigella strain SC300 was used for subsequent dose-response and time-course experiments. A significant increase in MICA surface expression was observed even for an infection as low as five bacteria per cell and was further enhanced as the bacteria–cell ratio increased (Fig. 2b). MICA expression stayed at a plateau from 1–4 h of infection (data not shown), and during these experiments the level of classical HLA class I molecules remained constant at the cell surface (Fig. 2b), therefore excluding a nonspecific effect of bacterial adherence on membrane protein organization.
Binding of Afimbrial Adhesin AfaE to Its Specific Cellular Receptor CD55 Mediates MICA Cell-Surface Expression Increase.
The main proteins encoded by the afa operon are bacterial adhesin AfaE and the invasin AfaD (13). To identify the bacterial product and its cellular counterpart involved in MICA surface expression, we used purified recombinant AfaE-III and AfaD proteins coated on polystyrene beads. AfaE beads reproduced host–bacteria effects on MICA expression at the same level achieved with live bacteria, whereas no effect was seen with AfaD beads on HeLa cells compared with control beads (Fig. 2c). Similar results were obtained with Caco-2 intestinal cell lines (data not shown). These data indicated that the AfaE-III adhesin was responsible for the induction of MICA surface expression.
DAF/CD55 is the cellular receptor for the AfaE family of adhesins. It contains four SCR domains; among those, SCR3 is the most critical for AfaE-I and AfaE-III binding (15). We attempted to block the AfaE-induced MICA surface expression in the presence of CD55-specific mAbs. BRIC 216 (Fig. 2d) and 1H4 (data not shown), two antibodies directed against SCR3, totally blocked MICA expression in the presence of AfaE beads on HeLa cells. BRIC 110 and 8D11 mAbs, directed against SCR2 and SCR4 domains, respectively, also blocked the effect of AfaE beads in agreement with the involvement of these two domains in AfaE-III binding (15). Conversely, IA10, directed against the SCR1 domain, which is not involved in the AfaE-III–CD55 interaction, did not change the increase of MICA expression induced by AfaE beads (Fig. 2d). Similar effects of antibodies directed against the SCR3 domain of CD55 were observed on adhesion of live bacteria SC300 producing AfaE-I (data not shown). CD55 crosslinking (26) did not induce MICA expression, implying that a specific conformational interaction between AfaE and CD55 was necessary for MICA surface expression (data not shown).
Effect of Bacteria-Induced MICA Expression on IFN-γ Production by NK Cells.
To assess the biological relevance of AfaE-induced MICA expression on intestinal epithelial cells, we developed a functional assay using NKL, an NK cell line cytotoxic against tumor cell lines because of the MIC/NKG2D interaction (5). MICA expression was induced by adhesion of SC300 for 2 h on Caco-2 cells followed by an overnight incubation with NKL. IFN-γ production in culture supernatants was measured by ELISA (Fig. 3). Under these conditions, SC300 as well as A30, a wild-type DAEC strain (not shown), induced a reproducible increase of IFN-γ production in at least five different experiments. There was no effect of BS176, the same Shigella strain as SC300 but devoid of the adhesin-encoding plasmid (not shown). Incubation with the CD55-specific mAb BRIC 216 inhibited IFN-γ production, showing the requirement for AfaE-mediated bacterial adhesion. The MICA-specific serum also abrogated the bacteria-induced IFN-γ production in favor of the direct dependency of MICA expression on the induction of cytokine production. Finally, wortmannin completely abolished the increase in IFN-γ production, consistent with the phosphatidylinositol 3kinase-dependent signaling pathway of NKL activation through NKG2D/DAP10 complexes (8).
Figure 3.

Effect of bacteria-induced MICA expression on NK cell IFN-γ production. Caco-2 cells were either uninfected (Caco-2 control) or infected with SC300 (Caco-2 SC300), a noninvasive S. flexneri strain expressing the adhesin AfaE. Before contact with NKL cells, Caco-2 target cells were fixed in PBS/1% paraformaldehyde. Supernatants were assayed for IFN-γ production in ELISA and given as the mean of triplicate measurements. Blocking experiments of IFN-γ release were carried out with preimmune or MICA immune serum (1:50) added to Caco-2 after a 2-h bacterial infection. For blockade of bacterial adhesion, CD55-specific mAb BRIC216 or an Ig-isotypic control (10 μg/ml) was incubated with targets before bacterial infection. In phosphatidylinositol 3-kinase inhibition experiments, NKL were treated for 1 h with 25 mM wortmannin before contact with 1% paraformaldehyde fixed Caco-2 cells.
