

Abstract
Parkinson's disease (PD) is the most common movement disorder characterized by dopaminergic dysfunction and degeneration. The cause of most PD cases is unknown, although postmortem studies have implicated the involvement of oxidative stress. The identification of familial PD-associated genes offers the opportunity to study mechanisms of PD pathogenesis in model organisms. Here, we show that DJ-1A, a Drosophila homologue of the familial PD-associated gene DJ-1, plays an essential role in oxidative stress response and neuronal maintenance. Inhibition of DJ-1A function through RNA interference (RNAi) results in cellular accumulation of reactive oxygen species, organismal hypersensitivity to oxidative stress, and dysfunction and degeneration of dopaminergic and photoreceptor neurons. To identify other genes that may interact with DJ-1A in regulating cell survival, we performed genetic interaction studies and identified components of the phosphatidylinositol 3-kinase (PI3K)/Akt-signaling pathway as specific modulators of DJ-1A RNAi-induced neurodegeneration. PI3K signaling suppresses DJ-1A RNAi phenotypes at least in part by reducing cellular reactive oxygen species levels. Consistent with the genetic interaction results, we also found reduced phosphorylation of Akt in DJ-1A RNAi animals, indicating an impairment of PI3K/Akt signaling by DJ-1A down-regulation. Together with recent findings in mammalian systems, these results implicate impairments of PI3K/Akt signaling and oxidative stress response in DJ-1-associated disease pathogenesis. We also observed impairment of PI3K/Akt signaling in the fly parkin model of PD, hinting at a common molecular event in the pathogenesis of PD. Manipulation of PI3K/Akt signaling may therefore offer therapeutic benefits for the treatment of PD.
Keywords: Parkinson's disease, PI3K/PTEN/Akt signaling, reactive oxygen species
Parkinson's disease (PD) is the most common movement disorder and the second most common neurodegenerative disease. The movement abnormality in PD arises from deficiency of brain dopamine (DA) contents and the degeneration of dopaminergic neurons in the substantia nigra. The most common forms of PD are sporadic with no known cause. Nevertheless, postmortem studies have identified common features associated with sporadic PD, including defects in mitochondrial complex I function, oxidative damage, and abnormal protein aggregation (1).
The contribution of genetic factors in the pathogenesis of PD, although initially controversial, has been firmly established by recent human genetic studies. At least 10 distinct loci (PARK1 to -11) have been linked to rare familial forms of PD (2). It is anticipated that understanding the molecular lesions associated with these familial PD (FPD) genes will shed light on the pathogenesis of the sporadic forms of the disease. To date, five unequivocal FPD genes have been molecularly cloned. These include α-Synuclein (α-Syn), Parkin, DJ-1, PINK-1, and dardarin. Biochemical and biophysical studies of α-Syn and Parkin have primarily linked dysfunction of these genes to aberrant protein folding and ubiquitin-proteasome dysfunction. Intriguingly, in vivo genetic and in vitro cell culture studies have revealed their connection to mitochondrial dysfunction and oxidative stress, reinforcing the involvement of these processes in PD pathogenesis in general (3).
DJ-1 encodes a conserved protein belonging to the ThiJ/PfpI/DJ-1 superfamily. The exact molecular function of DJ-1 is still unclear. Human DJ-1 was initially discovered as a candidate oncoprotein that could transform cells in cooperation with activated ras (4), and it was later found as a component of an RNA-binding protein complex and was associated with male infertility (4-6). Under oxidative stress conditions, DJ-1 was modified by oxidation, and the modified form associated with mitochondria in cultured cells (7-10). Knocking down DJ-1 expression with small interfering RNA (siRNA) resulted in susceptibility to oxidative stress, endoplasmic reticulum stress, and proteasome inhibition (11). Recent analyses of DJ-1 knockout mice have shed light on the physiological function of DJ-1 in mammals. DJ-1-deficient mice were found to have nigrostriatal dopaminergic dysfunction, motor deficits, and hypersensitivity to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress stimuli (12-14). In mammalian cells, DJ-1 was found to regulate the phosphorylation status of protein kinase B (PKB)/Akt through the tumor suppressor PTEN (15). The relevance of this novel finding of DJ-1 function to PD pathogenesis remains to be explored.
As an alternative approach to understanding the role of DJ-1 dysfunction in PD pathogenesis, we have used Drosophila as a model system. We inhibited the function of a Drosophila DJ-1 homologue (DJ-1A) by transgenic RNA interference (RNAi). DJ-1A RNAi flies show cellular accumulation of reactive oxygen species (ROS), hypersensitivity to oxidative stress, and degeneration of dopaminergic and photoreceptor neurons. Genetic interaction studies with candidate genes and pathways previously implicated in survival signaling led to the identification of genes in the PI3K/Akt-signaling pathway as specific modifiers of DJ-1A-associated cell death phenotype. Consistent with the genetic interaction results, PI3K signaling was found to regulate cellular ROS levels, and we found that DJ-1A down-regulation leads to impairment of PI3K/Akt signaling. Significantly, we found that dysfunction of parkin, another PD-associated gene, also led to impaired PI3K/Akt signaling. Our results implicate oxidative stress and impairment of PI3K/Akt signaling as a general feature of PD pathogenesis and suggest new avenues for therapeutic intervention.
