

Abstract
The x-ray crystal structure of a 417-nt ribonuclease P RNA from Bacillus stearothermophilus was solved to 3.3-Å resolution. This RNA enzyme is constructed from a number of coaxially stacked helical domains joined together by local and long-range interactions. These helical domains are arranged to form a remarkably flat surface, which is implicated by a wealth of biochemical data in the binding and cleavage of the precursors of transfer RNA substrate. Previous photoaffinity crosslinking data are used to position the substrate on the crystal structure and to identify the chemically active site of the ribozyme. This site is located in a highly conserved core structure formed by intricately interlaced long-range interactions between interhelical sequences.
Keywords: ribozyme, RNA crystallography, tRNA processing
RNase P catalyzes hydrolysis of a phosphodiester bond in precursors of transfer RNA (tRNA) to form the 5′-phosphorylated mature tRNA with the release of a 5′-precursor fragment (1, 2). RNase P homologs occur in all organisms, and the cellular RNase P always is a ribonucleoprotein that consists of one large RNA and one or more protein component. In bacteria, RNase P is typically comprised of a 350- to 400-nt RNA and one ≈120-aa basic protein. Although both RNA and protein components are necessary for cell viability, in vitro at high salt concentrations, bacterial RNase P RNA can act as a catalyst independently of protein (3). Bacterial RNase P is a ribozyme, an RNA-based enzyme.
Knowledge of the structure of RNase P RNA is essential for understanding its function, and structure has been the focus of numerous studies of the RNA. Phylogenetic comparative analyses of RNase P RNA sequences have established the secondary and some tertiary structure of the RNA in a broad diversity of organisms (4-8). Photochemical crosslinking studies provided structural information to orient the helical elements and identified nucleotides associated with the active site of the RNA (9, 10). There are two major structural types of bacterial RNase P RNA, A (ancestral) and B (Bacillus), which differ in a number of structural elements attached to a homologous conserved structure. About two-thirds of any bacterial RNase P RNA is shown by sequence covariations to be involved in Watson-Crick base-pairing interactions, but the interactions that form the global structure have been speculative.
To gain a better understanding of bacterial RNase P, we crystallized and solved the structure of a 417-nt B-type RNase P RNA from Bacillus stearothermophilus, a moderately thermophilic, low G+C Gram-positive bacterium. Although the structure does not yet explain the chemical mechanism of catalysis, it is in agreement with a wealth of available biochemical and comparative data, and it provides a structural context for the chemically active site of this ribozyme.
Materials and Methods
RNA Purification, Crystallization, and Data Collection. As detailed in supporting information, which is published on the PNAS web site, RNA was transcribed in vitro with T7 phage RNA polymerase from linearized plasmid pBstHH2. Transcribed RNA was purified by preparative electrophoresis in denaturing PAGE, followed by nondenaturing ion exchange chromatography, rapid dialysis, and concentration to 10-20 mg/ml. Diffraction quality crystals of annealed RNA were grown by vapor diffusion from agarose gels within 9 months. To prepare heavy atom derivatives, crystals were soaked in the cryoprotectant solution, containing Os(III) hexamine triflate or Pb(OAc)2 (see supporting information). Cryoprotected crystals were flash-cooled in liquid nitrogen before data collection. Diffraction data were collected at 100 K. Inverse beam strategy was used to collect anomalous data for Os and Pb derivatives. All data sets were processed with d*trek (11).
Structure Determination. Crystallographic phases were solved to 3.75-Å resolution by a three-wavelength multiwavelength anomalous diffraction experiment on the Os(III) hexamine derivative and extended to 3.3-Å resolution by density modification against native structure factors with cns (12). The initial model was constructed from phylogenetically predicted helical pieces fit to the relevant regions of the experimental map. As the quality of the combined electron density maps improved upon refinement (supporting information), the remaining bits of the structure were modeled and further refined. Typical cycles of the refinement, interspersed between the rounds of manual rebuilding with program o (13), included restrained simulated annealing by torsion dynamics, crystallographic conjugate gradient minimization, and grouped B-factor refinement. Table 1 outlines the quality of the refined model. See supporting information for details on model building and refinement.
Table 1. Refinement statistics.
| Resolution, Å | 27-3.30 |
| Number of reflections | |
| Overall, F/σ(F) > 2 (%) | 35,599 (97.8) |
| Test set (%) | 1,798 (5.1) |
| Rcryst,* % | 32.5 |
| Rfree,† % | 34.2 |
| Number of atoms | 6,383 |
| Bond length rms deviation, Å | 0.003 |
| Bond angles rms deviation | 0.80 |
| Average B-factor, Å2 | 154.4 |
| Crossvalidated coordinate error (Luzzati/σA),‡ Å | 0.72/0.80 |
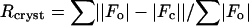 , where |Fo| = observed structure factor amplitude, |Fc| = calculated structure factor amplitude.
, where |Fo| = observed structure factor amplitude, |Fc| = calculated structure factor amplitude.
Rfree, R-factor based on the test set (data excluded from the refinement).
Calculated with cns.
Results
Overall Structure. Crystals of B. stearothermophilus RNase P RNA were developed as outlined in Materials and Methods and used in diffraction analysis. The phylogenetic structure model provided a foundation for building the structure into the electron density maps. Helices and some other structural features are evident in experimental electron density maps and could easily be matched with the phylogenetic model. The availability of the detailed phylogenetic model (14) thus obviated the need to use substituted RNAs to confirm the sequence register. Approximately one-fourth of the RNA could not be traced because of disorder in the crystal.
The structure of the traceable part of the RNA, which contains all of the elements required for catalysis, is shown in Fig. 1. The RNA forms six coaxially stacked helical subdomains held together by a variety of long-range interactions. An unusual feature of the RNase P RNA architecture is the remarkably flat face formed by the relative spatial arrangement of the coaxially stacked helical subdomains (Fig. 1). The long-range interactions that seem particularly important for structural integrity are discussed as follows and include: (i) contacts between loop L5.1†† and L15.1; (ii) the docking of loop L8 into the minor groove of helix P4; and (iii) a complicated structure formed by a number of interhelical joining regions (J3/4, J5/15, J15/15.1, J15.2/2, J19/4, and J4/1) that are drawn together from distant parts of the RNA sequence and form the catalytic core (below).
Fig. 1.