Immunohistochemical Localization of MIC in Surgically Resected Colonic Specimens of Patients with CD.
Because strains of E. coli with an adherent phenotype have been isolated from ileal lesions of patients with CD (16), a condition considered as a Th1-mediated disease (27), we performed MICA immunohistochemical staining of colonic specimens from patients and noninflammatory bowel disease controls. In colonic mucosa from controls (n = 6), a weak but specific expression of MICA was observed confined to the basal part of glandular epithelial cells (Fig. 4 a and b). In severely inflamed lesions of patients with CD (n = 18), we constantly detected an intense MICA staining of the whole epithelium (Fig. 4 c and d). MICA staining also extended more profoundly to the deeper part of glandular crypts in the case of CD compared with controls (data not shown). As reported by Berstad and Brandtzaeg (28), a strong CD55/DAF staining was observed on colonic biopsies of patients affected with Crohn's disease. A strong MHC class I signal was also observed in all specimens, whereas MHC class II molecules were expressed in the case of active Crohn's lesions (data not shown).
Figure 4.
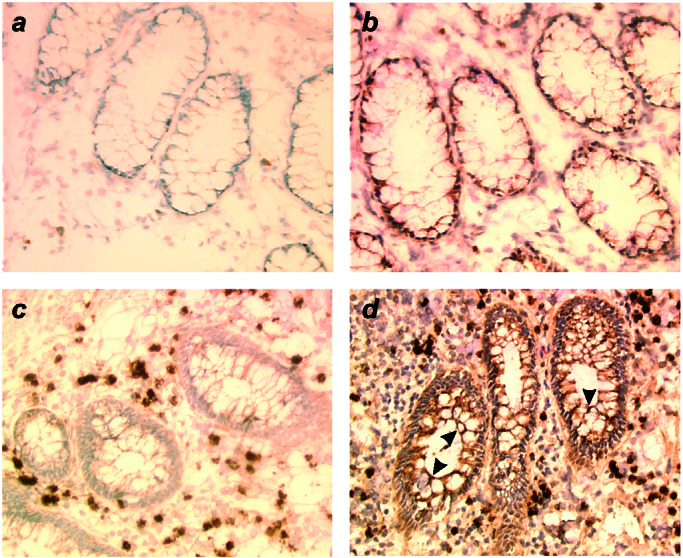
Indirect immunoperoxidase staining on cryocut sections of normal colonic samples and colonic samples of patient's with Crohn's disease. Primary antibodies were MICA preimmune (a and c) and MICA-specific rabbit antisera (b and d). On normal colonic glandular epithelial cells (a and b), MICA expression was weak and limited to the basal part of epithelial cells (b). MICA was expressed on colonic glandular epithelial cells in patients with CD (arrowheads on d) with no epithelial staining on the same sample with preimmune serum as negative control (c). Original magnification, 300×.
Discussion
Mucosal epithelial cells are the first line of contact of the host with environmental pathogens, and their role in transmitting signals to adjacent and underlying cells in the mucosa has been well documented (29). We describe here a pathway of host–pathogen interaction and the first direct evidence for the induction of MICA expression at the surface of epithelial cells during enteric bacterial infection. Surface MICA increase was observed in response to afa-expressing strains of E. coli that are associated with acute and persistent diarrhea in children in developing countries (14, 24). A high level of asymptomatic fecal carriage and an age-dependent susceptibility to these widespread E. coli strains have also been strongly suggested (24). The different members of the DAEC pathogenic group have in common a diffusely cell-adherent phenotype mediated by the interaction of the bacterial adhesin with CD55 (13, 15). It is conceivable that other human enteric pathogens, especially enteroviruses, which also use CD55 for cell entry such as coxsackieviruses or echoviruses, could behave similarly (30, 31). DAF/CD55 is a molecule already implicated in the control of innate immunity by inactivating complement C3 convertases and preventing C3b deposition on cell membranes. For that reason, transgenic pigs expressing human DAF have been developed to overcome complement-mediated hyperacute rejection in xenotransplantation (32). Our findings, which bring to light another function for DAF, should be considered in determining the safety of this approach.