Experimental Procedures
Drosophila Genetics. Fly culture and crosses were performed according to standard procedures and raised at indicated temperatures. All general fly stocks and GAL4 lines were obtained from the Bloomington Drosophila Stock Center. The other fly stocks have been described: UAS-Akt (16); UAS-PI3K p110 and UAS-PI3K p110 DN (17); UAS-PTEN (18); and UAS-tau V337M (19). To generate UAS-dsDJ-1A and UAS-dsDJ-1B transgenics, genomic DNA/cDNA hybrid constructs were generated as described (20). To make UAS-DJ-1A, UAS-DJ-1B, and UAS-hDJ-1 transgenics, corresponding full-length cDNA was cloned into the pUAST vector. Details of the cloning steps are available upon request. Approximately 9 μg of pUAST transgenic construct was mixed with 3 μg of helper plasmid in 20 μl of injection buffer. Standard procedures were followed for embryo injection and recovery of transgenic lines.
Molecular Biology. For RT/PCR analysis, 2nd to 3rd instar larvae from the cross between UAS-dsDJ-1A and Da-GAL4 were used to prepare total RNA by using an RNeasy Kit (Qiagen, Valencia, CA). Details of the quantitative RT/PCR procedure were essentially as described (21). Antibodies against DJ-1A and DJ-1B were elicited in rabbits with recombinant proteins purified from bacteria culture expressing pGEX-6P-1-DJ-1A or pGEX-6P-1-DJ-1B vectors, which contain corresponding full-length cDNA inserts. Western blot analysis using these antibodies was performed as described (21), with each primary antibody used at 1:5,000 dilution. For Western blot analysis of Akt, Da-GAL4/+ and Da-GAL4>DJ-1A RNAi animals were raised at 18°C from the larvae stage to obtain viable Da-GAL4>DJ-1A RNAi adult animals, because these animals die at larvae stage when raised at 25°C. Newly eclosed adult flies were transferred to 29°C to induce stronger RNAi. Da-GAL4>dParkin RNAi flies were raised at 29°C constantly. Fly head extracts were prepared for Western blot analysis with anti-Akt and anti-p-Akt (S505) antibodies (Cell Signaling Technology, Beverly, MA).
Histology and Immunohistochemistry. Sections of paraplast-embedded adult fly heads were prepared and processed as described (21). The sections were incubated in primary antibody overnight at 4°C, and subsequently processed by using the Vectastain Universal Elite ABC Kit (Vector Laboratories). The primary antibody used was anti-tyrosine hydroxylase (TH) polyclonal antibody (Pel-Freez Biologicals, 1:100). For the analysis of adult retina, eye sectioning and staining with toluidine blue was performed as described (22). Between four and five fly heads for each genotype per time point were examined, and each experiment was repeated at least once. The neuronal culture system was established and processed for immunofluorescence staining as described (21). For ROS staining of neuronal culture and adult fly brain, 2,7-dichlorofluorescein diacetate (DCFH-DA) (Molecular Probes) was used following the manufacturer's instructions.
DA Measurement. HPLC analysis of catecholamine levels was performed as described (23, 24). For sample preparation, adult male fly heads were dissected out and homogenized in 0.1 M perchloric acid (generally 50 μl per four or five heads) by using a motorized hand-held tissue grinder. The homogenate was frozen immediately on dry ice and stored at -80°C before HPLC analysis.
Oxidative Stress Assay. For oxidative stress assay, flies were kept in plastic vials with a piece of Kimwipe paper soaked with 1% H2O2 or 100 mM 3-amino-triazole (3-AT) in Schneider's Medium. The vials were kept at 25°C in a shielded box. Fresh H2O2 or 3-AT was added to the paper daily with a syringe. Mortality was recorded every 12 h or at shorter intervals.
Results
Specific Knockdown of Drosophila DJ-1A Expression by Transgenic RNAi. In the sequenced Drosophila genome, there are two previously uncharacterized genes, CG6646 and CG1349 (referred to as DJ-1A and DJ-1B, respectively), which are homologous to human DJ-1. Sequence alignment shows that DJ-1A contains the three conserved amino acids proposed to form a putative catalytic triad in human DJ-1 (25), whereas DJ-1B lacks one of the three amino acids. This finding suggests that DJ-1A may be more closely related to human DJ-1. As a first step toward addressing the function of DJ-1A in Drosophila, we used the transgenic RNAi approach to knockdown DJ-1A expression (20). To confirm that the expression of DJ-1A dsRNA resulted in a down-regulation of endogenous DJ-1A transcripts, we used quantitative RT-PCR to measure DJ-1A mRNA levels after ubiquitous induction of DJ-1A RNAi. A dramatic reduction of DJ-1A mRNA was observed, whereas DJ-1B mRNA was relatively unchanged (Fig. 1A). We next tested the effect of RNAi on endogenous DJ-1 protein expression using DJ-1A- and DJ-1B-specific antibodies. As shown in Fig. 1B, ubiquitous DJ-1A RNAi resulted in a significant reduction of endogenous DJ-1A protein expression on Western blots. In contrast, the level of DJ-1B protein was relatively unaffected. Taken together, these results show that RNAi causes a specific knockdown of DJ-1A RNA and protein expression.
Fig. 1.

