

Abstract
Recently, it was proposed that alternative splicing may act as a mechanism for opening accelerated paths of evolution, by reducing negative selection pressure, but there has been little evidence so far that this mechanism could produce adaptive benefit. Here, we use metrics of very different types of selection pressures [e.g., against amino acid mutations (Ka/Ks), against mutations at synonymous sites (Ks), and for protein reading-frame preservation] to address this question by genomewide analyses of human, chimpanzee, mouse, and rat. These data show that alternative splicing relaxes Ka/Ks selection pressure up to 7-fold, but intriguingly this effect is accompanied by a strong increase in selection pressure against synonymous mutations, which propagates into the adjacent intron, and correlates strongly with the alternative splicing level observed for each exon. These effects are highly local to the alternatively spliced exon. Comparisons of these four genomes consistently show an increase in the density of amino acid mutations (Ka) in alternatively spliced exons and a decrease in the density of synonymous mutations (Ks). This selection pressure against synonymous mutations in alternatively spliced exons was accompanied in all four genomes by a striking increase in selection pressure for protein reading-frame preservation, and both increased markedly with increasing evolutionary age. Restricting our analysis to a subset of exons with strong evidence for biologically functional alternative splicing produced identical results. Thus alternative splicing apparently can create evolutionary “hotspots” within a protein sequence, and these events have evidently been selected for during mammalian evolution.
Keywords: bioinformatics, exon, comparative genomics, Ka/Ks, RNA splicing
Alternative splicing recently has emerged as a major mechanism of functional regulation in the human genome and in other organisms (1-3), with up to 80% of human genes reported to be alternatively spliced (4). One area that has attracted much interest is comparing alternative splicing in different genomes. Several groups have sought to assess whether alternative splicing is more abundant in the human genome vs. other genomes (5-7). Another major focus has been to use sequence conservation (regions of high-percent identity) to discover motifs that are important for regulation and alternative splicing (8-11). These data indicate that such regulatory motifs are clustered near splice sites, in both exonic sequence and the flanking introns. For example, measurements of conservation by percent identity between human and mouse show an ≈20% increase in the 30 nt of intron sequence immediately adjacent to alternatively spliced exons, relative to that for constitutive exons (8). The sequence of alternatively spliced exons also appears to have slightly higher conservation than constitutive exons, perhaps by a few percentage points of identity in comparisons of human vs. mouse (11).
It has also been proposed that alternative splicing can greatly increase the rate of certain types of evolutionary alterations, such as exon creation, by reducing negative selection pressure against such events (12-14). Evidence from many groups has shown associations between alternative splicing and increases in different types of evolutionary change, including exon duplication (15, 16), Alu element-mediated exonization (17), exon creation/loss (13, 18), and introduction of premature protein termination codons (19). In all of these cases, alternative splicing is associated with reduced levels of conservation during genome evolution. These lines of evidence suggest that alternative splicing has played a significant role during mammalian evolution, by opening neutral pathways for more rapid evolutionary change. However, at least superficially, these data would appear to be inconsistent with reports that alternative splicing is associated with increased levels of conservation (8, 11).
These data raise several questions about the role of alternative splicing in evolution. First, is the hypothesis that alternative splicing reduces negative selection pressure a general phenomenon? For example, does it hold true even for alternatively spliced exons that are clearly functional, or is it limited to alternatively spliced exons that have no biological function? Several groups have presented evidence for a stringent criterion that an alternative splicing event is functional, based on independent observations of that specific alternative splicing event in two different organisms (e.g., human and mouse) (20-22). For this data set, evolutionary processes measured over this period have genuinely taken place under the influence of alternative splicing and should reflect its effects. We have therefore performed a genomewide analysis of exons observed to be alternatively spliced in both human and mouse transcripts, which we will refer to as “ancestral alternative exons.”
Second, if alternative splicing does reduce selection pressure in a general way, is there any evidence that this phenomenon is adaptive, i.e., that such events have been selected for during evolution? Questions such as these require a transition from a single metric of evolutionary change (such as percent identity) to multiple metrics that can distinguish different types of selection pressure, e.g., selection pressure against amino acid mutations, and selection pressure against synonymous nucleotide substitutions that disrupt important nucleotide motifs (e.g., binding sites for splicing factors), etc. We have therefore analyzed the well known selection pressure metrics Ka/Ks and Ks, which give empirical measures of these two selection pressures (23, 24). Nonsynonymous nucleotide sites experience the background nucleotide mutation level (whose density is symbolized by π), nucleotide selection pressure (which we will symbolize as ρ), and amino acid selection pressure (ω), whereas synonymous sites experience only the first two factors. Thus, in the standard formulation of Ka/Ks, the densities of observed mutations at nonsynonymous sites (Ka) and synonymous sites (Ks) are
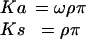 |
[1] |
and Ka/Ks = ω, with no dependence on π or ρ (23). Ka/Ks has been very widely used, because the normalization by Ks yields a metric of amino acid selection pressure that is independent of π [which varies enormously according to the total time of evolutionary divergence between a pair of genomes (25)]. A Ka/Ks ratio of 1 indicates neutral evolution (absence of selection pressure); by contrast, in most protein-coding regions Ka/Ks is significantly less than 1, indicating strong negative selection pressure against amino acid mutations (26).