It is noteworthy that CD14, another membrane-signaling receptor known to interact with bacterial products, especially lipopolysaccharide from Gram-negative bacteria (33), is also a glycophosphatidylinositol-anchored molecule. Clustering of glycophosphatidylinositol-anchored proteins in membrane microdomains has been observed in vivo and could be crucial for signal transduction and protein sorting in polarized cells (34). These two host/pathogen signaling systems are nonetheless distinct, as purified lipopolysaccharide from different strains of enteric bacteria did not increase MIC expression (data not shown).
Nonpeptide binding MHC-like molecules (for instance CD1c and CD1d) seem increasingly important in the control of innate immune responses. Ligands of these molecules have been defined. Human CD1c is recognized directly by Vδ1+ T cells in the absence of antigen (35), and CD1d presents the glycolipid α-galactosylceramide to NK T cells (36) as shown by tetramer staining (37). Despite the lack of MICA/B homologs in rodents, two murine NKG2D ligands, H-60 and Rae 1b, have been described (38, 39). In summary, there is an increasingly diverse combination of receptors and ligands involved in the control of innate immunity. Our data support the concept that MICA, with an expression highly restricted to intestinal epithelium in normal conditions, may signal the presence of bacterial infection to CD8+ T or NK cells expressing NKG2D receptors at an early stage of bacterial adhesion. This could be responsible for the rapid antigen-independent activation of CD8+ and NK T cells observed in response to bacterial pathogens (40). Although the mechanisms of MICA expression through bacterial adherence are quite different from those recently described by Groh et al. (11) in cytomegalovirus-infected cells and by Das et al. (12) during M. tuberculosis infection, the role of MICA–NKG2D interaction as a danger signal able to enhance immune responses against pathogens is fully consistent with our data. Indeed, we show that in the case of an innate immune response against bacteria, a MICA-mediated signal could be rapidly delivered to NK cells, in line with the costimulatory signal delivered to CD8αβ T cells in the case of an antigen-specific cognate immune recognition (11). The functional consequences of MICA–NKGD2 interaction could depend on the affinity of the association that has been recently linked to the allelic polymorphism at amino acid position 129 in the MICA α2 domain (7). In this view, the low IFN-γ increase observed in our experiments could be explained by the low affinity of MICA*008 (HeLa) and MICA*033 (Caco-2) for NKG2D.
The results presented here could also be relevant to human diseases, especially cancer and autoimmunity. MICA molecules are expressed on a variety of human epithelial tumors and can be recognized in the absence of antigen processing and in an unrestricted way leading to the cytotoxic lysis of tumor cells and IFN-γ production (9). However, among the many factors regulating tumor growth, those relevant to MICA expression are still unknown. CD55 expression level is actually increased in a range of tumors and especially colorectal carcinomas (41, 42), where it is considered to protect tumor cells from complement attack. With regard to our data, CD55 overexpression in tumors could facilitate adherence of E. coli strains, leading to increased MICA expression and a subsequent MICA-directed immune response. In support of this hypothesis, mucosal E. coli colonization has been associated with colorectal carcinoma (43). The role of E. coli colonization in tumor progression is still an unsolved matter that could be answered by the concomitant examination of biopsies for bacterial pathogens, MICA expression, and tumor-infiltrating lymphocytes.
At the opposite, a persistent response toward MICA molecules could take part in the pathogenesis of inflammatory bowel diseases in genetically susceptible individuals. Evidence has been accumulated to indicate that Th1 cytokines are predominantly produced in CD tissues (27). MICA–NKG2D could participate to the inflammatory process by increasing NK cell IFN-γ production and could also provide a costimulatory signal to T cells specific for a putative autoantigen. Numerous reports support the role of luminal bacteria in the pathogenesis of CD in association with genetic defects (44, 45). Particularly relevant to our work, adherent E. coli strains have been isolated in early and chronic ileal lesions at an increased frequency compared with controls (16, 17). In addition, expression of DAF/CD55 is up-regulated in tissue sections from inflammatory bowel disease specimens (28). Finally, expression of NKG2D can be up-regulated by the cytokine IL-15 released in the inflamed intestinal epithelium (46). Altogether, these data could help to define the interaction between multiple factors in CD pathogenesis, including genetic defects of monocyte response to bacterial components, as for instance NOD2 gene mutations (44, 45), persistence of bacterial products in situ leading to MICA cell-surface overexpression, and lymphocyte activation via NKG2D–MICA interaction.