Inhibition of DJ-1A expression by RNAi leads to photoreceptor neuron loss and eye degeneration. (A) Quantitative RT/PCR analysis of DJ-1A mRNA level after RNAi. DJ-1B and RP49 serve as controls. (B) Western blot analysis of DJ-1A protein level after RNAi. DJ-1B and actin serve as controls. (C-H) SEM images of GMR-GAL4/+ (C), GMR-GAL4>UAS-DJ-1A-RNAi (D), GMR-GAL4/Df(2R)CX1 (E), and GMR-GAL4>UAS-DJ-1A-RNAi/Df(2R)CX1 (F) eyes. Df(2R)CX1 is a chromosomal deficiency that deletes DJ-1A. G and H are magnified views of C and D, respectively. (I and J) Staining of photoreceptor neurons in GMR-GAL4/+ (I), and GMR-GAL4>UAS-DJ-1A-RNAi (J) eyes. Arrows in J mark ommatidia with photoreceptor loss. (K-N) Rescue of DJ-1A RNAi phenotypes by overexpressing DJ-1A or human DJ-1. All flies are homozygous for a recombinant GMR-GAL4;UAS-DJ-1A-RNAi chromosome and thus have a stronger phenotype than the one shown in B. In addition, the flies coexpress UAS-GFP (L), UAS-DJ-1A (M), UAS-hDJ-1 (N), or no other transgene (K).
Targeted Inhibition of DJ-1A in the Eye Results in Photoreceptor Loss. We next analyzed the physiological consequence of inhibiting DJ-1A function. Ubiquitous expression of DJ-1A dsRNA with actin-GAL4 or daughterless (Da)-GAL4 resulted in larval lethality. This finding suggests that DJ-1A is an essential gene in Drosophila. To circumvent the lethality problem, we used well characterized GAL4 drivers to inhibit DJ-1A expression in specific tissues and cell types and at different stages. Induction of DJ-1A RNAi in the developing eye using GMR-GAL4 driver produced a rough eye phenotype (Fig. 1 D and H). GMR-GAL4 directs gene expression in postmitotic cells posterior to the morphogenetic furrow and a small group of premitotic cells in the developing eye. Staining of eye sections revealed loss of photoreceptor neurons in some ommatidia (Fig. 1J), indicating that the rough eye phenotype is caused at least in part by photoreceptor cell loss. This RNAi effect is dosage-dependent, because increasing the copy number of GAL4 and UAS transgenes caused a more severe degeneration of the eye (Fig. 1K). Several lines of evidence suggest that this eye phenotype is caused by specific inhibition of DJ-1A. First, overexpression of white or dParkin control dsRNAs using the same GAL4 driver had no effect on eye morphology (data not shown), suggesting that the eye phenotype was not due to a nonspecific effect of dsRNA expression. Second, in a DJ-1A heterozygous genetic background, the eye phenotype was significantly enhanced (Fig. 1F), consistent with the RNAi effect being dosage dependent. Finally, we could rescue the RNAi phenotype with increased expression of DJ-1A. Given that the RNAi effect is dosage-dependent, we reasoned that, by raising the basal level of DJ-1A transcripts, the RNAi effect would be dampened. Indeed, coexpression of UAS-DJ-1A transgenes partially suppressed the eye degeneration phenotype induced by strong RNAi (Fig. 1, compare M with K). Coexpression of a human DJ-1 transgene could also partially rescue (Fig. 1, compare N with K), suggesting that human DJ-1 and fly DJ-1A may possess similar properties. In contrast, coexpression of a GFP transgene had no effect (Fig. 1, compare L with K), suggesting that the rescue is not due to titration of GAL4 by added expression of a UAS-transgene. We conclude that the abnormal eye phenotype is specifically caused by inhibition of DJ-1A expression.
Inhibition of DJ-1A in Dopaminergic Neurons Leads to Decreases of TH+ Neuron Number and Brain DA Content. We next analyzed the effects of inhibiting DJ-1A function in dopaminergic neurons by inducing RNAi with the Ddc-GAL4 driver. We focused on the dopaminergic neurons in the dorsomedial clusters (DMC), which are known to be susceptible under disease conditions (26). Immunostaining of paraffin brain sections of Ddc-GAL4>DJ-1A RNAi flies revealed an age-dependent reduction in the number of TH+ neurons in the DMC. In 1-day-old flies, a normal complement of TH+ neurons (≈18) was present (Fig. 2C), but, in 35-day and older flies, only 10-12 of these neurons could be detected immunochemically (Fig. 2 B and C). Control flies showed no significant change in the number of these neurons during aging (Fig. 2 A and C). Induction of DJ-1A RNAi with another dopaminergic GAL4 driver, TH-GAL4, or the pan-neuronal elav-GAL4 driver also resulted in reduction of TH+ neurons in the DM clusters (Fig. 2C).
Fig. 2.

Dopaminergic defects in DJ-1A RNAi flies. (A and B) TH immunostaining of DMC dopaminergic neurons in 35-day-old control Ddc-GAL4/+ (A) and Ddc-GAL4>DJ-1A RNAi (B) male flies. Sections containing most of the DMC dopaminergic neurons are shown. (C) Quantification of TH+ neurons in the DMC of control flies and DJ-1A RNAi flies directed with TH-GAL4, Ddc-GAL4, or elav-GAL4 drivers. The difference in cell count between 1-day-old and 35-day-old DJ-1A RNAi flies is significant. *, P < 0.01 in Student's t test. (D) Quantification of head DA levels in TH-GAL4/+ and TH-GAL4>DJ-1A RNAi flies. *, P < 0.01 in Student's t test.
To further confirm that loss of DJ-1A leads to dopaminergic dysfunction, we measured brain DA levels using head extracts prepared from control and DJ-1A RNAi flies. In newly eclosed flies, DA content was comparable between control and RNAi flies (Fig. 2D). However, 1 day after eclosion, DJ-1A RNAi flies showed significantly reduced DA level than control flies. At 4, 7, and 10 days of age, control and DJ-1A RNAi flies both showed age-dependent decline of DA, but DJ-1A RNAi flies consistently exhibited more reduction than the controls (Fig. 2D). Because a normal complement of TH+ dopaminergic neurons was present in 1-day old flies, the reduction of brain DA content at this early stage could not be attributed to neuronal loss. This result suggests that, in addition to promoting dopaminergic neuron survival, Drosophila DJ-1A may play an early role in regulating brain DA levels.
