

Abstract
Expression of the antiviral cytokines IFN-α/β is among the most potent innate defenses of higher vertebrates to virus infections, which is controlled by the inducible transcription factor IFN regulatory factor (IRF)3. Borna disease virus (BDV) establishes persistent noncytolytic infections in animals and tissue culture cells, indicating that it can circumvent this antiviral reaction by an unexplained activity. In this study, we identify the BDV P protein as microbial gene product that associates with and inhibits the principal regulatory kinase of IRF3, Traf family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK-1). We demonstrate that the P protein counteracts TBK-1-dependent IFN-β expression in cells and, hence, the establishment of an antiviral state. Furthermore, our data show that the BDV P protein itself is phosphorylated by TBK-1, suggesting that P functions as a viral decoy substrate that prevents activation of cellular target proteins of TBK-1. Thus, our findings provide evidence for a previously undescribed mechanism by which a viral protein interferes with the induction of the antiviral IFN cascade.
Keywords: Borna disease virus, innate immunity
Vertebrate cells and organisms respond to the presence of microorganisms with a variety of immediate defenses that aim to control the invasion and spread of harmful pathogens. One of the earliest and most potent innate immune reactions to virus infection is the transcriptional induction of IFN-α and -β, collectively referred to as type I IFN. These antiviral cytokines bind to a common IFN-α/β receptor and thereby activate signaling through the Janus kinase-signal transducer and activator of transcription (STAT) pathway, which in turn induces the expression of a number of IFN-dependent gene products that limit viral propagation (1). Type I IFN induction is orchestrated by amplification of an initial wave of IFN-β by an auto- and paracrine feedback loop that facilitates expression of several IFN-α genes (2). The activation of IFN-α and -β genes is chiefly controlled by the latent IFN regulatory factors (IRF)3 and -7 that localize to the cytoplasm in the inactive state (3, 4). The main stimulus of IRF3 in virus-infected cells is believed to consist of dsRNA, a byproduct of viral replication. IRF3 activation involves phosphorylation of a C-terminal serine-rich cluster, which facilitates nuclear accumulation, association with the CBP/p300 coactivator, and binding to promoter sites with an IRF consensus motif (5). Recent analyses have identified two noncanonical IκB kinases (IKK), the Traf family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK-1; NAK, T2K) and the inducible IKK (IKK-i, IKK-ε) as IRF3 kinases (6, 7). The constitutively expressed TBK-1 is particularly critical for IFN induction, because activation of IRF3 by virus infection and the accumulation of IFN-β mRNA is defective in murine fibroblasts lacking TBK-1 but not IKK-ε (8-10). The factors upstream of TBK-1 that recognize dsRNA and signal for the presence of a viral infection are not well defined but appear to include the caspase activation and recruitment domain-containing proteins RIG-I, mda-5, and FADD (9, 11, 12). Interestingly, TBK-1 is also elemental for IFN-β induction by ligands of the Toll-like receptor (TLR) family members TLR-3 and -4, which is mediated by the Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF, also known as TI-CAM), at least in fibroblasts (8, 13-16). Thus, distinct signaling pathways that are activated by viral or particular bacterial infections converge at the TBK-1/IRF3 module, emphasizing the central role of these factors in innate immune reactions (4).
A decisive factor for efficient replication and spread of many, if not all, viruses is the ability to subvert the cellular IFN defense, and many virus families accomplish this task by the expression of antagonistic proteins (17). Thus, viral proteins have been shown to compete for binding of secreted IFNs to their cognate receptors, to interfere with Janus kinase-STAT signaling, or to inhibit IFN-controlled antiviral gene products, such as protein kinase PKR. In addition, a group of viral pathogens of humans, including influenza, Ebola, and hepatitis C virus, have been shown to express unrelated gene products that inhibit the activation of IRF3 and repress transcriptional induction of IFN genes (18-20). However, in none of these cases has the molecular basis of this blockade been elucidated.
Borna disease virus (BDV) is a nonsegmented negative-strand RNA virus that is the causative agent of a fatal CNS disease affecting primarily horses and sheep (21). Further interest in BDV has been fueled by a potential linkage to the etiology of human neuropsychiatric diseases (22). Prominent features of BDV biology include a strong neurotropism and the ability to establish noncytolytic persistent infections in animals as well as in cell lines of neuronal and nonneural origin (21). The linear 8.9-kb RNA genome of BDV contains six major ORFs that encode a viral nucleocapsid protein N, a viral phosphoprotein P, the p10 protein, the matrix protein M, the surface glycoprotein G, and an RNA-dependent RNA polymerase, termed L (21). Several findings indicated that Bornaviruses suppress the induction of IFN-α/β, but the mechanism of this escape has not been uncovered. First, exogenous IFN protects cells from infection with BDV, similarly to other negative-strand RNA viruses (23, 24). Second, the levels of IFN-α mRNA remain unchanged in the rat brain during acute infection with BDV (25). Finally, the absence of type I IFN receptors does not influence replicative titers or spread of BDV in mice, suggesting that IFN is not sufficiently activated to contain the virus in vivo (26). In this report, we show that the BDV P protein inhibits the induction of the IFN-β promoter caused by either viral challenge or TBK-1 expression. P was found to physically associate with TBK-1 in cells and to inhibit its kinase activity. Finally, we observed that the P protein strongly reduced TBK-1-mediated secretion of IFN-β and induction of an antiviral state. Thus, these data establish the BDV P protein as a viral gene product that both binds to and regulates the kinase activity of TBK-1, a central factor in the induction of the antiviral IFN response.