In this article, we analyze Ka and Ks both for ancestral alternative exons that have strong evidence of functional alternative splicing and in genomewide comparisons of four mammalian genomes (human, chimpanzee, rat, and mouse) to evaluate how alternative splicing affected selection pressure over different evolutionary time scales. We use a standard metric for alternative splicing, the exon inclusion level, defined as the fraction of a gene's transcripts that include an exon rather than skipping it (13), and measure its impact on Ka and Ks selection pressures.
Methods
Alternative Splicing Analysis. We detected alternative splice forms in human and mouse by mapping mRNA and ESTs onto genomic sequences as described (27) by using the following data: (i) UniGene EST data (28) from June 2003 for human and mouse (ftp://ftp.ncbi.nih.gov/repository/UniGene) and (ii) genomic sequence data from June 2003 for human and mouse (ftp://ftp.ensembl.org). Internal exons were identified as genomic regions flanked by two splices, and all exon boundaries were confirmed by checking consensus splice site motifs. We computed the exon inclusion level for each alternatively spliced exon, defined as the number of ESTs that included an exon divided by total number of ESTs that either included or skipped this exon. Based on this ratio, we grouped alternatively spliced exons into three classes: major form (inclusion level above 2/3), medium form (inclusion level between 1/3 and 2/3), and minor form (inclusion level below 1/3).
We identified orthologous human-mouse exons as described (13), using orthologous gene information from HOMOLOGENE (29) (ftp://ftp.ncbi.nih.gov/pub/HomoloGene) downloaded in July 2003, including all orthologous pairs of genes that were successfully mapped onto genomic sequences during our splicing calculation. We defined a pair of human-mouse orthologous exons as ancestral alternative exons if the exon was alternatively spliced in both human and mouse transcripts. Similarly we defined a pair of human-mouse orthologous exons as “ancestral constitutive exons” if the exon was constitutively spliced in both organisms.
Ka/Ks and Ks Sequence Divergence Metrics. We computed the Ks rate and Ka/Ks ratio between orthologous exon pairs following the approach of Li and colleagues (30). Briefly, orthologous exon sequences from human and mouse were translated in all possible reading frames. Translations containing STOP codons were removed, and the remaining protein sequences were aligned in all possible combinations. We computed sequence identities in all resulting alignments by using the global sequence alignment program needle in the emboss software package (31). After excluding alignments between human and mouse protein sequences that were translated from different reading frames (indicated by a cut-off of 50% protein sequence identity), we selected the reading-frame pair with the highest amino acid identity, and then aligned these two protein sequences by using clustalw (32) under default parameters. This protein alignment was used to realign corresponding nucleotide sequences, and gaps in the alignment were trimmed. We estimated Ka and Ks from the codon-based nucleotide sequence alignment by using the Yang-Nielsen maximum-likelihood method, implemented in the yn00 program of the paml package (33). For each group of exons, we summed up the total numbers of nonsynonymous and synonymous substitutions/sites over all sequences to calculate overall Ka, Ks, and Ka/Ks.
For each pair of orthologous exons, we aligned the entire exons as well as 250-bp upstream and downstream intronic sequences, by using the program needle in the emboss software package (31). We computed the observed nucleotide substitution density (number of observed substitutions per site) in the alignment.
Genomewide Analyses of Conserved Constitutive and Alternative Exons in Human, Chimpanzee, Mouse, and Rat. We calculated Ka, Ks, and Ka/Ks for constitutive and alternative exons conserved between the genomic sequences of human and chimpanzee, or mouse and rat, or human and mouse. The exon inclusion level was estimated based on human EST data (for human vs. chimpanzee analysis and human vs. mouse) or based on mouse EST data (mouse vs. rat). We estimated Ka and Ks for each pair of human-mouse orthologous exons by using the Yang-Nielsen method as described above. For human vs. chimpanzee, we searched the entire chimpanzee genome (ftp://ftp.ensembl.org/pub/chimp-22.1) with each human exon, by using blastn (34), requiring an expectation score of 10-4 or less and a match length within at least 12 nt of the human exon's length. Using the best hit from the chimpanzee genome, we identified the best reading-frame pair as above, requiring 80% protein sequence identity. For mouse vs. rat, we searched the rat genome (ftp://ftp.ensembl.org/pub/rat-23.3c) for each mouse exon and processed hits in the same way. We also performed an additional mouse vs. human comparison by using the splicing microarray data by Pan and colleagues (18). Following their procedures, we calculated the overall inclusion level of each mouse alternative exon by averaging over 10 tissues and chose exons with confident overall inclusion levels (top 2,000 confidence ranks assigned by Pan et al.). This filter left us with 962 mouse alternative exons conserved in the human genome for further analyses.