DJ-1A RNAi Flies Show Elevated ROS Accumulation and Hypersensitivity to Oxidative Stress. We further characterized the DJ-1A RNAi animals to learn DJ-1A function in vivo. Human DJ-1 was previously found to respond to oxidative stress (8). This finding prompted us to analyze the response of DJ-1A RNAi flies under oxidative conditions. We used the elav-GAL4 driver to systematically induce DJ-1A RNAi in postmitotic neurons of transgenic flies and examined the response of these flies to treatment with exogenous H2O2. When treated with 1% H2O2, the time to reach 50% mortality was shortened by 27% in DJ-1A RNAi flies than control flies (Fig. 3A). This finding suggests that neuronal DJ-1A is important in fending off H2O2-induced lethality. To further confirm the sensitivity of DJ-1A RNAi flies to intracellular H2O2 levels, we treated DJ-1A RNAi flies with 3-AT, a known inhibitor of catalase, which converts H2O2 to H2O. DJ-1A RNAi flies were found to be more sensitive to 3-AT treatment than the control flies (Fig. 3B). To test whether DJ-1A may be actively involved in ROS scavenging, we also overexpressed DJ-1A ubiquitously with the Da-GAL4 driver and observed that DJ-1A overexpression was sufficient to confer resistance against 3-AT treatment (Fig. 6, which is published as supporting information on the PNAS web site).
Fig. 3.

DJ-1A RNAi leads to ROS accumulation and hypersensitivity to oxidative stress. (A and B) Comparison of survival curves of elav-GAL4/+ flies with elav-GAL4>DJ-1A RNAi flies that are treated with 1% H2O2 (A) or 100 mM 3-AT (B). (C and D) DCFH-DA staining of cultured Da-GAL4/+ (C) and Da-GAL4>DJ-1A RNAi (D) neurons. (Upper) Fluorescent DCFH-DA staining in green. (Lower) Black and white images of the neuronal culture being analyzed. (E) Recombinant DJ-1A protein exhibits detectable in vitro H2O2 scavenging activity, whereas the control proteins BSA, GST, and PAR-1 have no such activity.
If DJ-1A normally plays a critical role in sensing cellular ROS levels and eliciting protective responses to remove these toxic agents, one would predict that inhibiting DJ-1A function would lead to elevated levels of endogenous ROS. We tested this possibility by staining cultured neurons with DCFH-DA, which is an indicator of hydroxyl free radicals. Compared with control neuronal culture, which only showed weak ROS staining in a small percentage of neurons, DJ-1A RNAi neuronal culture had more neurons stained by this dye, and the staining intensity was much higher (Fig. 3, compare D with C).
Human DJ-1 protein was previously shown to be able to eliminate H2O2 in vitro by oxidizing itself at specific Cys residues (10, 27). To test whether Drosophila DJ-1A has similar H2O2 scavenging activity, we incubated bacterially expressed recombinant DJ-1A protein with H2O2 in test tubes and measured the conversion of H2O2. DJ-1A protein was found to have a specific activity in scavenging H2O2, whereas a control BSA protein has no such activity (Fig. 3E). This activity may not be simply attributed to nonspecific reaction of H2O2 with Cys residues, because the amino acid composition of BSA has a higher percentage of Cys residues than DJ-1A. Instead, this result indicates that DJ-1A may possess a specific activity in eliminating H2O2. In addition to BSA, which is normally resides in an extracellular environment, two intracellular proteins, GST and the Ser/Thr protein kinase PAR-1, also showed no H2O2 scavenging activity. It should be noted that the H2O2 scavenging activity of DJ-1A was two orders of magnitude lower than that of catalase in the same assay (20 units/mg vs. 2,300 units/mg), suggesting that degrading H2O2 may not be the main function of DJ-1A.
Modulation of DJ-1A RNAi-Induced Cell Death by the PI3K/Akt-Signaling Pathway. In an effort to understand how DJ-1A dysfunction leads to neuronal death, we tested possible genetic modification of DJ-1A RNAi-induced eye phenotypes by candidate genes and signaling pathways previously implicated in cell survival regulation. To see the genetic interaction more clearly, we used the weak RNAi phenotype induced by one copy each of the GMR-GAL4 and DJ-1A RNAi transgenes to score for enhancement; the stronger RNAi phenotype induced by two copies of the GMR-GAL4 and DJ-1A RNAi transgenes was used to score for suppression whenever possible.
The EGF receptor (EGFR)/Ras1/mitogen-activated protein kinase (MAPK) signaling pathway has previously been shown to directly target the Drosophila proapoptotic gene head involution defective (hid) in the eye through MAPK-mediated phosphorylation and inactivation of HID (28). By using loss-of-function and gain-of-function alleles of rolled (MAPK) and loss-of-function alleles of hid, we did not detect clear genetic interaction with DJ-1A RNAi flies (data not shown). Similarly, we could not detect clear genetic interaction between DJ-1A RNAi and loss-of-function or overexpression alleles of genes in the JNK pathway, which has also been shown to induced cell death in the eye when activated (29).
