

Abstract
Molecular wires comprising a Ru(II)- or Re(I)-complex head group, an aromatic tail group, and an alkane linker reversibly inhibit the activity of the copper amine oxidase from Arthrobacter globiformis (AGAO), with Ki values between 6 μM and 37 nM. In the crystal structure of a Ru(II)-wire:AGAO conjugate, the wire occupies the AGAO active-site substrate access channel, the trihydroxyphenylalanine quinone cofactor is ordered in the “off-Cu” position with its reactive carbonyl oriented toward the inhibitor, and the “gate” residue, Tyr-296, is in the “open” position. Head groups, tail-group substituents, and linker lengths all influence wire-binding interactions with the enzyme.
Keywords: diimine, topaquinone, metalloenzyme, active site
Copper and quinone containing amine oxidases (EC 1.4.3.6) catalyze the oxidative deamination of primary amines to the corresponding aldehydes with concomitant generation of ammonia and hydrogen peroxide.
 |
Each subunit of these homodimeric enzymes contains a deeply buried active site comprised of a single type II (“non-blue,” square-pyramidal) copper atom and an organic cofactor, 2,4,5-trihydroxyphenylalanine quinone (topaquinone or TPQ) (1, 2). The finding that the human vascular adhesion protein (HVAP-1) is a copper amine oxidase (CuAO) has heightened interest in the mechanism and inhibition of these enzymes (3). With the potential for therapeutic applications, research has focused on elucidation of the factors that govern inhibitor sensitivity and selectivity.
We are exploring the potential of channel-blocking metal-diimine wire complexes to function as highly selective inhibitors of CuAOs. We chose phenylethylamine oxidase from Arthrobacter globiformis for initial study, owing to its ease of expression and purification as a C-terminal Strep-tag II fusion protein (4). Our choice of metal-diimine wires was based on the results of extensive investigations of their conjugates with cytochrome P450cam, which have revealed structural features of conformational states that likely are involved in steps of the catalytic cycle of the enzyme (5-9). Similar molecular wires have been used in attempts to measure the reduction potentials of deeply buried protein cofactors; indeed, in experiments of relevance here, a diethylaniline-tipped triphenylene wire coupled to a gold electrode allowed electrochemical characterization of Arthrobacter globiformis amine oxidase (AGAO) cofactor TPQ (10). Binding of the wire in the active-site channel was not established independently but could be inferred from the efficiency of electron tunneling from the electrode to the buried cofactor.
We have designed and synthesized a series of highly potent channel-blocking inhibitors of AGAO. The crystal structure of a Ru-wire:AGAO conjugate clearly demonstrates that the wire resides in the active-site channel; it also reveals key aspects of active-site topology and conformational mobility. Furthermore, variations in binding in response to changes in wire sensitizer, substrate, and linker compositions have led to particularly powerful AGAO inhibitors.
Materials and Methods
Syntheses. The synthesis of the [Ru(II)(bpy)2(phen)] complexes is shown in Scheme 1, where bpy stands for 2,2′-bipyridine and phen stands for 1,10-phenanthroline. The details for a representative complex, [Ru(II)(bpy)2(phen)-C4-DMA] (5a), where DMA stands for dimethylaniline, and its precursors are given below. The syntheses of the other compounds reported in this work and their precursors may be found in Supporting Text, which is published as supporting information on the PNAS web site. All syntheses were conducted under a nitrogen atmosphere by using standard Schlenk techniques and degassed solvents. Dimethylformamide and tetrahydrofuran were dried before use by passing through activated silica columns. All other solvents were obtained in omnisolv grade from EM Biosciences (San Diego) and used as received. We prepared 4-methyl-4′-chloromethyl-bpy and 2,2′,3,3′,4,5,5′,6,6′-nonafluoro-4′-(phenylethynyl)-1,1′-biphenyl (11) according to procedures described in refs. 11 and 12. Cesium carbonate was dried by heating at 250°C under vacuum just before use. All other reagents were used as received from Aldrich unless otherwise noted. All product work-up procedures were performed in air. All metal complexes decompose over several days on standing in air/light. They were stored in the dark under argon at -5°C until just before use.
Scheme 1.