Frame Preservation Analysis. We defined an exon as “frame preserving” if the length of the exon was a multiple of 3 nt and as “frame switching” if not (35). Inclusion or exclusion of a frame-preserving exon by alternative splicing leaves the downstream protein reading frame unchanged; for this reason, frame preservation has been proposed by several groups as evidence that an alternative splicing event is functional (21, 35-37). We calculated the frame-preservation ratio for a given set of exons as the number of frame-preserving exons divided by the number of frame-switching exons (35).
Results
Ka/Ks Analysis. To understand in detail how alternative splicing affects selection pressure, we performed a genomewide analysis of exons observed to be alternatively spliced in both human and mouse transcripts. Our results showed that ancestral alternative exons had much higher Ka/Ks values compared with ancestral constitutive exons. The overall Ka/Ks for 137 ancestral alternative exons was 0.170, significantly higher than that for 10,255 ancestral constitutive exons (0.071).
To make our analysis more quantitative, we used a standard metric for alternative splicing, exon inclusion level (13, 38), defined as the number of transcripts observed to include the exon, divided by the total number of transcripts that either include or skip it. We categorized ancestral alternative exons into three groups based on this ratio measured from human transcript data. We found a striking negative correlation between the exon inclusion level θ and mean Ka/Ks ratio (Table 1). Exons with high inclusion levels (θ > 2/3, defined as major-form exons) had a low Ka/Ks ratio (0.131), whereas exons with low inclusion levels (θ < 1/3, defined as minor-form exons) had an overall Ka/Ks ratio (0.500) >7-fold higher than constitutive exons (0.071). Thus, alternative splicing appears to relax negative selection against amino acid changes, even when there is strong evidence that these alternative splicing events are functional (they were observed in both mouse and human transcripts). Moreover, the degree of relaxation depends quantitatively on the amount of alternative splicing in these exons.
Table 1. Analysis of Ka, Ks for ancestral alternatively spliced exons between human and mouse.
| Human-mouse ancestral | No. of exons. (avg. length, nt) | No. of synonymous substitutions | No. of synonymous sites | Ks | No. of nonsynonymous substitutions | No. of nonsynonymous sites | Ka | Ka/Ks |
|---|---|---|---|---|---|---|---|---|
| Constitutive | 10,255 (131) | 259,908 | 347,356 | 0.748 | 52,524 | 982,424 | 0.053 | 0.071 |
| Major | 83 (109) | 966 | 2,401 | 0.402 | 342 | 6,497 | 0.053 | 0.131 |
| Medium | 28 (86) | 114 | 604 | 0.189 | 86 | 1,568 | 0.055 | 0.291 |
| Minor | 26 (76) | 74 | 631 | 0.117 | 85 | 1,451 | 0.059 | 0.500 |
Ks Analysis. The Ka/Ks metric divides the observed density of amino acid substitutions (Ka) against the observed density of synonymous nucleotide substitutions (Ks). In mammals, it has generally been assumed that synonymous substitutions are selectively neutral (39), i.e., that Ks simply reflects the background mutation rate of a gene. Consistent with this view, genes with relaxed selection pressure levels typically have been found to be associated with increases in Ka, without significant changes in Ks (40, 41), reflecting the ubiquitous importance of protein-level selection pressure.
However, contrary to this expectation, when we measured Ka and Ks rates separately for ancestral alternatively spliced exons, we found that increased Ka/Ks levels were associated with a large drop in the Ks rate in minor-form exons (Table 1). The overall Ks rate (Yang-Nielsen estimates) for constitutive exons was 0.748, but dropped to 0.402 for major-form exons and 0.117 for ancestral minor-form exons, a >6-fold reduction.
Control Tests vs. Neighboring Exons and Introns. To control for gene-specific effects such as gene expression level, we also repeated our Ks analysis for constitutive exons within the same genes as these minor-form exons. Ancestral alternative exons experience a significant reduction in the rate of synonymous divergence, even compared with neighboring exons within the same genes (Fig. 1). This finding suggests that the Ks rate at these exons is no longer proportional to the background mutation rate. Instead these silent sites appear to be under purifying selection, and the degree of selection is strongest at ancestral minor-form exons.
Fig. 1.

Ancestral minor-form exons have a much reduced Ks rate compared with their surrounding constitutive exons. Ks of alternatively spliced exons versus neighboring constitutive exons (empty bars) within the same gene. Ks was measured by using the Yang-Nielsen method. Error bars indicate the 95% confidence interval for the mean Ks computed by nonparametric bootstrapping.