In contrast, a clear genetic interaction was detected between DJ-1A and the Drosophila PI3K/Akt pathway genes. A dramatic enhancement of DJ-1A RNAi-induced eye degeneration was observed when PTEN was coexpressed with the DJ-1A RNAi transgene (Fig. 4D). The resulting fly eyes were dramatically reduced in size, with collapsed and fused ommatidia and necrotic spots, which were not present in DJ-1A RNAi only flies. Staining of photoreceptor neurons revealed a near complete loss of photoreceptor neurons in PTEN coexpression fly eyes (data not shown). Overexpression of this UAS-PTEN transgene alone with GMR-GAL4 driver had little effect on the regular organization of the ommatidia and photoreceptor number per ommatidium, although the overall size of the eye was moderately reduced (Fig. 4L). When we tested with a mutant form of human PTEN that contains an inactivating C124S mutation (18), no effect on DJ-1A RNAi phenotype was observed (data not shown). Similar to the effect of Drosophila PTEN, an enhancement of the DJ-1A RNAi phenotype was observed when a dominant-negative (DN) form of PI3K catalytic subunit Dp110 (PI3K DN) was coexpressed (17) (Fig. 4C), although expression of this PI3K DN transgene alone had little effect on eye morphology (Fig. 4K).
Fig. 4.

Modification of DJ-1A RNAi phenotypes by altered expression of genes in the PI3K/Akt pathway. (A-D) SEM eye images of DJ-1A RNAi flies coexpressing UAS-Akt (A), UAS-PI3K Dp110 (B), UAS-PI3K Dp110DN (C), or UAS-PTEN (D). (E-H) SEM eye images of human tauV337M transgenic flies coexpressing UAS-GFP (E), UAS-PI3K Dp110 (F), UAS-PI3K Dp110DN (G), or UAS-PTEN (H). (I-L) SEM eye images of flies expressing UAS-Akt (I), UAS-PI3K Dp110 (J), UAS-PI3K Dp110DN (K), or UAS-PTEN (L) transgenes alone. GMR-GAL4 was used to direct UAS transgene expression in all panels.
A clear suppression of DJ-1A RNAi phenotype was observed when the wild-type form of PI3K catalytic subunit Dp110 was coexpressed. The eyes were restored to normal size, and the organization of the ommatidia was significantly improved (Fig. 4B). Overexpression of a UAS-Akt transgene had similar effect as PI3K in suppressing DJ-1A RNAi-induced toxicity in the eye (Fig. 4A), consistent with Akt/PKB being a key downstream effecter component in the PI3K-signaling pathway (16).
Given the known pleiotropic function of the PI3K-signaling pathway in regulating cell size and cell number in Drosophila and its potential role in regulating cell survival, we next tested the effect of manipulating PI3K pathway gene activity on an eye degeneration phenotype caused by a different mechanism. Overexpression of human tau in the fly eye also led to a reduction in eye size and loss of photoreceptor neurons (19, 22). In contrast to the DJ-1A RNAi situation, coexpression of wild-type PI3K, PI3K DN, or PTEN showed little effect on human tau-induced toxicity in the eye (Fig. 4, compare F, G, and H, respectively, with E). The genetic interaction between DJ-1A and PI3K pathway genes in the eye thus seems to be rather specific.
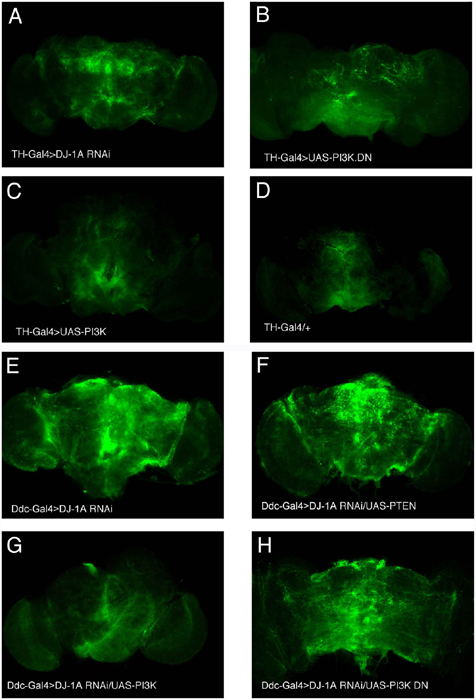
We next tested the effects of modulating PI3K signaling on the dopaminergic degeneration phenotype induced by DJ-1A RNAi. Coexpression of PI3K completely suppressed the reduction of TH+ DA neuron phenotype induced by DJ-1A RNAi. The number of DA neurons in the DMCs was maintained at the wild-type level in all of the transgenic flies and at all ages examined (Fig. 5A), indicating that coexpression of PI3K blocked DJ-1A RNAi-induced age-dependent dopaminergic degeneration. Conversely, coexpression of PI3K DN showed a statistically significant enhancement of DJ-1A RNAi toxicity in dopaminergic neurons (Fig. 5A).
Fig. 5.

Modification of DJ-1A RNAi-induced dopaminergic phenotype by altered expression of PI3K/Akt pathway genes and Western blot analysis showing reduced Akt phosphorylation after DJ-1A or Parkin down-regulation. (A) Quantification of TH+ DA neurons in the DMC of Ddc-GAL4/+ control flies, DJ-1A RNAi flies, PI3K or PI3K DN single overexpression flies, and DJ-1A RNAi flies coexpressing PI3K or PI3K DN transgenes. *, P < 0.01 in Student's t test. Ddc-GAL4 was used to direct transgene expression. (B and C) Western blot analysis of fly head extracts prepared from Da-GAL4/+ and Da-GAL4>DJ-1A RNAi (B) or Da-GAL4/+ and Da-GAL4>dParkin RNAi (C) genotyped flies. Blots were probed with anti-phospho-Akt and anti-Akt antibodies, respectively.