Spectra. NMR spectra were obtained by using a Varian Mercury 300 spectrometer. Mass spectra were obtained by using a LCQ quadropole ion trap mass spectrometer (Finnigan-MAT, San Jose, CA). Electronic spectra were obtained by using a Hewlett-Packard 8452 diode array spectrometer. Steady-state fluorescence spectra were collected by using a K2 fluorimeter (ISSS, Champaign, IL) with excitation at 450 nm for Ru(II) complexes and 355 nm for 10.
m-Br-(CH2)3-OC6H4NMe2 (3a). A stirred mixture of 1.0 g (7.3 mmol) of m-dimethylaminophenol, 6.5 g (20 mmol) of Cs2CO3, and 5.0 ml (10 g, 50 mmol) of 1,3-dibromopropane in 20 ml of dimethylformamide was heated at 45°C for 3 h. Solvent and excess 1,3-dibromopropane were removed under vacuum. The product was separated from the residue by using silica gel chromatography with CH2Cl2/hexanes (25/75 vol/vol) as the eluent. Removal of solvent under vacuum gave the product as a colorless solid. Yield: 1.6 g (85%). 1H NMR (300 MHz, CD2Cl2): δ 2.29 (tt, J = 6.6, 5.7 Hz, 2H), 2.92 (s, 6H), 3.62 (t, J = 6.6 Hz, 2H) 4.08 (t, J = 5.7 Hz, 2H), 6.21-6.27 (m, 2H), 6.35 (m, 1H), 7.11 (m, 1H). ESI-MS (MeOH) m/z: 258 (MH+).
4-(m-(CH2)4-OC6H4NMe2)-phen (4a). A solution of 1.2 g (6.0 mmol) of 4-methylphenanthroline in 25 ml of tetrahydrofuran was treated with dropwise addition of 3.0 ml of 2.0 M Li(NiPr2) in tetrahydrofuran/heptane/ethylbenzene. The resulting black solution was allowed to stir for 30 min and then was added dropwise to 1.54 g (6.0 mmol) of 3a over 20 min. The reaction was allowed to proceed for 12 h and then quenched by addition of 5 ml of EtOH and 3 ml of H2O. Solvent was removed under vacuum, and the residue was chromatographed on silica by using 10/90 MeOH/CH2Cl2 eluant. In some cases, a second silica column (10/90 MeOH/CH2Cl2) was used to further purify the product. Fractions containing pure product were pooled to give the crude product as a waxy solid. The product was extracted with 25 ml of Et2O to remove impurities and give the product as a white powder. Yield: 0.75 g (33%). 1H NMR (300 MHz, CD2Cl2): δ 1.9-2.1 (m, 4H), 2.90 (s, 6H), 3.3 (m, 2H, partially obscured by CD2HOD signal), 4.05 (m, 2H), 6.26 (m, 2H), 6.36 (m, 1H), 7.05 (m, 1H), 7.62 (d, 1H), 7.74 (dd, 1H), 7.91 (d, 1H), 8.17 (d, 1H), 8.41 (dd, 1H), 8.94 (d, 1H), 9.07 (d, 1H). ESI-MS (MeOH) m/z: 372 (MH+).
{Ru(bpy)2[4-(m-(CH2)4-OC6H4NMe2)-phen]}(NO3)2 (5a). A mixture of 0.48 g (1.0 mmol) of cis-Ru(bpy)2Cl2 and 370 mg (1.0 mmol) of 4-m-(CH2)4-OC6H4NMe2-phen (4a) in 10 ml of 20/80 EtOH/H2O was heated at reflux for 12 h. Solvent was removed under vacuum, and the residue was purified by silica chromatography (2 × 40 cm) using KNO3 saturated 15/15/70 (vol/vol/vol) H2O/EtOH/MeCN eluent. For 5d, the crude product required further purification, which was effected by two additional silica gel chromatography steps by using KNO3 saturated H2O/EtOH/MeCN (5/5/90 vol/vol/vol) eluent. This process resulted in partial separation of the desired product. Fractions containing “pure” product, as determined by TLC and electrospray ionization-MS, were pooled, and solvent was removed under vacuum. The product was extracted from the resulting residue by using 20 ml of 20/80 (vol/vol) MeOH/CH2Cl2. Removal of solvent gave the product as a red powder. The reported yield is low because a significant quantity of product was also present in the impure product fractions from the chromatographic separations. Yield: 0.27 g (31%). 1H NMR (300 MHz, CD3OD): δ 1.85-2.05 (m, 6H), 2.9 (s, 6H), 4.06 (t, J = 5.7 Hz, 2H), 6.21-6.24 (m, 2H), 6.35 (ddd, J = 7.8, 1.8, 0.5 Hz, 1H), 7.05 (m, 1H), 7.30 (m, 2H), 7.53 (m, 2H), 7.61 (m, 2H), 7.72 (d, J = 5.5 Hz, 1H), 7.81 (dd, J = 8, 5 Hz, 1H), 7.93 (d,J = 5.5 Hz, 2H), 8.0-8.1 (m, 3H), 8.12-8.2 (m, 3H), 8.29 (d, J = 9 Hz, 1H), 8.51 (d, J = 9 Hz, 1H), 8.58 (dd, J = 7, 0.7 Hz, 1H), 8.7 (m, 3H). ESI-MS (MeOH) m/z: 847 (M-NO3+), 784 (M-2NO3-H+).