Materials and Methods
Animals, Viruses, and Cells. Normal and persistently with BDV-infected Madin-Darby canine kidney cells (MDCK) and 293T cells were grown as described (27, 28). Stocks of the mutant influenza A and B viruses lacking the NS1 genes (A/delNS1 and B/delNS1) were prepared as detailed elsewhere (27). Stocks of vesicular stomatitis virus (VSV) were grown in baby hamster kidney cells. Six-week-old female Lewis rats were obtained from the animal breeding facility at the Friedrich Loeffler Institute and infected intracerebrally in the left brain hemisphere with 0.05 ml of the BDV He/80 strain corresponding to 5 × 103 focus-forming units. The animals were killed 18 days postinfection, and the brain was fixed in 4% paraformaldehyde.
Plasmids. Plasmid pCA-P was prepared by inserting the BDV cDNA encoding the viral P protein into pCAGGS. P cDNA in which the internal ATG start codon of the P′ isoform had been converted to TTC was also inserted into pcDNA3 (Invitrogen). Plasmids expressing the BDV N and M proteins in mammalian cells were constructed by subcloning the corresponding cDNAs into pcDNA3. A bacterial P expression vector was constructed in pGEX-5X-1 (Amersham Pharmacia). Vectors encoding the BDV p10 protein, Ebola virus VP35 protein, TBK-1-Flag, IRF3-hemagglutinin (HA), and EGFP-IRF3 have been described (18, 28-31). The activation of the IFN-β promoter, IRF3-dependent promoters, and of IFN-stimulated response elements (ISRE)-controlled gene expression was assessed by use of the firefly luciferase reporter plasmids p125-Luc (11), p4x(PRD)I/III-Luc (30), and pISRE-Luc (Stratagene), respectively.
Transfection and Luciferase Reporter Gene Assays. In reporter gene assays, 5 × 105 cells were transiently transfected with 50 ng of the indicated reporter construct together with 5 ng of the pRL-TK-Luc plasmid (Stratagene) by using Lipofectamine 2000 (Invitrogen). To assess the activities of viral proteins, cells were cotransfected with the indicated amounts of expression plasmid. Total levels of transfected DNA were kept constant with empty vector plasmid. For stimulation, cells were infected 24 h posttransfection with influenza A/del NS1 virus and were lysed 8 h postinfection in reporter lysis buffer (Promega). For stimulation of the IFN-β promoter by TBK-1, cells were cotransfected with 50 ng of pcDNA-TBK-1-Flag together with the indicated amounts of effector plasmid. Reporter activity was determined with Promega's dual luciferase assay system. Firefly luciferase values were normalized for transfection efficiency by means of the Renilla luciferase activity that is constitutively expressed by pRL-TK-luc. The reporter activation by virus infection was expressed in comparison with mock-infected cells that had been transfected with the same set of plasmids.
Immunohistochemistry and Immunofluorescence Analysis. Sections from fixed brain tissue of BDV-infected Lewis rats were stained with hematoxylin/eosin. Immunohistochemistry was carried out on serial sections with the BDV N-specific mAb 38/17C1 to detect virus infection (32) and an IRF3-specific rabbit antibody (Santa Cruz Biotechnology, sc 9082), respectively. Staining reactions were enhanced by the use of a biotinylated secondary antibody and an ABC kit (Vector Laboratories) was used for detection of BDV- and IRF3-specific signals. To localize IRF3 in tissue culture cells, they were transfected with pEGFP-C1-hIRF3 and seeded on glass coverslips. Cells were prepared for immunofluorescence analysis after 24 h, as described (28), and were incubated with a P-specific monoclonal antibody followed by staining with a secondary Alexa594-coupled goat anti-mouse Ig (IgG) (Molecular Probes). As a positive control, MDCK cells expressing EGFP-IRF3 were infected with influenza B/delNS1 virus. The samples were analyzed under a Nikon Axiophot300 fluorescence microscope, and images were recorded with a SPOT RT digital camera (Visitron Systems, Puchheim, Germany).