Evidence of selection pressure on silent sites is often attributed to factors such as codon usage bias (42), which can cause reduced Ks and an artificial increase in Ka/Ks. Might this explain our results? Because intronic sequences, by definition, are not translated and are thus free from selection on codon usage, we sought to test this hypothesis by measuring the rate of nucleotide divergence at intronic sequences flanking alternative exons. Again we observed a striking reduction in the observed mutation frequency specifically for intron sequences flanking minor-form exons (Fig. 2). For the 50-nt intronic region upstream of constitutive exons, the density of observed substitutions was 0.414, versus 0.334 for majorform exons and 0.198 for minor-form exons, a >2-fold increase in selection pressure. The same trend was observed for the 50-nt region downstream of each exon. This selection pressure diminished beyond 150 nt from the exon and beyond 250 nt returned to the background level observed in constitutive exons. This finding is consistent with previous reports for an increased selection pressure against mutations in introns flanking alternative exons (8, 36, 43). Our data further indicate that the strength of such selection pressure is associated with the efficiency of the splicing reaction, being strongest for minor-form exons.
Fig. 2.

Intronic nucleotide substitution density as a function of alternative splicing and distance to intronexon junctions. Intronic nucleotide substitution density increases as a function of increasing exon inclusion levels for alternatively spliced exons and was highest in constitutive exons. The greatest difference in the intronic nucleotide substitution density between minor-form and constitutive exons was observed in the 50-nt intronic regions immediately adjacent to the intron-exon junctions. (A) Upstream introns. x axis, distance from the upstream intron-exon junction; y axis, intronic nucleotide substitution density. (B) Downstream introns. x axis, distance from the downstream exon-intron junction; y axis, intronic nucleotide substitution density.
Analysis of Ka and Ks in Human, Chimpanzee, Mouse, and Rat Genomes. The appearance of Ks in the denominator of the term Ka/Ks might seem to imply that changes in Ks can change the value of Ka/Ks, but this is not true in the standard formulation of Ka/Ks, because Ks is also present in the numerator of Ka/Ks (see Eq. 1 and Introduction). Indeed Ks is included in the denominator of Ka/Ks solely to cancel its presence from the numerator, to obtain a measure of protein-level selection pressure separate from the baseline nucleotide substitution frequency (23).
To test our interpretation completely independent of this assumption, we have analyzed the observed density of amino acid substitutions (Ka) in several genome comparisons ranging in time scale from human vs. chimpanzee [5.4 million years (My)], to mouse vs. rat (41 My), and to human vs. mouse (91 My) (44). For ancestral alternatively spliced exons (human vs. mouse), we observed a marginal increase (11%) in Ka for minor-form exons compared with major-form exons. In our genomewide analyses, we observed no increase in human vs. mouse, a 41% increase in mouse vs. rat, and a nearly 3-fold increase in human vs. chimpanzee (Fig. 3 and Table 2). Thus, even the absolute density of amino acid substitutions, without any correction made for the underlying nucleotide substitution density, shows a reproducible increase in alternatively spliced exons and correlates with the level of alternative splicing for each exon (i.e., its exon skipping frequency). We obtained similar results by comparing exons with similar sizes (data not shown).
Fig. 3.

Increased Ka/Ks and decreased Ks is a general phenomenon associated with alternative splicing during recent mammalian evolution. The ratios for minor-form exons over major-form exons calculated for Ka (Middle, ▪), Ks (Bottom, ♦), and Ka/Ks (Top, •) are shown. Reduced Ks and elevated Ka/Ks is observed in all three genome comparisons: human vs. chimpanzee, mouse vs. rat, and human vs. mouse. Ka, Ks, and Ka/Ks were estimated by using the Yang-Nielsen method.
Table 2. Analysis of Ka, Ks for conserved alternatively spliced exons in human, chimpanzee, mouse, and rat genomes.
| Genome | No. of exons (avg. length, nt) | No. of synonymous substitutions | No. of synonymous sites | Ks | No. of nonsynonymous substitutions | No. of nonsynonymous sites | Ka | Ka/Ks |
|---|---|---|---|---|---|---|---|---|
| Human-chimpanzee | ||||||||
| Constitutive | 56,108 (134) | 28,743 | 1,979,260 | 0.0145 | 16,256 | 5,421,368 | 0.0030 | 0.206 |
| Major | 701 (117) | 268 | 22,030 | 0.0122 | 130 | 58,583 | 0.0022 | 0.182 |
| Medium | 425 (121) | 156 | 13,846 | 0.0113 | 132 | 36,845 | 0.0036 | 0.318 |
| Minor | 147 (79) | 30 | 3,379 | 0.0089 | 51 | 8,024 | 0.0064 | 0.716 |
| Mouse-rat | ||||||||
| Constitutive | 16,843 (133) | 112,635 | 594,461 | 0.189 | 29,311 | 1,592,266 | 0.018 | 0.097 |
| Major | 466 (116) | 2,135 | 14,522 | 0.147 | 663 | 38,395 | 0.017 | 0.117 |
| Medium | 205 (111) | 699 | 6,155 | 0.114 | 328 | 16,048 | 0.020 | 0.180 |
| Minor | 50 (92) | 59 | 1,278 | 0.046 | 77 | 3,192 | 0.024 | 0.523 |
| Human-mouse | ||||||||
| Constitutive | 12,886 (135) | 341,211 | 451,438 | 0.756 | 80,276 | 1,270,034 | 0.063 | 0.084 |
| Major | 757 (121) | 14,054 | 23,287 | 0.604 | 3,821 | 67,270 | 0.057 | 0.094 |
| Medium | 283 (129) | 5,003 | 9,863 | 0.507 | 1,813 | 26,263 | 0.069 | 0.136 |
| Minor | 43 (82) | 158 | 997 | 0.158 | 137 | 2,439 | 0.056 | 0.354 |
Is the reduction in Ks observed in ancestral alternatively spliced exons reproducible across these multiple genome comparisons? In all cases, Ks showed a clear correlation with the exon inclusion level, with highest values for constitutive exons and lowest values for minor-form exons (Table 2). In all cases the difference between constitutive vs. minor-form exons was statistically significant, with the smallest difference in human vs. chimpanzee (a 58% difference, P = 3.7 × 10-3) and the largest difference in human vs. mouse ancestral alternatively spliced exons (a >5-fold difference, P = 6.6 × 10-16).