Crystallization of the [Ru(II)phen-C4-DMA]-AGAO Complex. AGAO was purified as described in ref. 4. Crystals of AGAO were grown by the hanging-drop vapor diffusion method. Each drop comprised protein (2 μl, ≈10 mg/ml) mixed with an equal volume of well solution (700 mM ammonium sulfate/150 mM Na citrate, pH 6.5). Crystals appeared after several weeks at room temperature. Cryo-protection and addition of wire were performed simultaneously by progressively soaking a crystal in well solutions (30 μl) containing [Ru(II)phen-C4-DMA] (5 mM, ≈25-fold molar excess) and increasing concentrations of glycerol (≈2.5% increments to 30% vol/vol) over a 24-h period. The concentration of wire was kept constant throughout these changes. The crystal was flash-frozen in a 100 K N2 gas stream.
X-Ray Data. Diffraction images were recorded on a Mar345 image plate detector with x-rays from a RU-200 rotating anode generator (Rigaku, Tokyo) (Cu Kα, 1.5418 Å) with mirror optics from Osmic (Auburn Hills, MI). Diffraction data were indexed and scaled by using the hkl software suite (13). Details are set out in Table 1.
Table 1.
Crystallographic data and structure refinement statistics for the conjugate of [Ru(II)phen-C4-DMA] with AGAO
| Crystallographic data | |
| Space group | C2 |
| Unit cell dimensions | |
| a, Å | 158.06 |
| b, Å | 62.91 |
| c, Å | 92.10 |
| β, ° | 112.11 |
| Data collection temperature, K | 100 |
| Resolution, Å | 1.73-15.0 (1.73-1.76)† |
| Mosaicity, ° | 0.38 |
| Observations, total | 316,679 |
| Observations, unique | 82,062 |
| Redundancy | 3.9 (2.7) |
| Rmerge‡ | 0.041 (0.105) |
| I/σ(I) | 22.5 (5.8) |
| Completeness, % | 94.4 (85.2) |
| Refinement statistics | |
| Resolution range, Å | 1.73-15.0 (1.73-1.78) |
| Nonhydrogen atoms used in refinement | 5,510 |
| Components of model | |
| Protein | Residues 9-628 |
| Metal atoms | Cu, Na, Ru |
| Water molecules | 508 |
| Other | [Ru(II)phen-C4-DMA], 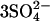 , 6 glycerol , 6 glycerol |
| Reflections in working set | 77,947 (5,056) |
| Reflections in free set | 4,106 (266) |
| Rwork§ | 0.154 (0.183) |
| Rfree¶ | 0.171 (0.213) |
| Rtotal | 0.155 |
| rmsd bond lengths, Å | 0.011 |
| rmsd bond angles, ° | 1.5 |
| B value from Wilson plot, Å2 | 20.3 |
| Average B value, Å2 | 15.6 |
| ESU,∥ Å | 0.053 |
| Ramachandran plot residues in†† | |
| Most favored regions, % | 90.1 |
| Additionally allowed regions, % | 8.7 |
| Generously allowed regions, % | 0.4 (Lys-242 and Phe-142) |
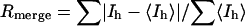
Crystal Structure Refinement. The starting model for refinement was the native AGAO structure refined at 1.60 Å resolution (D.B.L., A.P.D., H.C.F., and J.M.G., unpublished results). No solvent molecules, metal ions, sulfate ions, or glycerol molecules were included. Residues with multiple conformers in the native structure were assigned zero occupancy, and the active-site TPQ (residue 382) was replaced by an Ala residue. Initial low-resolution (15 to 4 Å) rigid-body refinement was followed by rounds of restrained refinement with translation, libration, and screw-motion parameterization (14), alternating with the inspection of electron-density maps and manual adjustment of the model. Solvent atoms and the Cu atom were added to the model gradually, consistent with difference electron density and reasonable stereochemistry. The TPQ side chain, which was clearly defined, was modeled in the “off-Cu” conformation. Solvent atoms in the active-site channel were added last. At a late stage of the refinement, there were two significant difference electron-density features in the active-site channel. One such feature was adjacent to the TPQ and could be modeled as the DMA group of the [Ru(II)phen-C4-DMA] wire (see Fig. 2c). The other, more intense, difference electron-density feature was located at the entrance to the active-site channel and could be resolved as two peaks separated by ≈2.9 Å. Two large anomalous difference peaks occurred at the same positions (see Fig. 2c), the larger being equal to an anomalous difference peak at the Cu atom site and the other being ≈50% smaller. These peaks were interpreted as alternative sites for the Ru atom, the positions of the phen and bpy ligand groups being inferred from known molecular geometries. The chiral Ru(II) head group was arbitrarily modeled as the Δ enantiomer.