Immunoprecipitation and Immunoblotting Analyses. To analyze the TBK-1/P interaction, normal or BDV-infected 293T cells (3 × 106) were transfected with the indicated amounts of pcDNA-TBK1-Flag, pCA-P, and pCA-IRF3-HA. Cell extracts were prepared 24 h posttransfection in 300 μl of lysis buffer (20 mM Tris·HCl, pH 7.5/0.2% Nonidet P-40/100 mM NaCl/1 mM EDTA/10 mM β-glycerophosphate/1 mM Na3VO4/1 mM Pefabloc, Roth, Karlsruhe, Germany) and cleared by centrifugation. The lysates were incubated for 3 h at 4°C with 1 μg of M2 anti-Flag (Sigma) or anti-Myc (9E10, Santa Cruz Biotechnology) mAb. Immunocomplexes were collected and washed on protein-G-agarose beads (Roche, Mannheim, Germany), and the precipitated proteins were dissolved in SDS sample buffer. The association of endogenous IRF3 and was analyzed in extracts from 293T cells (107) that had been transfected with 10 μg of pcDNA-TBK-1-Flag and pCA-P. Cell lysates were prepared and incubated with 1 μg of IRF3-specific mAb SL12.1 (BD PharM-ingen). Immunocomplexes were collected on protein-G-agarose, and the precipitated proteins were analyzed by immunoblotting with suitable primary [M2 anti-Flag; rabbit anti-BDV P serum; rabbit anti-CREB-binding protein (Santa Cruz Biotechnology A22); rabbit anti-HA (Upstate Biotechnology, Lake Placid, NY)] and secondary horseradish peroxidase-conjugated antibodies (DAKO Diagnostika) by using an enhanced chemiluminiscence protocol (Pierce).
Immunecomplex Kinase Assay. BDV-infected 293T cells (1 × 107) were transfected with 1 μg of pcDNA-TBK-1-Flag or empty vector. Normal 293T cells were transfected in parallel with 1 μg of pcDNA-TBK-1-Flag and, if indicated, cotransfected with increasing amounts of pCA-P. In a second set of experiments, 293T cells were also transfected with pcDNA plasmid-expressing kinase-inactive TBK-1 (TBK-1 dn). Extracts were prepared 24 h posttransfection in 1 ml of Triton lysis buffer (30), and TBK-1 was immunoprecipitated from cleared extracts with anti-Flag M2 mAb. Immunecomplexes were washed and incubated with recombinant GST-IRF3 (380-427) or GST-IκBα (1-72) as substrates in the presence of 100 μM unlabeled ATP and 5 μCi of [γ32P]-ATP (1 Ci = 37 GBq), as described (30). Samples were separated by SDS/PAGE, followed by transfer onto a nitrocellulose membrane and visualized by autoradiography. Band intensities of the phosphorylated substrates were quantified by densitometry by using imagegauge 3.01 software, and values were normalized to the amounts of immunoprecipitated kinase in the respective sample. Recombinant GST-P fusion protein was purified from bacterial lysates by affinity chromatography and was used as substrate in TBK-1 immunecomplex kinase assays under the same assay conditions as described above.
Analysis of IFN-β Induction and TBK-1-Dependent Antiviral Activity by ELISA, RT-PCR, and VSV Plaque Assay. The supernatants of untreated and transfected 293T cells were analyzed for IFN-β levels by an ELISA kit (Fujirebio, Tokyo). Total RNA was isolated from the corresponding cells by using the RNeasy kit (Qiagen, Hilden, Germany). The samples were analyzed with the OneStepRT-PCR kit (Qiagen) by using 1 μg of RNA as template with primer pairs specific for human IFN-β or β-actin transcripts in a total volume of 50 μl. Five microliters of each reaction was subjected to agarose gel electrophoresis for transcript analysis. To analyze TBK-1-mediated antiviral activity, 293T cells were transfected with the indicated amounts of pcDNA-TBK-1-Flag and pCA-P expression plasmids. One day posttransfection, supernatants were transferred onto cultures of A549 cells for 16 h at 37°C before infection with VSV at a multiplicity of 0.1. Viral titers were determined at the indicated time points by standard plaque assay on MDCK cells.
Results
BDV Infection Does Not Activate IRF3. The BDV has a strong preference to replicate in cells of the CNS in animals but can also multiply in cells of various origin including epithelial cells. Fig. 1 and Fig. 6, which is published as supporting information on the PNAS web site, show that BDV does neither induce activation of the transcription factor IRF3 in the cortex of an acutely infected Lewis rat or in persistently infected tissue culture cells, as judged by the purely cytoplasmic localization of IRF3. Thus, BDV differs remarkably from other members of the Mononegaviruses such as measles or vesicular stomatitis virus that are strong activators of IRF3 (33).
Fig. 1.