These multiple genome comparison data also provide some basis for assessing whether our observed increase in Ka/Ks is real or an artifact of decreasing Ks. Specifically, are these data consistent with the standard formulation of Ka/Ks (in which Ka/Ks is independent of Ks), or do they support an alternative model, in which decreases in Ks can cause increases in Ka/Ks? To assess this question in our alternative splicing data set, we calculated the minor-form/majorform ratio for Ks, Ka, and Ka/Ks in the three different genome comparisons (Fig. 3). These different data sets display substantial shifts in Ks (shifts ranging from 37% to ≈4-fold), giving some opportunity to see the impact of changes in Ks on changes in Ka/Ks. Strikingly, the large shifts in Ks produced no corresponding shift in Ka/Ks, which remained approximately constant in all three data sets, because the observed shifts in Ka exactly followed the trend of shifts in Ks. These results are exactly what is expected under the standard formulation of Ka/Ks and are not consistent with the hypothesis that decreasing Ks causes increased Ka/Ks in our data.
Finally, to estimate the exon inclusion levels independently of the EST data, we used the mouse splicing microarray data provided by Pan and colleagues (18). We analyzed 962 mouse alternative exons that were conserved in the human genome. Our analysis showed a 3.5-fold increase in Ka/Ks and a 2.5-fold decrease in Ks for minor-form exons compared with major-form exons (see Table 3, which is published as supporting information on the PNAS web site, for details), consistent with the trend observed in other analyses based on EST data.
Minor-Form Exons Display Increased Selection Pressure for Frame Preservation. We previously defined exons whose length is an exact multiple of 3 nt as frame preserving, because inclusion or skipping of the exon will not alter the protein reading frame of subsequent exons (35). It has been previously observed that exons that were observed to be alternatively spliced in both human and mouse ESTs show an increased ratio of frame-preserving vs. nonframe-preserving exons (21, 35), implying selection pressure for frame preservation. We have therefore measured evidence for such selection pressure as a function of exon inclusion level, across the genomewide comparisons between human vs. chimpanzee, mouse vs. rat, and human vs. mouse (see Fig. 4). These data show a reproducible increase in frame-preservation ratio specifically in minor-form alternatively spliced exons, up to a maximum value of 2.6 (vs. an average value of 0.6 in constitutive exons).
Fig. 4.

Protein reading-frame preservation as a function of alternative splicing. The frame-preservation ratio (ratio of frame-preserving exons over frame-switching exons) was highest in minor-form exons and near the value expected by random chance (0.5) in constitutive exons.
Older Alternatively Spliced Exons Show Increased Evidence of RNA Selection Pressure. Over the wide range of evolutionary time scales we have analyzed (5 My to 90+ My), the effect of alternative splicing on Ka/Ks was strikingly consistent. For example, the ratio of Ka/Ks in minor-form vs. major-form exons was approximately constant in all of these genome comparisons (see Fig. 3). At least over this range of time scales, the effect of alternative splicing on Ka/Ks does not appear to be a sensitive function of time or to have changed substantially over the last 100 My of mammalian evolution.
By contrast, the effect of alternative splicing on Ks showed a very clear increasing trend with increasing age of evolutionary conservation (Fig. 3), with the smallest difference between minor-form vs. major-form Ks observed in human vs. chimpanzee (37%) and the largest difference in human vs. mouse (3.8-fold). These data suggest that older alternatively spliced exons, conserved over longer periods of evolutionary history, display much stronger evidence of RNA selection pressure.
It is interesting to note that selection pressure for frame preservation displayed a similar increasing trend as a function of increasing age of evolutionary conservation (Fig. 4). The ratio between minor form vs. constitutive frame preservation was lowest in the human vs. chimpanzee comparison (1.6), intermediate in the mouse vs. rat comparison (3.6), and highest in the human vs. mouse comparison (4.0).