Fig. 2.

Crystal structure of the wire-AGAO complex. (a) The AGAO molecule, showing the [Ru(II)phen-C4-DMA] wire inserted in the active-site channel of each subunit. The two subunits of the homodimeric AGAO molecule are colored gray and brown, respectively. (b) The interaction between the [Ru(II)phen-C4-DMA] wire and the AGAO molecule. The [Ru(II)(phen)(bpy)2] head group lies at the protein-solvent interface and fills the opening of the active-site channel. The DMA group makes close (≈3 Å) contacts with the TPQ cofactor. The -(CH2)4-linker is disordered; it is shown in one of its modeled conformations. (c) Electron-density contours and the model of the [Ru(II)phen-C4-DMA]-AGAO complex. The wire is fitted to Fo - Fc electron density (green) contoured at 3σ. The TPQ cofactor is shown in 2Fo - Fc electron density (gray) contoured at 1.5σ. Anomalous difference electron density (blue) is contoured at 4.5σ.
The hypothesis that there were two slightly different Ru positions implied that the wire has at least two conformers. No significant electron density was observed between the DMA and Ru ends of the wire (see Fig. 2c). Nevertheless, it was possible to fit plausible models of the -(CH2)4- linker into the active-site channel (see Fig. 2b).
The occupancies of the atoms at the DMA and Ru ends of the wire were varied systematically until the residual electron density was optimized. In the final model, the occupancy of the DMA group was 1.0, whereas the occupancies of the two Ru atoms and their ligand groups were 0.3 and 0.2, respectively.
The x-ray data and refinement statistics are presented in Table 1. The coordinates and structure factors of the complex have been deposited in the Protein Data Bank (PDB ID code 2BT3).
Crystallographic Software. The monomer library for the [Ru(II)-phen-C4-DMA] wire was constructed by using a combination of prodrg (15), the ccp4 molecular library sketcher (16), and manual parameter adjustment. Solvent atoms were placed at positions indicated by arp/warp (17) and/or evidence from difference density peaks. Least-squares refinement was carried out with refmac5 (18). The final structure was validated by using procheck (19) and molprobity (20). Figs. 2 and 3 were produced by means of pymol (21) and ligplot (22), respectively.
Fig. 3.

A Ligplot cartoon (19) representing the contacts between [Ru(II)-phen-C4-DMA] and residues in the AGAO active-site channel. The atoms or residue involved in a contact are indicated by “hairs” on the relevant circle. Residue Leu 358* is marked with an asterisk to indicate that it belongs to the second subunit of the homodimeric molecule.
Inhibition Experiments. Amine oxidase activity was determined by monitoring benzaldehyde production over the course of 3 min at 25°C by using ε250 = 12,800 M-1·cm-1 (23). All kinetics experiments were carried out in 0.1 M potassium phosphate buffer (pH 7.2). Kinetics analysis involved first equilibrating each enzyme with a given amount of inhibitor for 1 min under magnetic stirring, followed by addition of substrate benzylamine to initiate each assay. Assays were run in duplicate or triplicate at several inhibitor concentrations. Data were fit to the Michaelis-Menten equation by using origin software (Version 7.0, Microcal, Amherst, MA). Steady-state kinetics data were collected on a Hewlett-Packard 8453 diode-array spectrophotometer equipped with a thermostated cell chamber connected to an Endocal RTE-5 circulating water bath. Solutions of the Ru wires were prepared in H2O by using ε455 = 14,500 M-1·cm-1. A stock solution of [Ru(II)phen-C4-DMA] was prepared in absolute ethanol by using ε252 = 29,000 M-1·cm-1. N,N-dimethyl-m-anisidine (Acros Organics, Geel, Belgium) was distilled before use, and a stock solution was made up in acetonitrile. A stock solution of C11-DMA (see Fig. 1; compound 11) was prepared in dimethylformamide. Because of the limited solubility of several inhibitors, it was necessary to run control assays in the presence of the stock-solution solvents to exclude the possibility that these solvents act as inhibitors. Ethanol, acetonitrile, and dimethylformamide were shown not to affect the rate of amine oxidation at concentrations comparable with those used during kinetics experiments.