BDV infection does not activate IRF3. (A) Serial sections of brain from an experimentally BDV-infected Lewis rat (18 days postinfection) were stained by immunohistochemistry with IRF3- and BDV N-specific antibodies, as indicated. The arrows mark cells in the cortex that express viral antigen and show cytoplasmic staining for IRF3. Arrowheads indicate an uninfected cell. The sections were counterstained with hematoxylin/eosin (×400). (B) The cellular localization of a transfected IRF3-EGFP fusion protein was analyzed in normal and persistently with BDV-infected MDCK cells. The cells were also stained with a P-specific antibody and analyzed by immunofluorescence microscopy (×400). As an activation control, IRF3-EGFP-expressing MDCK cells were infected with the influenza B/delNS1 virus.
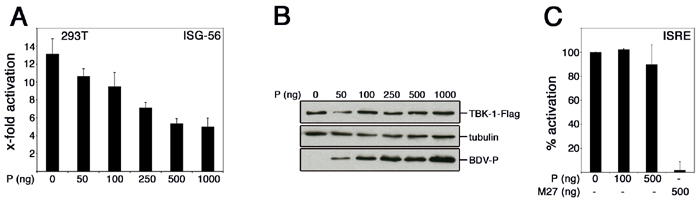
Identification of the BDV P Protein as an Inhibitor of Virus- and TBK-1-Induced IFN Promoter Induction. The lack of activation in infected cells suggested that BDV either prevents or avoids induction of IRF3 by an undefined mechanism. To probe the capacity of BDV to influence IRF3-dependent gene expression, we screened single viral gene products for the ability to suppress activation of the IFN-β promoter by influenza A mutant virus lacking the IFN-antagonistic NS1 protein (29). Among the candidates tested, only the expression of the BDV P gene inhibited IFN-β promoter induction (Fig. 2A), similarly as observed for the Ebola virus VP35 protein, a known IFN antagonist (18). Because induction of IFN-β and other antiviral response genes by virus infection critically depends on the activation of the transcription factor IRF3 by the kinase TBK-1 (8, 10), we examined whether expression of the BDV P protein affected TBK-1-mediated induction of IRF3-controlled gene expression. In Figs. 2 B-D and 7A, it is shown that the BDV P protein efficiently inhibited the strong activation by coexpressed TBK-1 of the IFN-β promoter (Fig. 2B) as well as of promoters that are controlled by oligomerized IRF3-binding sites (Fig. 2C) or by ISRE (Fig. 2D and Fig. 7 A and B, which is published as supporting information on the PNAS web site). A mutational analysis indicated that the N-terminal domain of the P protein is important for efficient down-regulation of the IFN-β promoter (Fig. 8, which is published as supporting information on the PNAS web site). Notably, P expression hardly affected stimulation of an ISRE promoter by exogenous IFN-α (Fig. 7C).
Fig. 2.

The BDV P protein inhibits activation of the IFN-β promoter induced by virus infection and TBK-1 expression. (A Left) MDCK cells were transfected each with 2 μg of plasmid expressing the indicated BDV -P, -M, -N, and -p10 or the Ebola virus VP35 protein together with 50 ng of vector encoding a luciferase reporter gene for IFN-β and the constitutively expressed pRL-TK-luc for normalization. Twenty-four hours posttransfection, cells were mock-treated or infected with influenza A/delNS1 virus for 8 h followed by dual luciferase assay analysis. Normalized reporter activation in infected cells was expressed in comparison with values obtained from mock-infected cells that had received the same set of plasmids. The inhibitory effects of the P and VP35 proteins were also observed in an identical experimental setting in 293T cells (A Center). Assays were run in duplicates, and average values from two independent sets of experiments are shown with indicated standard deviations. The expression of BDV proteins N, P, p10, and M in transfected and virus-infected MDCK cells was compared by immunoblot analyses by using monospecific antisera (A Right). (B-D) 293T cells were cotransfected with or without 50 ng of pcDNA-TBK-1-Flag together with the indicated amounts of pCA-P expression plasmid and 50 ng of reporter plasmid controlled by the IFN-β promoter (B), IRF3-binding sites (C), or ISRE elements (D). Coexpression of the BDV p10 protein was analyzed for a control. (E) The intracellular accumulation of the BDV P protein by increasing amounts of transfected pCA-P was verified by immunoblotting.