Acknowledgments
We thank Dr. P. Sansonetti and Dr. A. Phalippon for providing the S. flexneri strains and for helpful discussions; Dr. P. Bouvet for the S. typhimurium strain; Dr. D. M. Lublin for the IH4 and 8D11 monoclonal antibodies; and Dr. G. Raposo, Dr. N. Mooney, and Dr. A. Haziot for helpful discussions and for a critical reading of the manuscript. This work was supported by grants from Programme de Recherche Fondamentale en Microbiologie et Maladies Infectieuses et Parasitaires-Ministère de l'Education Nationale, de la Recherche et de la Technologie (to C.L.B.) and from Assistance Publique–Hôpitaux de Paris (PHRC AOM96120) (to A.J.).
Abbreviations
- NK
natural killer
- DAEC
diffusely adherent E. coli
- Afa
afimbrial adhesin
- DAF
decay-accelerating factor
- CD
Crohn's disease
- SCR
short consensus repeat
Footnotes
See commentary on page 2584.
References
- 1.Bahram S, Bresnahan M, Geraghty D E, Spies T. Proc Natl Acad Sci USA. 1994;91:6259–6263. doi: 10.1073/pnas.91.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersdorf E W, Shuler K B, Longton G M, Spies T, Hansen J A. Immunogenetics. 1999;49:605–612. doi: 10.1007/s002510050655. .E. W.K. B.G. M.H. J. [DOI] [PubMed] [Google Scholar]
- 3.Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Proc Natl Acad Sci USA. 1996;93:12445–12450. doi: 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li P, Willie S T, Bauer S, Morris D L, Spies T, Strong R K. Immunity. 1999;10:577–584. doi: 10.1016/s1074-7613(00)80057-6. [DOI] [PubMed] [Google Scholar]
- 5.Bauer S, Groh V, Wu J, Steine A, Phillips J H, Lanier L L, Spies T. Science. 1999;285:727–729. [PubMed] [Google Scholar]
- 6.Li P, Morris D L, Willcox B E, Steinle A, Spies T, Strong R K. Nat Immunol. 2001;2:443–451. doi: 10.1038/87757. [DOI] [PubMed] [Google Scholar]
- 7.Steinle A, Li P, Morris D L, Groh V, Lanier L L, Strong R K, Spies T. Immunogenetics. 2001;53:279–287. doi: 10.1007/s002510100325. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Song Y, Bakker A B H, Bauer S, Spies T, Lanier L L, Phillips J H. Science. 1999;285:730–732. doi: 10.1126/science.285.5428.730. [DOI] [PubMed] [Google Scholar]
- 9.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein K H, Spies T. Proc Natl Acad Sci USA. 1999;96:6879–6884. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groh V, Steinle A, Bauer S, Spies T. Science. 1998;279:1737–1740. doi: 10.1126/science.279.5357.1737. [DOI] [PubMed] [Google Scholar]
- 11.Groh V, Rhinehart R, Randolph-Habecker J, Topp M S, Riddell S R, Spies T. Nat Immunol. 2001;2:255–260. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 12.Das H, Groh V, Kuijl C, Sugita M, Morita C T, Spies T, Bukowski J F. Immunity. 2001;15:83–93. doi: 10.1016/s1074-7613(01)00168-6. [DOI] [PubMed] [Google Scholar]
- 13.Jouve M, Garcia M I, Courcoux P, Labigne A, Gounon P, Le Bouguenec C. Infect Immun. 1997;65:4082–4089. doi: 10.1128/iai.65.10.4082-4089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Bouguenec C. Clin Microbiol Rev. 1999;12:180–181. doi: 10.1128/cmr.12.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowicki B, Hart A, Coyne K E, Lublin D M, Nowicki S. J Exp Med. 1993;178:2115–2121. doi: 10.1084/jem.178.6.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel J F. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 17.Masseret E, Boudeau J, Colombel J F, Neut C, Desreumaux P, Joly B, Cortot A, Darfeuille-Michaud A. Gut. 2001;48:320–325. doi: 10.1136/gut.48.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia M I, Labigne A, Le Bouguenec C. J Bacteriol. 1994;176:7601–7613. doi: 10.1128/jb.176.24.7601-7613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nhieu G T, Sansonetti P J. Curr Opin Microbiol. 1999;2:51–55. doi: 10.1016/s1369-5274(99)80009-5. [DOI] [PubMed] [Google Scholar]
- 20.Labigne-Roussel A F, Lark D, Schoolnik G, Falkow S. Infect Immun. 1984;46:251–259. doi: 10.1128/iai.46.1.251-259.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zwirner N W, Fernandez-Vina M A, Stastny P. Immunogenetics. 1998;47:139–148. doi: 10.1007/s002510050339. [DOI] [PubMed] [Google Scholar]