Fig. 1.

AGAO inhibitors.
Treatment of kinetics data for a competitive inhibitor was performed according to Segel (24). Inhibition constants are calculated from a linear plot of KMapparent vs. [I] where the x-intercept is equal to -Ki.
Results and Discussion
The 10 wires are shown in Fig. 1. The initial wire design incorporated a DMA substrate to anchor the probe in the active site. Alkane linkers optimize conformational flexibility, thereby allowing the wires to fit well in the substrate channel. The linker length was varied from 1 to 11 methylene units, and [Ru(II)(bpy)2(phen)]2+, [Re(I)(CO)2(N-MeIm)(phen)]+, and CH3 head groups were investigated. The role of the dimethylamino group on inhibitor binding was probed in experiments by using the phenyl analogue of m-DMA.
The ability of the Ru(II) and Re(I) complexes to act as fluorescence probes of AGAO binding was evaluated by both steady-state and time-resolved emission spectroscopy. Wire emission properties (see Table 3, which is published as supporting information on the PNAS web site) are similar to those for [Ru(II)(bpy)2(phen)]2+ and [Re(I)(CO)3(N-MeIm)(phen)]+, indicating that there is no internal quenching by the DMA group in any of the complexes. As expected from the lack of overlap between wire emission and TPQ absorption, the emission spectrum of each wire:AGAO conjugate is virtually identical with the corresponding wire spectrum.
Crystal Structure of the [Ru(II)phen-C4-DMA]-AGAO Complex. [Ru(II)-phen-C4-DMA] occupies the active-site channel of each subunit of the homodimeric AGAO molecule. There is no deviation from the crystallographic twofold symmetry of the molecule, and the present crystallographic data do not permit us to conclude whether the two channels are occupied symmetrically. Within the limits of precision, the structure of the protein in the wire complex is identical to the structure of the native protein (25). The TPQ ring, which is disordered in many CuAO structures and is bound to the Cu atom in some of them, is fully ordered in the off-Cu position. It is directed toward the active-site channel, as would be required for interaction with a substrate. In agreement with other AGAO structures where the TPQ is in the off-Cu position (D.B.L., unpublished results), the so-called gate residue Tyr-296 is in the “open” position.
The DMA group lies close to the TPQ cofactor in a pocket lined by nine hydrophobic residues (see Figs. 2 and 3). There are contacts of ≈3 Å between the two DMA C(methyl) atoms and the TPQ O5 atom, and the aromatic ring plane of the DMA group lies at ≈70° from the plane of the TPQ ring (Fig. 2c). The position of the DMA group in relation to the TPQ is the same as that of the methylphenoxy moiety in the covalent adduct of AGAO with the inhibitor 4-(4-methylphenoxy)-2-butyn-1-amine (15). In other complexes of AGAO with covalently bound aromatic inhibitors, the aromatic ring lies in the same position (D.B.L., A.P.D., H.C.F., and J.M.G., unpublished results).
The [Ru(II)(phen)(bpy)2] head group of the wire nestles into the opening of the active-site channel (Fig. 2), making relatively few contacts with the protein. Evidence from difference electron density and anomalous difference electron-density maps indicates that the Ru complex is distributed over two locations. It is possible that these locations correspond to the enantiomers (Λ and Δ) of the [Ru(II)(phen)(bpy)2] head group. Experiments should be conducted to test this hypothesis. If the Ru complex is distributed over two sites, then the portion of the wire linking the Ru complex and the DMA group must have at least two conformations. The inferred disorder is consistent with the following observations: the -(CH2)4-linker is not represented by significant electron density; the refined occupancies of the two Ru sites (0.3, 0.2) are significantly lower than the occupancy of the more tightly anchored DMA group (1.0); and the Ru complex is relatively unrestrained by contacts with the protein.