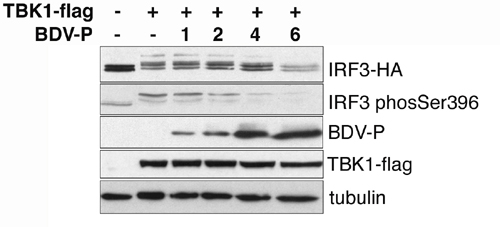
The BDV P Protein Associates with TBK-1 and Inhibits Its Kinase Activity. The regulation of TBK-1-mediated gene expression by the BDV P protein raised the intriguing possibility that this viral gene product forms a complex with the IRF3 kinase. Physical binding of TBK-1 to the P protein was investigated in BDV-infected and transfected cells expressing Flag-tagged TBK-1. When TBK-1 was immunoprecipitated with Flag-specific antibody from cell lysates, both the viral and ectopical expressed P proteins were detected in the precipitated complexes (Fig. 3A). The degree of P coimmunoprecipitation in transfected cells was reproducibly higher compared with infected cells, which might be due to the known involvement of the P protein in complexes with other viral proteins in infected cells (21). Interestingly, when TBK-1 was coexpressed with P, we noted the appearance of a slowly migrating form of the P protein on SDS gels, which was enriched in the fraction associated with TBK-1 (Fig. 3A, lanes IP, αFlag, and +TBK-1). Treatment of the Flag immunoprecipitate with phosphatase strongly reduced the slowly migrating band, indicating that the induced modification was due to phosphorylation (data not shown). We then addressed the hypothesis whether the viral P and the cellular IRF3 proteins would compete for binding to and/or phosporylation by TBK-1. Fig. 3B shows that the expression of increasing amounts of IRF3 reduced the slowly migrating fraction of P in the lysate and led to a dose-dependent decrease in TBK-1/P coprecipitation. IRF3 did not coprecipitate with TBK-1 in the presence or absence of P, suggesting that the association of the kinase with the viral P protein is more stable than with its natural substrate IRF3 (Fig. 3B). Furthermore, expression of the BDV P protein strongly reduced the TBK-1-stimulated phosphorylation of IRF3 and its association with the coactivator CBP (Fig. 3C and Fig. 9, which is published as supporting information on the PNAS web site), indicating the inhibitory function of the viral protein on IRF3 activation.
Fig. 3.

The BDV P protein interacts with TBK-1 and interferes with activation of IRF3. (A) Normal (Upper) and BDV-infected 293T cells (Lower) were transfected with 4 μg of pCA-P and 2 μg of pcDNA-TBK-1-Flag, as indicated. Cell lysates were prepared and immunoprecipitated with anti-Flag or anti-Myc antibodies. Proteins in 5 μl of cell lysate and in the immunoprecipitates were analyzed by immunoblotting with Flag- and BDV P-specific antibodies, as indicated. (B) 293T cells were cotransfected with the indicated expression vectors for TBK-1-Flag, BDV-P, and IRF3-HA. Lysates were used for immunoprecipitations with anti-Flag or -Myc antibodies. Proteins in 5 μl of lysate (Upper) and in the precipitates (Lower) were analyzed by immunoblotting with anti-Flag and pooled HA- and P-specific antibodies, respectively. (C) 293T cells were transfected without or with pcDNA-TBK-1-Flag together with increasing amounts of pCA-P, as indicated. Complex formation between endogenous IRF3 and CBP proteins was studied by immunoblot analysis of anti-IRF3 precipitates from cell extracts by the useof CBP- (upper row) and IRF3-specific antibodies (lower row).
To characterize the effect of the viral protein on TBK-1 function, we first compared the kinase activity of TBK-1 in the absence or presence of BDV P by immune complex kinase assay. Flag-tagged TBK-1 was transfected in BDV-infected cells or in cells expressing increasing amounts of the P protein (Fig. 4A). It was observed that TBK-1 activity in Flag immunoprecipitates was strongly decreased both in BDV-infected cells and in cells ectopically expressing the P gene. Subsequently, it was shown that immunoprecipitated TBK-1 phosphorylated recombinant P protein in vitro (Fig. 4B). Collectively, these findings suggest that the BDV P protein is both a target and an inhibitor of TBK-1, and that P is able to interfere with TBK-1-mediated phosphorylation and, hence, activation of IRF3.
Fig. 4.

The BDV P protein inhibits kinase activity of TBK-1 and can be phosphorylated by TBK-1 in vitro. (A) 293T cells were transfected with TBK-1-Flag expression vector and increasing amounts of P expression plasmid. TBK-1 was precipitated from lysates, and kinase activity was analyzed with recombinant GST-IRF3 (380-427) as a substrate (upper row). The presence of TBK-1 in the precipitates was verified by immunoblot analysis with Flag-specific antibody (lower row). Relative kinase activities are given as percentage of the activity in the TBK-1 only control sample that was arbitrarily set to 100% (see Materials and Methods). (B) Empty vector or expression plasmids for wild-type or dominant-negative (dn) TBK-1-Flag were transfected into 293T cells. Lysates were Flag-precipitated, and the immobilized proteins were tested for kinase activity by incubation with [γ32P]-ATP and recombinant GST, GST-P, and GST-IκBα (1-72) proteins as substrates. Samples were separated by SDS gel electrophoresis and transferred onto a nitrocellulose membrane. Radiolabeled proteins were visualized by autoradiography (Upper) and the recombinant substrate proteins were detected by immunoblotting with a GST-specific antibody (Lower).