Discussion
Our Ks data provide specific evidence of extensive “RNA-level” selection pressure that is distinct from protein-level selection. Several studies have reported increased percent identity around alternatively spliced exons (8-10, 45). Our analysis extends this finding in several ways. Increased conservation in protein coding regions is ordinarily attributed to amino acid selection pressure. However, our data indicate amino acid selection pressure cannot explain the increased conservation in alternatively spliced exons. First, we observed up to 6-fold reduction in mutation density at synonymous sites, which do not alter amino acid sequence. This effect cannot be attributed to codon usage bias, as it extends into the flanking intronic sequence (where by definition codon usage bias is impossible), in agreement with previous studies (8, 36, 43). Second, within these same alternatively spliced exons, amino acid selection pressure was actually weakened, as indicated by an up to 7-fold increase in Ka/Ks. Third, the fact that the Ks reduction correlates strongly with the efficiency of the splicing reaction for that exon (i.e., its inclusion level) directly indicates that this reflects selection pressure on the splicing reaction itself.
It is interesting that conserved minor-form alternatively spliced exons (included only a small fraction of the gene's transcripts) show a dramatically higher level of RNA sequence selection pressure than major-form exons (included in the vast majority of the gene's transcripts). This observation may suggest that minor-form exons require more regulatory signals, and that their splicing may be more highly regulated, whereas major-form exons may represent a “default” splicing pattern. Intriguingly, our data show that this increased RNA selection takes time to evolve: for exons that are at least 5 My old (conserved in human vs. chimp), Ks for minor-form exons was only 37% lower than major-form; by contrast, for exons that are at least 90 My old (conserved in human vs. mouse), Ks for minor-form exons was 4-fold lower. This result is validated by a completely different selection pressure metric (frame preservation) that follows an almost identical trend. Together, these data show the gradual evolution of strong selection pressure on minor-form exons.
Our hypothesis of RNA selection pressure associated with alternative splicing is abundantly supported by evidence in the literature for molecular mechanisms that could give rise to such RNA-level selection. In a very recent study Pagani et al. (46) systematically introduced synonymous mutations to exon 12 of CFTR and investigated their effects on the splicing of the exon (46). They found that 31% of the synonymous substitutions being tested severely induced exon skipping and resulted in an inactive protein. This study unambiguously demonstrates that synonymous substitutions can affect splicing and are not neutral in evolution, providing direct evidence for such “RNA-level selection pressure.” In fact, in many disease-related genes (e.g., ATM, NF1, CFTR, SMN2, and others), synonymous mutations are known to disrupt existing splicing signals or introduce new splicing signals and significantly alter the splicing patterns of the gene leading to various human diseases (45). Recent analyses of splicing regulatory motifs show an enriched density of these motifs at alternatively spliced exons and their surrounding introns (47-49), suggesting the requirement of multiple splicing signals and their cooperation for precise and combinatorial regulations of alternative splicing [e.g., the brain-specific alternative splicing of GRIN1 CI cassette exon (49)]. Indeed there is a known case in BRCA1 where alternatively spliced exons with no codon usage bias were found to have greatly increased Ka/Ks and reduced Ks where several splicing regulatory elements were detected (50, 51).
Our data also suggest that alternative splicing can relax amino acid selection pressure in a strongly local fashion, without affecting neighboring constitutive exons in the same gene. Thus alternative splicing can create evolutionary hotspots in which one part of a protein sequence is allowed to accumulate amino acid mutations at a much higher rate than the rest of the protein. Multiple analyses of the human, chimpanzee, mouse, and rat genomes showed that alternative splicing was associated with an increase not only in the normalized ratio of amino acid mutations (Ka/Ks), but also in the absolute density of amino acid mutations (Ka), computed without even taking into consideration the underlying density of nucleotide substitution (Ks). The increase in Ka/Ks associated with alternative splicing was observed to be constant over time scales ranging from 5 My (human vs. chimp) to 90 My (human vs. mouse). Thus, this effect cannot be attributed to newly created, nonfunctional exons. For example, this effect was also observed in the human vs. mouse ancestral alternative splicing data set, which has not only been conserved for >90 My of evolution, but its pattern of alternative splicing has been conserved as well (i.e., these exons were independently observed to be both included and skipped in human transcripts and mouse transcripts); this criterion has been widely used, indicating that an alternative splicing event is functional (20-22).