Inhibition of the Enzyme. Over the past few years, there has been increasing interest in the development and assessment of amine oxidase inhibitors (26-34). The results of these studies have underscored the importance of structural differences among the active sites of CuAOs in governing reactivity with a given inhibitor (29-32). Other recent results also suggest that it may be feasible to design selective mechanism-based CuAO inhibitors. In the crystal structure of AGAO, the active-site channel and pocket are lined by 13 hydrophobic residues (25). When an inhibitor, 4-(2-naphthyloxy)-2-butyn-1-amine, was modeled into the AGAO active site as a Schiff base derivative, it made significant contacts with 5 of these residues (Phe-105, Trp-168, Tyr-302, Tyr-307, and Trp-359), and the possibility of π-stacking interactions with the side chains of Phe-105 and Tyr-302 was noted (29). Structural and kinetics studies of the interaction between AGAO and 4-(aryloxy)-2-butynamines have provided further details of the active-site residues that may affect substrate binding (31).
All eight metal-diimine wires strongly inhibit AGAO (Table 2). Inhibition is completely reversible with dialysis resulting in full recovery of enzymatic activity. Representative Michaelis-Menten and Ki plots are given in Fig. 4. All inhibitors exhibit clean competitive inhibition with respect to substrate amine, indicating that they block the AGAO active-site channel or otherwise interfere with binding of substrate amine to the free enzyme. Active-site blocking is demonstrated by the structural analysis of [Ru(II)phen-C4-DMA]-AGAO (Fig. 2). The inhibitor is capable of making contacts of 3-5 Å with up to nine residues in the active-site channel and pocket (Fig. 3), including the five near-neighbor residues identified earlier in simulations of 4-(2-naphthyloxy)-2-butyn-1-amine-AGAO active-site complexes (29).
Table 2. AGAO inhibition constants.
| Inhibitor | Ki |
|---|---|
| (2) [Ru(II)bpy-C1-DMA] | 300 ± 18 nM |
| (5a) [Ru(II)phen-C4-DMA] | 37 ± 3 nM |
| (5b) [Ru(II)phen-C5-DMA] | 43 ± 1.5 nM |
| (5c) [Ru(II)phen-C6-DMA] | 80 ± 11 nM |
| (5d) [Ru(II)phen-C9-DMA] | 92 ± 9 nM |
| (5e) [Ru(II)phen-C11-DMA] | 92 ± 11 nM |
| (8) [Ru(II)phen-C4-OPh] | 680 ± 35 nM |
| (10) [Re(I)phen-C4-DMA] | 195 ± 8 nM |
| (11) C11-DMA | 6 ± 1 μM |
| N,N-dimethyl-m-anisidine | 8 ± 1 μM |
Fig. 4.

Inhibition of AGAO by [Ru(II)phen-C4-DMA]. (a) Kinetics raw data fit to a Michaelis-Menten model. Legend shows the concentration of inhibitor used for each curve. Error bars represent standard deviation of the rate at the given benzylamine concentration. (b) The Ki plot for [Ru(II)phen-C4-DMA]. Error bars represent the error associated with the hyperbolic fits in a.
[Ru(II)phen-C4-DMA] is the most potent inhibitor (Ki = 37 nM). When the [Ru(II)(bpy)2(phen)]2+ head group is replaced by [Re(I)(CO)3(N-MeIm)(phen)]+ (Fig. 1; compound 10) the Ki increases ≈5-fold to 195 nM. If the head group is omitted altogether in a DMA wire with an 11-carbon linker (Scheme 1; compound 11), the Ki increases further to 6 μM. A similar result is obtained with the ligand N,N-dimethyl-m-anisidine, which has a Ki of 8 μM. It is possible that the head group interacts with the protein surface in a way that stabilizes the protein-inhibitor complex. The interaction may be either electrostatic or hydrophobic or both. In AGAO, the molecular surface surrounding the entrance to the active-site channel is predominantly negative (25). A wire with a Ru(II) head group (charge +2) should bind better than one with a Re(I) head group (charge +1) or a wire with no head group, as observed. The hydrophobic surfaces of the head groups decrease in the same order as the charges, which also may provide an explanation for the trend in binding. In the crystal structure of the [Ru(II)phen-C4-DMA]:AGAO conjugate, contacts between the head group and the protein are clearly important (Fig. 3c), but the quality of the electron-density maps does not enable us to discriminate between electrostatic and other types of interaction.
The importance of the TPQ-contacting tail group is illustrated by the increase in Ki when the dimethylamine group of DMA is replaced by a hydrogen atom: the Ki of [Ru(II)phen-C4-OPh] (680 nM) is 18-fold greater than that of [Ru(II)phen-C4-DMA] (37 nM). However, the finding that [Ru(II)phen-C4-OPh] inhibits AGAO at a submicromolar level shows that the dimethylamine group is not essential for binding.