The BDV P Protein Inhibits the Induction of IFN-β by TBK-1 and Its Antiviral Activity. Expression of TBK-1 has been shown to activate the IFN-β gene and to decrease viral replication (6). We therefore analyzed whether the BDV P protein compromised the capacity of TBK-1 to induce this cytokine and to counteract its antiviral activity. Fig. 5A shows that IFN-β cytokine and mRNA levels in TBK-1-transfected cells were lowered to baseline values upon coexpression of increasing amounts of P. The impact of P expression on the antiviral activity conferred by TBK-1 was assessed by analyzing replication of VSV. When the supernatants of transfected 293T cells expressing TBK-1 and various amounts of P were used to condition A549 cells, it was observed that the extent of VSV replication correlated directly with the amount of transfected P expression plasmid (Fig. 5B). Taken together, these data establish the BDV P protein as an IFN antagonist that directly inhibits the IFN-α/β-inducing TBK-1.
Fig. 5.

Expression of the BDV P protein impairs TBK-1-mediated IFN-β induction and antiviral activity. (A) 293T cells were transfected with empty vector or expression plasmids for TBK-1-Flag (500 ng) and BDV P, as indicated. Secreted IFN-β in the cell supernatants was measured 24 h later by ELISA (Upper). Total RNA preparations derived from cells of the same experiment were used to analyze IFN-β and β-actin transcripts by RT-RCR followed by agarose gel electrophoresis and ethidium bromide staining. (B) Supernatants of 293T cells that had been transfected with 500 ng of TBK-1-Flag expression plasmid together with empty vector (♦) or 500 ng (□) or 2,500 ng of pCA-P (Δ) were used to condition A549 cells for 16 h. Subsequently, cells were infected with VSV, and viral replication was monitored by determination of viral plaque titers at the indicated time points postinfection.
Discussion
Mutational changes in the genes of viral IFN antagonists can drastically reduce the replication and pathogenicity of a virus, highlighting that countermeasures against the cellular IFN defense are critical chain links in the processes that determine viral virulence (17). Thus, the characterization of the viral gene products that exert these “antihost” functions can foster our understanding of viral virulence and may eventually lead to the identification of novel antiviral targets. Expression of IFN-β and other immediate defense genes is tightly controlled by the latent transcription factor IRF3 and its upstream kinase, the ubiquitous TBK-1, which together built a molecular switch for antiviral activity. We have identified the BDV P protein as a viral gene product that physically associates with TBK-1 and represses its IFN-inducing kinase activity. Thus, IRF3 is not activated in BDV-infected cells, which is different from infections with many other RNA viruses, such as measles virus and West Nile virus, which rather interfere with IFN-dependent signaling events at later stages. Our data also demonstrate the biological consequences of this viral sabotage, because the BDV P protein was found to suppress IRF3-dependent gene activation as well as the induction of IFN-β secretion and TBK-1-mediated limitation of viral replication. It is tempting to speculate that the control of this cellular immune function is an important aspect of BDV's capability to establish persistent infections in vitro and in vivo.
How can P's mode of action be envisioned? The BDV phosphoprotein is a central cofactor of viral RNA synthesis that interacts with the catalytic viral RNA polymerase as well as with the viral N and p10 proteins (21, 28). A previous study had characterized target sites in P for the protein kinases Cε and casein kinase II at serine residues 26/28 and 70/86, respectively (34). However, the roles of these modifications have remained elusive. Our analysis indicated that the P protein can be phosphorylated by TBK-1 in cells and in vitro. Interestingly, the BDV phosphoprotein exhibits a conserved Ser-XXX-Ser recognition motif for mitogen-activated protein kinases at positions 8-11 with high similarity to established TBK-1 phosphorylation sites in IRF3 and IκBα (35). Strikingly, in TBK-1 immunoprecipitates, we could detect the BDV P protein but not the natural target IRF3, indicating that the viral protein binds more tightly to the kinase. Moreover, coexpression of the P protein strongly compromised kinase activity when TBK-1 was precipitated from cells and analyzed for phosphorylation of an in vitro substrate. Thus, the P protein may act as a viral decoy substrate for TBK-1, which remains in physical contact with the kinase after phosphorylation and prevents recognition of cellular substrates such as IRF3. This hypothesis is supported by the finding that an increase in the intracellular concentration of IRF3 competed for the formation of precipitable TBK-1/P complexes. Alternatively, phosphorylation of P may occur only as a side effect of the interaction, and the binding of P could inhibit the catalytic activity of TBK-1 also by other means.