We next examined the effect of PI3K signaling on DJ-1A RNAi-induced ROS accumulation. We found that, in adult fly brain, induction of DJ-1A RNAi within dopaminergic neurons led to an elevation of ROS levels, consistent with neuronal culture studies described earlier (Fig. 7A, which is published as supporting information on the PNAS web site). Inhibition of PI3K signaling in these neurons by means of overexpression of PI3K DN also led to elevation of ROS levels (Fig. 7B), whereas flies overexpressing wild-type PI3K showed basal ROS levels (Fig. 7C). Strikingly, in DJ-1A RNAi flies coexpressing PI3K, cellular ROS levels are reduced to baseline levels as in wild-type controls (Fig. 7G). Together, these results indicate that PI3K signaling specifically suppresses DJ-1A RNAi-induced neurotoxicity and that this suppression is correlated with a reduction of cellular ROS levels.
DJ-1A RNAi Flies and Parkin Mutant Flies Exhibit Impaired PI3K/Akt Signaling. The fact that increased PI3K/Akt signaling specifically suppressed DJ-1A RNAi-induced cell death suggests that the cell death in DJ-1A RNAi animals may be caused by a reduction of PI3K/Akt signaling. To test this possibility, we examined the phosphorylation status of Akt, an indicator of PI3K/Akt signaling, in DJ-1A RNAi animals. Head extracts from Da-GAL4/+ and Da-GAL4>DJ-1A RNAi animals were analyzed by Western blot analysis by using anti-phospho-Akt and anti-Akt antibodies. As shown in Fig. 5B, although the level of total Akt protein was comparable between control and DJ-1A RNAi fly heads, the amount of phospho-Akt was significantly reduced in DJ-1A RNAi animals. This result indicates that DJ-1A down-regulation leads to hypophosphorylation of Akt and impairment of PI3K/Akt signaling in the fly brain. To test whether impairment of PI3/Akt signaling is a general feature of PD models, we analyzed the Drosophila parkin model. As shown in Fig. 5C, inhibition of Parkin function also led to a reduction of phospho-Akt levels. These results implicate reduced PI3K/Akt signaling as a common molecular event in the pathogenic cascade of PD.
Discussion
Loss-of-function mutations in human DJ-1 are linked to familial Parkinson's disease. The exact molecular function of DJ-1 that is relevant to disease pathogenesis is not well understood. Our results suggest that Drosophila DJ-1A plays an important role in cellular ROS homeostasis and protection against oxidative stress. This conclusion is consistent with previous studies in mammalian cell culture and DJ-1 knockout mice (13, 27, 30). Human DJ-1 was found to have H2O2-scavenging activity in vitro. (10) (27). Our analysis of Drosophila DJ-1A protein supported this notion. However, the H2O2-converting activity of DJ-1A is rather low compared with catalase, suggesting that the main molecular function of DJ-1 may not be limited to degrading H2O2. It is possible that the ability to react with H2O2 by means of oxidation-sensitive Cys residues may allow DJ-1 to act as a “sensor” of cellular ROS levels, and the oxidized DJ-1 may subsequently acquire a new function to defend against ROS-induced cellular damages. This result would be analogous to the switch of two yeast peroxiredoxins from peroxidase to molecular chaperone under oxidative stress (31). The recent description of human DJ-1 gaining molecular chaperone activity in vitro under oxidative conditions is consistent with this model (32).
To understand the cellular processes that mediated DJ-1A dysfunction-induced cell death, we performed genetic interaction studies with genes and signaling pathways that are involved in cell survival and identified components of the PI3K/PTEN/Akt pathway as modulators of DJ-1 RNAi-induced cell death phenotype. Increase of PI3K/Akt-signaling capacity showed suppression, whereas decreased PI3K/Akt signaling enhanced DJ-1A RNAi phenotypes. The effects of modulating PI3K/Akt signaling on DJ-1A RNAi-induced toxicity hold true in both photoreceptor neurons in the retina and dopaminergic neurons in the central brain, suggesting that the connection between DJ-1 and PI3K/Akt signaling is a general phenomenon. The finding that DJ-1A RNAi animals showed decreased phosphorylation of Akt indicate that DJ-1 is a regulator of PI3K/Akt signaling. A recent study by Kim et al. (15) identified DJ-1A as a suppressor of PTEN function in the fly eye, and the authors further extended this finding to mammalian cells and showed that DJ-1 knockdown by small interfering RNA results in decreased phosphorylation of PKB/Akt, whereas DJ-1 overexpression leads to PKB/Akt hyperphosphorylation and increased cell survival. This finding led to the proposal that DJ-1 acts as a novel regulator of PTEN. Our genetic and biochemical studies are consistent with this notion. It is not clear at this point how DJ-1A and the PI3K/PTEN/Akt-signaling pathway may interact. It is possible that the function of DJ-1 in regulating cellular ROS homeostasis or as a redox-sensitive molecular chaperone may facilitate PI3K/PTEN/Akt signaling, because many signal transduction pathways are known to be sensitive to cellular ROS levels or require chaperone activities (33, 34), and modulation of PTEN activity by ROS has been reported before (35). Alternatively, the genetic interaction between DJ-1A and PI3K-signaling pathway may be mediated by a direct role of PI3K signaling in cellular defense against ROS accumulation and related damages. Our data are consistent with both possibilities. Given that hyperactivation of DJ-1 could be oncogenic, whereas its deficiency leads to neuronal dysfunction and degeneration, future studies aimed at understanding the mechanisms by which DJ-1 and PI3K/PTEN/Akt pathway interact will have far-reaching implications for understanding disease mechanisms and developing therapeutic strategies.