In contrast to head (Ki ratios 200:1) and tail (Ki ratios 18:1) groups, the linker length plays a comparatively minor role (Fig. 5). The potency decreases only slightly as the linker length increases from -(CH2)4- to -(CH2)11-. The greatest variations within this range occur from -(CH2)4- to -(CH2)6-. We assume that the wires with shorter linkers have more ordered structures and that in wires with more than six methylene groups the head group is too far from the molecular surface to affect the interaction. Only the wire with a -(CH2)1-linker, which is too short to permit the DMA group to approach TPQ when the Ru(II) complex is at the surface of the protein molecule, is a relatively poor inhibitor.
Fig. 5.

Inhibition constant vs. linker length for Ru(II)-diimine wires.
Our channel-blocking wires include the most effective reversible CuAO inhibitors reported to date. What is more, we have made progress in understanding some of the structural subtleties that determine inhibitor potency. Most importantly, our wires target the active-site channel tracing the path of a substrate from the solvent to the active site. In these AGAO-wire conjugates, the tertiary amine terminus is in close proximity to the off-Cu cofactor TPQ. Because active-site channel residues vary substantially among CuAOs, carefully designed metallowires could function as highly selective inhibitors. Based on findings with different head groups, tail-group substituents, and linkers, we predict that selective CuAO inhibitors with picomolar affinities will be available in the near future.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants GM65011 (to S.M.C.), DK19038 and GM070868 (both to H.B.G.), and GM27659 (to D.M.D.) and Australian Research Council Grant DP0208320 (to H.C.F. and J.M.G.).
Abbreviations: AGAO, Arthrobacter globiformis amine oxidase; CuAO, copper amine oxidase; bpy, 2,2′-bipyridine; phen, 1,10-phenanthroline; DMA, dimethylaniline; TPQ, 2,4,5-trihydroxyphenylalanine quinone.
Data deposition: The coordinates and structure factors of the complex have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2BT3).
References
- 1.Klinman, J. P. (2003) Biochim. Biophys. Acta 1647, 131-137. [DOI] [PubMed] [Google Scholar]
- 2.McGuirl, M. A. & Dooley, D. M. (1999) Curr. Opin. Chem. Biol. 3, 138-144. [DOI] [PubMed] [Google Scholar]
- 3.Salmi, M. & Jalkanen, S. (2001) Trends Immunol. 22, 211-216. [DOI] [PubMed] [Google Scholar]
- 4.Juda, G. A., Bollinger, J. A. & Dooley, D. M. (2001) Protein Expression Purif. 22, 455-461. [DOI] [PubMed] [Google Scholar]
- 5.Dmochowski, I. J., Dunn, A. R., Wilker, J. J., Crane, B. R., Green, M. T., Dawson, J. H., Sligar, S. G., Winkler, J. R. & Gray, H. B. (2002) Methods Enzymol. 357, 120-133. [DOI] [PubMed] [Google Scholar]
- 6.Dunn, A. R., Dmochowski, I. J., Bilwes, A. M., Gray, H. B. & Crane, B. R. (2001) Proc. Natl. Acad. Sci. USA 98, 12420-12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn, A. R., Hays, A.-M. A., Goodin, D. B., Stout, C. D., Chiu, R., Winkler, J. R. & Gray, H. B. (2002) J. Am. Chem. Soc. 124, 10254-10255. [DOI] [PubMed] [Google Scholar]
- 8.Hays, A.-M. A., Dunn, A. R., Chiu, R., Gray, H. B., Stout, C. D. & Goodin, D. B. (2004) J. Mol. Biol. 344, 455-469. [DOI] [PubMed] [Google Scholar]
- 9.Wilker, J. J., Dmochowski, I. J., Dawson, J. H., Winkler, J. R. & Gray, H. B. (1999) Angew. Chem. Int. Ed. 38, 90-92. [Google Scholar]
- 10.Hess, C. R., Juda, G. A., Dooley, D. M., Amii, R. N., Hill, M. G., Winkler, J. R. & Gray, H. B. (2003) J. Am. Chem. Soc. 125, 7156-7157. [DOI] [PubMed] [Google Scholar]
- 11.Brownstein, S. & Zhou, M. (2003) Magn. Reson. Chem. 41, 1041-1044. [Google Scholar]
- 12.Wiles, M. R. & Massey, A. G. (1967) Tetrahedron Lett. 51, 5137-5138. [Google Scholar]
- 13.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 267, 307-326. [DOI] [PubMed] [Google Scholar]
- 14.Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001) Acta Crystallogr. D 57, 122-133. [DOI] [PubMed] [Google Scholar]
- 15.van Aalten, D. M. F., Bywater, R., Findlay, J. B. C., Hendlich, M., Hooft, R. W. W. & Vriend, G. (1996) J. Comput. Aided Mol. Des. 10, 255-262. [DOI] [PubMed] [Google Scholar]
- 16.Evans, P. R., Sawyer, L., Isaacs, N. & Bailey, S. (1994) Acta Crystallogr. D 53, 760-763. [Google Scholar]
- 17.Lamzin, V. S. & Wilson, K. S. (1993) Acta Crystallogr. D 49, 129-149. [DOI] [PubMed] [Google Scholar]
- 18.Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (2004) Acta Crystallogr. D 60, 240-255. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 20.Lovell, S. C., Davis, I. W., Arenda, W. B., III, de Bakker, P. I. W., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003) Proteins Struct. Funct. Genet. 50, 437-450. [DOI] [PubMed] [Google Scholar]
- 21.DeLano, W. L. (2002) ThepymolMolecular Graphics System (DeLano Scientific, San Carlos, CA).