A number of viral pathogens have evolved gene products that inhibit activation of IRF3, such as the NS1 proteins of influenza A and B viruses, the Ebola virus VP35 protein, the NS1/NS2 proteins of respiratory syncytial virus, and the hepatitis C virus (HCV) NS3/4a proteins (18-20, 27, 36). However, for none of these viruses has the molecular basis of the IFN blockade been pinpointed. Although there is no recognizable sequence homology to the BDV P protein, it appears reasonable to suggest that the IFN antagonists expressed by other pathogens may also interfere with the activity of TBK-1 or other upstream factors that are involved in IFN induction. In fact, very recent studies have described proteins encoded by vaccinia virus, rabies virus, and HCV as inhibitors of TBK-1-dependent IFN-β promoter induction (37-39), although it has not been reported whether they also affect TBK-1 kinase activity. Further, it was found that the conserved V proteins of several paramyxoviruses associate with the inducible RNA helicase mda-5 that, in the presence of intracellular dsRNAs, may signal for IFN induction by an unknown mechanism (12). Finally, we suggest that the precise delineation of interactions between factors of the innate immune system and inhibitory microbial gene products may guide the search for novel antivirals that neutralize viral antagonistic functions and thereby support the body's own defenses to eliminate the infectious agent.
Supplementary Material
Acknowledgments
We are grateful to W. Garten (Institute of Virology, Marburg, Germany) for antiserum and to E. Mühlberger, (Institute of Virology, Marburg, Germany), S. Jennings (Department of Virology, University of Freiburg, Freiburg, Germany), T. Fujita (Department of Tumor Cell Biology, Tokyo Metropolitan Institute of Medical Science, Tokyo Metropolitan Organization for Medical Research, Tokyo), M. Nakanishi (Department of Biochemistry, Nagoya City University Medical School, Nagoya, Japan), S. Akira (Department of Host Defense, Research Institute for Microbial Diseases, Osaka University, Osaka), and A. García-Sastre (Department of Microbiology, Mount Sinai School of Medicine, New York) for plasmid constructs. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Wo554/2-1; Lu477/4-5) and is also part of the activities of the VIRGIL European Network of Excellence on Antiviral Drug Resistance, supported by a grant (LSHM-CT-2004-503359) from the 6th Framework Program of the European Union.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BDV, Borna disease virus; TANK, Traf family member-associated NF-κB activator; TBK-1, TANK-binding kinase 1; IKK, inhibitor of κB kinase; IRF, IFN regulatory factor; MDCK, Madin-Darby canine kidney cells; ISRE, IFN-stimulated response elements; VSV, vesicular stomatitis virus; HA, hemagglutinin.
References
- 1.Samuel, C. E. (2001) Clin. Microbiol. Rev. 14, 778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taniguchi, T. & Takaoka, A. (2002) Curr. Opin. Immunol. 14, 111-116. [DOI] [PubMed] [Google Scholar]
- 3.Honda, K., Yanai, H., Negishi, H., Asagiri, M., Sato, M., Mizutani, T., Shimada, N., Ohba, Y., Takaoka, A., Yoshida, N., et al. (2005) Nature 434, 772-777. [DOI] [PubMed] [Google Scholar]
- 4.Pitha, P. M. (2004) Proc. Natl. Acad. Sci. USA 101, 695-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiscott, J., Pitha, P., Genin, P., Nguyen, H., Heylbroeck, C., Mamane, Y., Algarte, M. & Lin, R. (1999) J. Interferon Cytokine Res. 19, 1-13. [DOI] [PubMed] [Google Scholar]
- 6.Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R. & Hiscott, J. (2003) Science 300, 1148-1151. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E., Golenbock, D. T., Coyle, A. J., Liao, S. M. & Maniatis, T. (2003) Nat. Immunol. 4, 491-496. [DOI] [PubMed] [Google Scholar]
- 8.Hemmi, H., Takeuchi, O., Sato, S., Yamamoto, M., Kaisho, T., Sanjo, H., Kawai, T., Hoshino, K., Takeda, K. & Akira, S. (2004) J. Exp. Med. 199, 1641-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balachandran, S., Thomas, E. & Barber, G. N. (2004) Nature 432, 401-405. [DOI] [PubMed] [Google Scholar]
- 10.McWhirter, S. M., Fitzgerald, K. A., Rosains, J., Rowe, D. C., Golenbock, D. T. & Maniatis, T. (2004) Proc. Natl. Acad. Sci. USA 101, 233-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., Taira, K., Akira, S. & Fujita, T. (2004) Nat. Immunol. 5, 730-737. [DOI] [PubMed] [Google Scholar]
- 12.Andrejeva, J., Childs, K. S., Young, D. F., Carlos, T. S., Stock, N., Goodbourn, S. & Randall, R. E. (2004) Proc. Natl. Acad. Sci. USA 101, 17264-17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoebe, K., Du, X., Georgel, P., Janssen, E., Tabeta, K., Kim, S. O., Goode, J., Lin, P., Mann, N., Mudd, S., et al. (2003) Nature 424, 743-748. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto, M., Sato, S., Hemmi, H., Hoshino, K., Kaisho, T., Sanjo, H., Takeuchi, O., Sugiyama, M., Okabe, M., Takeda, K., et al. (2003) Science 301, 640-643. [DOI] [PubMed] [Google Scholar]
- 15.Oshiumi, H., Matsumoto, M., Funami, K., Akazawa, T. & Seya, T. (2003) Nat. Immunol. 4, 161-167. [DOI] [PubMed] [Google Scholar]
- 16.Perry, A. K., Chow, E. K., Goodnough, J. B., Yeh, W. C. & Cheng, G. (2004) J. Exp. Med. 199, 1651-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Sastre, A. (2004) Curr. Top Microbiol. Immunol. 283, 249-280. [DOI] [PubMed] [Google Scholar]
- 18.Basler, C. F., Mikulasova, A., Martinez-Sobrido, L., Paragas, J., Muhlberger, E., Bray, M., Klenk, H. D., Palese, P. & Garcia-Sastre, A. (2003) J. Virol. 77, 7945-7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foy, E., Li, K., Wang, C., Sumpter, R., Jr., Ikeda, M., Lemon, S. M. & Gale, M., Jr. (2003) Science 300, 1145-1148. [DOI] [PubMed] [Google Scholar]
- 20.Talon, J., Horvath, C. M., Polley, R., Basler, C. F., Muster, T., Palese, P. & Garcia-Sastre, A. (2000) J. Virol. 74, 7989-7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de la Torre, J. C. (2001) in Fields Virology, ed. Howley, P. M. (Lippincott, Williams & Wilkins, Philadelphia), pp. 1669-1677.