Oxidative stress and mitochondrial dysfunction are being increasingly recognized as common pathological features of neurodegenerative diseases including PD and Alzheimer's disease (2, 36). Previous genetic studies in Drosophila and mice have implicated Parkin, an E3 ubiquitin ligase associated with autosomal recessive juvenile parkinsonism, in these processes (37-40). In flies and mice, parkin mutants show defects in mitochondrial function and oxidative stress response. Like DJ-1, loss of Drosophila Parkin function also affects the viability of mutant animals. This result contrasts with the situation in mammals where loss of DJ-1 or Parkin is nonlethal. The differential effects on the viability of humans and flies are probably caused by fundamental differences in the anti-oxidant defense systems of these two species (41). The exact cellular mechanism by which Parkin dysfunction leads to susceptibility to oxidative stress and cell death remains to be established. Our finding that, similar to DJ-1A inactivation, inhibition of parkin also leads to impairment of PI3K/Akt signaling implicates these two genes in a common pathway that promotes neuronal survival. We speculate that impairment of PI3K/Akt signaling may be a common feature of familial as well as sporadic PD cases and that manipulation of this signaling pathway may provide a rational strategy for the therapeutic intervention of PD.
Supplementary Material
Acknowledgments
We are grateful to Drs. Morris Birnbaum (University of Pennsylvania, Philadelphia), Mel Feany (Harvard University, Boston), Ernst Hafen (University of Zurich, Zurich), Raj Sohal (University of Southern California, Los Angeles), Bertrand Mollereau (The Rockefeller University, New York), Hermann Steller (The Rockefeller University), Tian Xu (Yale University, New Haven, CT), and the Bloomington Drosophila Stock Center for fly stocks; Dr. Kazuaki Yoshikawa for his generous support; the Center for Research and Education of Osaka University School of Medicine for help with SEM and eye sectioning; Dr. Su Guo for reading the manuscripts; Dr. Ting-ting Huang for help with H2O2 assay; and Dr. Kyung-Tai Min for communicating unpublished results. Special thanks go to Jennifer Quach and Yali Zhang for excellent technical support and members of the B.L. laboratory for discussions. This work was supported by the McKnight, Beckman, and Sloan Foundations (to B.L.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DA, dopamine; DMC, dorsomedial cluster; PD, Parkinson's disease; RNAi, RNA interference; ROS, reactive oxygen species; TH, tyrosine hydroxylase; PKB, protein kinase B; PI3K, phosphatidylinositol 3-kinase; Da, daughterless; 3-AT, 3-amino-triazole; DCFH-DA, 2,7-dichlorofluorescein diacetate; DN, dominant-negative.
References
- 1.Dunnett, S. B. & Bjorklund, A. (1999) Nature 399, A32-A39. [DOI] [PubMed] [Google Scholar]
- 2.Dawson, T. M. & Dawson, V. L. (2003) Science 302, 819-822. [DOI] [PubMed] [Google Scholar]
- 3.Shen, J. & Cookson, M. R. (2004) Neuron 43, 301-304. [DOI] [PubMed] [Google Scholar]
- 4.Nagakubo, D., Taira, T., Kitaura, H., Ikeda, M., Tamai, K., Iguchi-Ariga, S. M. & Ariga, H. (1997) Biochem. Biophys. Res. Commun. 231, 509-513. [DOI] [PubMed] [Google Scholar]
- 5.Hod, Y., Pentyala, S. N., Whyard, T. C. & El-Maghrabi, M. R. (1999) J. Cell Biochem. 72, 435-444. [PubMed] [Google Scholar]
- 6.Takahashi, K., Taira, T., Niki, T., Seino, C., Iguchi-Ariga, S. M. & Ariga, H. (2001) J. Biol. Chem. 276, 37556-37563. [DOI] [PubMed] [Google Scholar]
- 7.Canet-Aviles, R. M., Wilson, M. A., Miller, D. W., Ahmad, R., McLendon, C., Bandyopadhyay, S., Baptista, M. J., Ringe, D., Petsko, G. A. & Cookson, M. R. (2004) Proc. Natl. Acad. Sci. USA 101, 9103-9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitsumoto, A. & Nakagawa, Y. (2001) Free Radical Res. 35, 885-893. [DOI] [PubMed] [Google Scholar]
- 9.Mitsumoto, A., Nakagawa, Y., Takeuchi, A., Okawa, K., Iwamatsu, A. & Takanezawa, Y. (2001) Free Radical Res. 35, 301-310. [DOI] [PubMed] [Google Scholar]
- 10.Kinumi, T., Kimata, J., Taira, T., Ariga, H. & Niki, E. (2004) Biochem. Biophys. Res. Commun. 317, 722-728. [DOI] [PubMed] [Google Scholar]
- 11.Yokota, T., Sugawara, K., Ito, K., Takahashi, R., Ariga, H. & Mizusawa, H. (2003) Biochem. Biophys. Res. Commun. 312, 1342-1348. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., Tong, Y., Martella, G., Tscherter, A., Martins, A., et al. (2005) Neuron 45, 489-496. [DOI] [PubMed] [Google Scholar]