- 22.Parham P, Barnstable C J, Bodmer W F. J Immunol. 1979;123:342–349. [PubMed] [Google Scholar]
- 23.Pende D, Cantoni C, Rivera P, Vitale M, Castriconi R, Marcenaro S, Nanni M, Biassoni R, Bottino C, Moretta A, Moretta L. Eur J Immunol. 2001;31:1076–1086. doi: 10.1002/1521-4141(200104)31:4<1076::aid-immu1076>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 24.Nataro J P, Kaper J B. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labigne-Roussel A, Falkow S. Infect Immun. 1988;56:640–648. doi: 10.1128/iai.56.3.640-648.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shenoy-Scaria A M, Kwong J, Fujita T, Olszowy M W, Shaw A S, Lublin D M. J Immunol. 1992;149:3535–3541. [PubMed] [Google Scholar]
- 27.Fuss I J, Neurath M, Boirivant M, Klein J S, de la Motte C, Strong S A, Fiocchi C, Strober W. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 28.Berstad A E, Brandtzaeg P. Gut. 1998;42:522–529. doi: 10.1136/gut.42.4.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagnoff M F, Eckmann L. J Clin Invest. 1997;100:6–10. doi: 10.1172/JCI119522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vuorinen T, Vainionpaa R, Heino J, Hyypia T. J Gen Virol. 1999;80:921–927. doi: 10.1099/0022-1317-80-4-921. [DOI] [PubMed] [Google Scholar]
- 31.Lea S M, Powell R M, McKee T, Evans D J, Brown D, Stuart D I, van der Merwe P A. J Biol Chem. 1998;273:30443–30447. doi: 10.1074/jbc.273.46.30443. [DOI] [PubMed] [Google Scholar]
- 32.Cozzi E, Tucker A W, Langford G A, Pino-Chavez G, Wright L, O'Connell M J, Young V J, Lancaster R, McLaughlin M, Hunt K, et al. Transplantation. 1997;64:1383–1392. doi: 10.1097/00007890-199711270-00002. [DOI] [PubMed] [Google Scholar]
- 33.Wright S D, Ramos R A, Tobias P S, Ulevitch R J, Mathison J C. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 34.Friedrichson T, Kurzchalia T V. Nature (London) 1998;394:802–805. doi: 10.1038/29570. [DOI] [PubMed] [Google Scholar]
- 35.Spada F M, Grant E P, Peters P J, Sugita M, Melian A, Leslie D S, Lee H K, van Donselaar E, Hanson D A, Krensky A M, et al. J Exp Med. 2000;191:937–948. doi: 10.1084/jem.191.6.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, Kronenberg M. J Exp Med. 1998;188:1521–1528. doi: 10.1084/jem.188.8.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benlagha K, Weiss A, Beavis A, Teyton L, Bendelac A. J Exp Med. 2000;191:1895–1904. doi: 10.1084/jem.191.11.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diefenbach A, Jamieson A M, Liu S D, Shastri N, Raulet D H. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 39.Cerwenka A, Bakker A B, McClanahan T, Wagner J, Wu J, Phillips J H, Lanier L L. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 40.Lertmemongkolchai G, Cai G, Hunter C A, Bancroft G J. J Immunol. 2001;166:1097–1105. doi: 10.4049/jimmunol.166.2.1097. [DOI] [PubMed] [Google Scholar]
- 41.Niehans G A, Cherwitz D L, Staley N A, Knapp D J, Dalmasso A P. Am J Pathol. 1996;149:129–142. [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Spendlove I, Morgan J, Durrant L G. Br J Cancer. 2001;84:80–86. doi: 10.1054/bjoc.2000.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. Gastroenterology. 1998;115:281–286. doi: 10.1016/s0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 44.Hugot J P, Chamaillard M, Zouali H, Lesage S, Cezard J P, Belaiche J, Almer S, Tysk C, O'Morain C A, Gassull M, et al. Nature (London) 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 45.Ogura Y, Bonen D K, Inohara N, Nicolae D L, Chen F F, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr R H, et al. Nature (London) 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 46.Roberts A I, Lee L, Schwarz E, Groh V, Spies T, Ebert E C, Jabri B. J Immunol. 2001;167:5527–5530. doi: 10.4049/jimmunol.167.10.5527. [DOI] [PubMed] [Google Scholar]