- 22.Lipkin, W. I., Hornig, M. & Briese, T. (2001) Trends. Microbiol. 9, 295-298. [DOI] [PubMed] [Google Scholar]
- 23.von Rheinbaben, F., Stitz, L. & Rott, R. (1985) J. Gen. Virol. 66, 2777-2780. [DOI] [PubMed] [Google Scholar]
- 24.Hallensleben, W. & Staeheli, P. (1999) Arch. Virol. 144, 1209-1216. [DOI] [PubMed] [Google Scholar]
- 25.Shankar, V., Kao, M., Hamir, A. N., Sheng, H., Koprowski, H. & Dietzschold, B. (1992) J. Virol. 66, 992-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staeheli, P., Sentandreu, M., Pagenstecher, A. & Hausmann, J. (2001) J. Virol. 75, 8216-8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dauber, B., Heins, G. & Wolff, T. (2004) J. Virol. 78, 1865-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolff, T., Pfleger, R., Wehner, T., Reinhardt, J. & Richt, J. A. (2000) J. Gen. Virol. 81, 939-947. [DOI] [PubMed] [Google Scholar]
- 29.Basler, C. F., Wang, X., Muhlberger, E., Volchkov, V., Paragas, J., Klenk, H. D., Garcia-Sastre, A. & Palese, P. (2000) Proc. Natl. Acad. Sci. USA 97, 12289-12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrhardt, C., Kardinal, C., Wurzer, W. J., Wolff, T., von Eichel-Streiber, C., Pleschka, S., Planz, O. & Ludwig, S. (2004) FEBS Lett. 567, 230-238. [DOI] [PubMed] [Google Scholar]
- 31.Tojima, Y., Fujimoto, A., Delhase, M., Chen, Y., Hatakeyama, S., Nakayama, K., Kaneko, Y., Nimura, Y., Motoyama, N., Ikeda, K., et al. (2000) Nature 404, 778-782. [DOI] [PubMed] [Google Scholar]
- 32.Bourteele, S., Oesterle, K., Pleschka, S., Ehrhardt, C., Wolff, T., Ludwig, S. & Planz, O. (2005) J. Virol. 79, 6043-6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Servant, M. J., ten Oever, B., LePage, C., Conti, L., Gessani, S., Julkunen, I., Lin, R. & Hiscott, J. (2001) J. Biol. Chem. 276, 355-363. [DOI] [PubMed] [Google Scholar]
- 34.Schwemmle, M., De, B., Shi, L., Banerjee, A. & Lipkin, W. I. (1997) J. Biol. Chem. 272, 21818-21823. [DOI] [PubMed] [Google Scholar]
- 35.tenOever, B. R., Sharma, S., Zou, W., Sun, Q., Grandvaux, N., Julkunen, I., Hemmi, H., Yamamoto, M., Akira, S., Yeh, W. C., et al. (2004) J. Virol. 78, 10636-10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bossert, B., Marozin, S. & Conzelmann, K. K. (2003) J. Virol. 77, 8661-8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiPerna, G., Stack, J., Bowie, A. G., Boyd, A., Kotwal, G., Zhang, Z., Arvikar, S., Latz, E., Fitzgerald, K. A. & Marshall, W. L. (2004) J. Biol. Chem. 279, 36570-36578. [DOI] [PubMed] [Google Scholar]
- 38.Brzozka, K., Finke, S. & Conzelmann, K. K. (2005) J. Virol. 79, 7673-7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otsuka, M., Kato, N., Moriyama, M., Taniguchi, H., Wang, Y., Dharel, N., Kawabe, T. & Omata, M. (2005) Hepatology 41, 1004-1012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}