- 22.Wallace, A. C., Laskowski, R. A. & Thornton, J. M. (1995) Protein Eng. 8, 127-134. [DOI] [PubMed] [Google Scholar]
- 23.Tabor, C. W., Tabor, H. & Rosenthal, S. M. (1954) J. Biol. Chem. 208, 645-661. [PubMed] [Google Scholar]
- 24.Segel, I. H. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems (Wiley, Chichester, U.K.).
- 25.Wilce, M. C. J., Dooley, D. M., Freeman, H. C., Guss, J. M., Matsunami, H., McIntire, W. S., Ruggiero, C. E., Tanizawa, K. & Yamaguchi, H. (1997) Biochemistry 36, 16116-16133. [DOI] [PubMed] [Google Scholar]
- 26.Medda, R., Padiglia, A., Pedersen, J. Z., Agro, A. F., Rotilio, G. & Floris, G. (1997) Biochemistry 36, 2595-2602. [DOI] [PubMed] [Google Scholar]
- 27.Padiglia, A., Floris, G., Longu, S., Schinina, M. E., Pedersen, J. Z., Agro, A. F., De Angelis, F. & Medda, R. (2004) Biol. Chem. 285, 323-329. [DOI] [PubMed] [Google Scholar]
- 28.Yegutkin, G. G., Salminen, T., Koskinen, K., Kurtis, C., McPherson, M. J., Jalkanen, S. & Salmi, M. (2004) Eur. J. Immunol. 34, 2276-2285. [DOI] [PubMed] [Google Scholar]
- 29.Shepard, E. M., Smith, J., Elmore, B. O., Kuchar, J. A., Sayre, L. M. & Dooley, D. M. (2002) Eur. J. Biochem. 269, 3645-3658. [DOI] [PubMed] [Google Scholar]
- 30.Shepard, E. M., Heggem, H., Juda, G. A. & Dooley, D. M. (2003) Biochim. Biophys. Acta 1647, 252-259. [DOI] [PubMed] [Google Scholar]
- 31.O'Connell, K. M., Langley, D. B., Shepard, E. M., Duff, A. P., Jeon, H.-B., Sun, G., Freeman, H. C., Guss, J. M., Sayre, L. M. & Dooley, D. M. (2004) Biochemistry 43, 10965-10978. [DOI] [PubMed] [Google Scholar]
- 32.Lee, Y., Ling, K.-Q., Lu, X., Silverman, R. B., Shepard, E. M., Dooley, D. M. & Sayre, L. M. (2002) J. Am. Chem. Soc. 124, 12135-12143. [DOI] [PubMed] [Google Scholar]
- 33.Bertini, V., Buffoni, F., Ignesti, G., Picci, N., Trombino, S., Iemma, F., Alfei, S., Pocci, M., Lucchesini, F. & De Munno, A. (2005) J. Med. Chem. 48, 664-670. [DOI] [PubMed] [Google Scholar]
- 34.Di Paolo, M.-L., Lunelli, M., Scarpa, M. & Rigo, A. (2004) Biochem. J. 384, 551-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murshudov, G. N. & Dodson, E. J. (1997) CCP4 Newslett., No. 33.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.