Such localized effects on protein evolution require careful interpretation. It is customary to view poor protein sequence conservation (i.e., neutral or near-neutral Ka/Ks values) as evidence of reduced functional importance. However, although it is natural to interpret a high Ka/Ks value for an entire gene sequence as evidence that it is not functional (e.g., a pseudogene), this assumption seems much less safe when the zone of high Ka/Ks is confined to a short segment of a protein. Recalling the definition of Ka/Ks, it should be emphasized that high values of Ka/Ks simply mean rapid change, not necessarily lack of function. For example, specific regions with high Ka/Ks have often been shown to be functionally very important [e.g., the antigen presentation cleft of MHC proteins (52) and drug resistance mutations in HIV (53)]. In many such cases the regions with highest Ka/Ks are the most important functional sites in the protein (such as the antigen binding site in MHC or drug resistance mutations in HIV protease). Subsegments of elevated Ka/Ks, often corresponding to individual exons that are alternatively spliced, appear to have been important in both the evolution and function of many proteins, such as BRCA1 (50) and CD45 (54). Our Ks data and frame-preservation results provide systematic evidence that such rapid evolution of a protein subsequence is not necessarily indicative of loss of function. This finding suggests that such alternative splicing-accelerated evolution has produced adaptive functions that have been selected for during recent evolution.
Supplementary Material
Acknowledgments
We thank Q. Pan and B. Blencowe for the mouse microarray data; D. Black, G. Chanfreau, B. Modrek, and F. Kondrashov for discussions and comments on this work; P. Green and S. Eddy for discussions on the possible interactions of Ks and Ka/Ks; and three anonymous reviewers for their comments. C.L. was supported by National Institutes of Health Grant U54-RR021813 and Department of Energy Grant DE-FC02-02ER63421.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Frontiers in Bioinformatics: Unsolved Problems and Challenges,” held October 15-17, 2004, at the Arnold and Mabel Beckman Center of the National Academies of Sciences and Engineering in Irvine, CA. Papers from this Colloquium will be available as a collection on the PNAS web site. See the introduction to this Colloquium on page 13355. The complete program is available on the NAS web site at www.nasonline.org/bioinformatics.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: My, million years.
References
- 1.Mironov, A. A., Fickett, J. W. & Gelfand, M. S. (1999) Genome Res. 9, 1288-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brett, D., Hanke, J., Lehmann, G., Haase, S., Delbruck, S., Krueger, S., Reich, J. & Bork, P. (2000) FEBS Lett. 474, 83-86. [DOI] [PubMed] [Google Scholar]
- 3.Modrek, B. & Lee, C. (2002) Nat. Genet. 30, 13-19. [DOI] [PubMed] [Google Scholar]
- 4.Kampa, D., Cheng, J., Kapranov, P., Yamanaka, M., Brubaker, S., Cawley, S., Drenkow, J., Piccolboni, A., Bekiranov, S., Helt, G., et al. (2004) Genome Res. 14, 331-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brett, D., Pospisil, H., Valcarcel, J., Reich, J. & Bork, P. (2002) Nat. Genet. 30, 29-30. [DOI] [PubMed] [Google Scholar]
- 6.Kim, H., Klein, R., Majewski, J. & Ott, J. (2004) Nat. Genet. 36, 915-916 and author reply 916-917. [DOI] [PubMed] [Google Scholar]
- 7.Valenzuela, A., Talavera, D., Orozco, M. & de la Cruz, X. (2004) J. Mol. Biol. 335, 495-502. [DOI] [PubMed] [Google Scholar]
- 8.Sorek, R. & Ast, G. (2003) Genome Res. 13, 1631-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W. J., Mattick, J. S. & Haussler, D. (2004) Science 304, 1321-1325. [DOI] [PubMed] [Google Scholar]
- 10.Fairbrother, W. G., Holste, D., Burge, C. B. & Sharp, P. A. (2004) PLoS Biol. 2, E268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itoh, H., Washio, T. & Tomita, M. (2004) RNA 10, 1005-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boue, S., Letunic, I. & Bork, P. (2003) BioEssays 25, 1031-1034. [DOI] [PubMed] [Google Scholar]
- 13.Modrek, B. & Lee, C. (2003) Nat. Genet. 34, 177-180. [DOI] [PubMed] [Google Scholar]
- 14.Lareau, L. F., Green, R. E., Bhatnagar, R. S. & Brenner, S. E. (2004) Curr. Opin. Struct. Biol. 14, 273-282. [DOI] [PubMed] [Google Scholar]
- 15.Kondrashov, F. A. & Koonin, E. V. (2001) Hum. Mol. Genet. 10, 2661-2669. [DOI] [PubMed] [Google Scholar]
- 16.Letunic, I., Copley, R. R. & Bork, P. (2002) Hum. Mol. Genet. 11, 1561-1567. [DOI] [PubMed] [Google Scholar]