- 13.Kim, R. H., Smith, P. D., Aleyasin, H., Hayley, S., Mount, M. P., Pownall, S., Wakeham, A., You-Ten, A. J., Kalia, S. K., Horne, P., et al. (2005) Proc. Natl. Acad. Sci. USA 102, 5215-5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, L., Cagniard, B., Mathews, T., Jones, S., Koh, H. C., Ding, Y., Carvey, P. M., Ling, Z., Kang, U. J. & Zhuang, X. (2005) J. Biol. Chem. 280, 21418-21426. [DOI] [PubMed] [Google Scholar]
- 15.Kim, R. H., Peters, M., Jang, Y., Shi, W., Pintilie, M., Fletcher, G. C., DeLuca, C., Liepa, J., Zhou, L., Snow, B., et al. (2005) Cancer Cell 7, 263-273. [DOI] [PubMed] [Google Scholar]
- 16.Verdu, J., Buratovich, M. A., Wilder, E. L. & Birnbaum, M. J. (1999) Nat. Cell Biol. 1, 500-506. [DOI] [PubMed] [Google Scholar]
- 17.Leevers, S. J., Weinkove, D., MacDougall, L. K., Hafen, E. & Waterfield, M. D. (1996) EMBO J. 15, 6584-6594. [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, H., Potter, C. J., Tao, W., Li, D. M., Brogiolo, W., Hafen, E., Sun, H. & Xu, T. (1999) Development (Cambridge, U.K.) 126, 5365-5372. [DOI] [PubMed] [Google Scholar]
- 19.Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M. & Feany, M. B. (2001) Science 293, 711-714. [DOI] [PubMed] [Google Scholar]
- 20.Kalidas, S. & Smith, D. P. (2002) Neuron 33, 177-184. [DOI] [PubMed] [Google Scholar]
- 21.Yang, Y., Nishimura, I., Imai, Y., Takahashi, R. & Lu, B. (2003) Neuron 37, 911-924. [DOI] [PubMed] [Google Scholar]
- 22.Nishimura, I., Yang, Y. & Lu, B. (2004) Cell 116, 671-682. [DOI] [PubMed] [Google Scholar]
- 23.Ito, S., Kato, T. & Fujita, K. (1988) Biochem. Pharmacol. 37, 1707-1710. [DOI] [PubMed] [Google Scholar]
- 24.Beal, M. F., Kowall, N. W., Swartz, K. J. & Ferrante, R. J. (1990) Neurosci. Lett. 108, 36-42. [DOI] [PubMed] [Google Scholar]
- 25.Tao, X. & Tong, L. (2003) J. Biol. Chem. 278, 31372-31379. [DOI] [PubMed] [Google Scholar]
- 26.Feany, M. B. & Bender, W. W. (2000) Nature 404, 394-398. [DOI] [PubMed] [Google Scholar]
- 27.Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S. M., Takahashi, K. & Ariga, H. (2004) EMBO Rep. 5, 213-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergmann, A., Agapite, J., McCall, K. & Steller, H. (1998) Cell 95, 331-341. [DOI] [PubMed] [Google Scholar]
- 29.Kuranaga, E., Kanuka, H., Igaki, T., Sawamoto, K., Ichijo, H., Okano, H. & Miura, M. (2002) Nat. Cell Biol. 4, 705-710. [DOI] [PubMed] [Google Scholar]
- 30.Martinat, C., Shendelman, S., Jonason, A., Leete, T., Beal, M. F., Yang, L., Floss, T. & Abeliovich, A. (2004) PloS Biol. 2, e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang, H. H., Lee, K. O., Chi, Y. H., Jung, B. G., Park, S. K., Park, J. H., Lee, J. R., Lee, S. S., Moon, J. C., Yun, J. W., et al. (2004) Cell 117, 625-635. [DOI] [PubMed] [Google Scholar]
- 32.Shendelman, S., Jonason, A., Martinat, C., Leete, T. & Abeliovich, A. (2004) PLoS Biol. 2, e362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibata, Y., Branicky, R., Landaverde, I. O. & Hekimi, S. (2003) Science 302, 1779-1782. [DOI] [PubMed] [Google Scholar]
- 34.Morey, M., Serras, F., Baguna, J., Hafen, E. & Corominas, M. (2001) Dev. Biol. 238, 145-156. [DOI] [PubMed] [Google Scholar]
- 35.Leslie, N. R., Bennett, D., Lindsay, Y. E., Stewart, H., Gray, A. & Downes, C. P. (2003) EMBO J. 22, 5501-5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albers, D. S. & Beal, M. F. (2000) J. Neural Transm. 59, Suppl., 133-154. [DOI] [PubMed] [Google Scholar]
- 37.Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y. & Shimizu, N. (1998) Nature 392, 605-608. [DOI] [PubMed] [Google Scholar]
- 38.Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B. & Pallanck, L. J. (2003) Proc. Natl. Acad. Sci. USA 100, 4078-4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pesah, Y., Pham, T., Burgess, H., Middlebrooks, B., Verstreken, P., Zhou, Y., Harding, M., Bellen, H. & Mardon, G. (2004) Development (Cambridge, U.K.) 131, 2183-2194. [DOI] [PubMed] [Google Scholar]
- 40.Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Klose, J. & Shen, J. (2004) J. Biol. Chem. 279, 18614-18622. [DOI] [PubMed] [Google Scholar]
- 41.Kanzok, S. M., Fechner, A., Bauer, H., Ulschmid, J. K., Muller, H. M., Botella-Munoz, J., Schneuwly, S., Schirmer, R. & Becker, K. (2001) Science 291, 643-646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}