- 17.Sorek, R., Ast, G. & Graur, D. (2002) Genome Res. 12, 1060-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan, Q., Shai, O., Misquitta, C., Zhang, W., Saltzman, A. L., Mohammad, N., Babak, T., Siu, H., Hughes, T. R., Morris, Q. D., et al. (2004) Mol. Cell 16, 929-941. [DOI] [PubMed] [Google Scholar]
- 19.Xing, Y. & Lee, C. (2004) Trends Genet. 20, 472-475. [DOI] [PubMed] [Google Scholar]
- 20.Kan, Z., States, D. & Gish, W. (2002) Genome Res. 12, 1837-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thanaraj, T. A., Clark, F. & Muilu, J. (2003) Nucleic Acids Res. 31, 2544-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorek, R., Shamir, R. & Ast, G. (2004) Trends Genet. 20, 68-71. [DOI] [PubMed] [Google Scholar]
- 23.Yang, Z. & Bielawski, J. P. (2000) Trends Ecol. Evol. 15, 496-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, W. H. (1993) J. Mol. Evol. 36, 96-99. [DOI] [PubMed] [Google Scholar]
- 25.Hurst, L. D. (2002) Trends Genet. 18, 486-487. [DOI] [PubMed] [Google Scholar]
- 26.Makalowski, W. & Boguski, M. S. (1998) Proc. Natl. Acad. Sci. USA 95, 9407-9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modrek, B., Resch, A., Grasso, C. & Lee, C. (2001) Nucleic Acids Res. 29, 2850-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuler, G. (1997) J. Mol. Med. 75, 694-698. [DOI] [PubMed] [Google Scholar]
- 29.Wheeler, D. L., Church, D. M., Edgar, R., Federhen, S., Helmberg, W., Madden, T. L., Pontius, J. U., Schuler, G. D., Schriml, L. M., Sequeira, E., et al. (2004) Nucleic Acids Res. 32, D35-D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nekrutenko, A., Chung, W. Y. & Li, W. H. (2003) Nucleic Acids Res. 31, 3564-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rice, P., Longden, I. & Bleasby, A. (2000) Trends Genet. 16, 276-277. [DOI] [PubMed] [Google Scholar]
- 32.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang, Z. (1997) Comput. Appl. Biosci. 13, 555-556. [DOI] [PubMed] [Google Scholar]
- 34.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403-410. [DOI] [PubMed] [Google Scholar]
- 35.Resch, A., Xing, Y., Alekseyenko, A., Modrek, B. & Lee, C. (2004) Nucleic Acids Res. 32, 1261-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Philipps, D. L., Park, J. W. & Graveley, B. R. (2004) RNA 10, 1838-1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorek, R., Shemesh, R., Cohen, Y., Basechess, O., Ast, G. & Shamir, R. (2004) Genome Res. 14, 1617-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hide, W. A., Babenko, V. N., van Heusden, P. A., Seoighe, C. & Kelso, J. F. (2001) Genome Res. 11, 1848-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharp, P. M., Averof, M., Lloyd, A. T., Matassi, G. & Peden, J. F. (1995) Philos. Trans. R. Soc. London B 349, 241-247. [DOI] [PubMed] [Google Scholar]
- 40.Thomas, M. A., Weston, B., Joseph, M., Wu, W., Nekrutenko, A. & Tonellato, P. J. (2003) Mol. Biol. Evol. 20, 964-968. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, L. & Li, W. H. (2004) Mol. Biol. Evol. 21, 236-239. [DOI] [PubMed] [Google Scholar]
- 42.Iida, K. & Akashi, H. (2000) Gene 261, 93-105. [DOI] [PubMed] [Google Scholar]
- 43.Sugnet, C. W., Kent, W. J., Ares, M., Jr. & Haussler, D. (2004) Pac. Symp. Biocomput. 66-77. [DOI] [PubMed]
- 44.Hedges, S. B. (2002) Nat. Rev. Genet. 3, 838-849. [DOI] [PubMed] [Google Scholar]
- 45.Cartegni, L., Chew, S. L. & Krainer, A. R. (2002) Nat. Rev. Genet. 3, 285-298. [DOI] [PubMed] [Google Scholar]
- 46.Pagani, F., Raponi, M. & Baralle, F. E. (2005) Proc. Natl. Acad. Sci. USA 102, 6368-6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minovitsky, S., Gee, S. L., Schokrpur, S., Dubchak, I. & Conboy, J. G. (2005) Nucleic Acids Res. 33, 714-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang, Z., Rolish, M. E., Yeo, G., Tung, V., Mawson, M. & Burge, C. B. (2004) Cell 119, 831-845. [DOI] [PubMed] [Google Scholar]
- 49.Han, K., Yeo, G., An, P., Burge, C. B. & Grabowski, P. J. (2005) PLoS Biol. 3, E158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orban, T. I. & Olah, E. (2001) Trends Genet. 17, 252-253. [DOI] [PubMed] [Google Scholar]
- 51.Hurst, L. D. & Pal, C. (2001) Trends Genet. 17, 62-65. [DOI] [PubMed] [Google Scholar]
- 52.Bernatchez, L. & Landry, C. (2003) J. Evol. Biol. 16, 363-377. [DOI] [PubMed] [Google Scholar]
- 53.Chen, L., Perlina, A. & Lee, C. J. (2004) J. Virol. 78, 3722-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Filip, L. C. & Mundy, N. I. (2004) Mol. Biol. Evol. 21, 1504-1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.