Overview of the B. stearothermophilus RNase P RNA structure. The elements of phylogenetically refined secondary structure are colored according to the coaxially stacked helical domains in the ribbon representations of the structure. The nucleotides colored in gray in the secondary structure (parts of P9, P10.1, P12, L15, and P19) could not be modeled because of disorder in the crystal. See supporting information for an expanded view of the secondary structure.
Docking Between Loops L5.1 and L15.1. The long-range interaction between loops L5.1 and L15.1, a characteristic of B-type RNase P RNAs, was proposed first on the basis of phylogenetic comparative analysis (15, 16). The crystal structure confirms the interaction between these loops, but the details of the interaction are unexpectedly more complex than simple base pairing. As shown in Fig. 2A, L15.1 forms an irregular antiparallel duplex for most of its length, with 12 of the 16 nucleotides engaged in cross-strand purine stacks (17) and canonical, sheared, and N7-amino A-A base pairs. The base of A291 is flipped out of the L15.1 stack and inserted into the L5.1 stack of bases between G73 and A74 (Fig. 2A). The conformation adopted by loop L15.1 poises the bases of nucleotides A290, A295, and A296 to dock into the distorted minor groove of L5.1 by means of A-minor interactions (18). Additional contacts are provided by the backbones of A295, A296, and C69 and U70, which form a canonical ribose zipper (19). All nucleotides that appear to be critical for this interaction (G73, A74, A290, A291, A295, and A296) are highly conserved in the B-type RNase P RNA (15).
Fig. 2.

Long-range interactions in the RNase P RNA. (A) Dock between P15.1 (blue) and P5.1 (green). Nucleotides A290, A295, and A296 in P15.1 involved in A-minor interactions with P5.1 are colored in yellow. Nucleotide A291, involved in a long-range cross-strand stacking interaction, is colored in red. (B) Dock between P8 (blue) and P4 (green). Nucleotides A99, A100, A106, and A107 in P8 are involved in A-minor interactions with P4 and are colored in yellow. See supporting information for an expanded view of the secondary structure.
Docking of L8 into the Minor Groove of P4. Interaction between loop L8 and the minor groove of highly conserved helix P4 also was predicted on the basis of phylogenetic analysis and modeling (15). Again, however, this long-range interaction observed in the crystal structure is more complicated than predicted. As shown in Fig. 2B, the conformation of L8 allows the bases of A99, A100, A106, and A107 to form a continuous stack and dock into the minor groove of helix P4 by A-minor interactions (18). As is often the case with A-minor interactions, the backbones of all four docked adenines are involved in ribose zippers: A99 and A100 of L8 form a single ribose zipper with A392 and U393 of P4, whereas A106 and A107 of L8 form a canonical ribose zipper with C54 and A55 of P4 (19 for ribose zipper nomenclature).
Interhelical Joining Regions. Helix P1, which draws together the 5′ and 3′ ends of the RNA, has been predicted to stack on helix P2 (20) or alternatively on helix P4 (15). In contrast to those predictions, helix P1 in the crystal structure is coaxially stacked on and caps a complicated structure that draws together interhelical joining regions and contains the most highly conserved nucleotides in the RNA. As illustrated in Fig. 3A, nucleotide G14, at the interior end of helix P1, sets up the complex by formation of a dinucleotide platform-containing base triple (21) with U400 and A401 from joining region J4/1 and is stacked on a column of bases drawn together from J19/4 (A385, C386, A387, and G388), J3/4 (A45), and J15.2/2 (U337, A336). U400 in the base-triple starts another stack with A399 (J4/1), A48 (J3/4) and continues with highly conserved helix P4. These two long base stacks interact with each other by a canonical ribose zipper (nucleotides A399, U400 and A385, C386) and through a variety of noncanonical interactions, with a third smaller stack of bases comprised of G47, G46, and A49 of J3/4. On the opposing side of the latter stack, two short stretches of nucleotides (A255, A257 of J5/15 and G275, C276 of J15/15.1) connect helix P15 to helix P4. Two more stacks of bases are formed by nucleotides A389 (J19/4), A330, G331 (J15.2/2), A277 (J15/15.1), and A390 (J19/4), A332, and U333 (J15.2/2) and contribute additional complexity. Individual conformations adopted by the backbones of the joining regions vary from fairly stretched (J19/4 and J4/1) to sharp turns (J3/4, J5/15, and J15/15.1); J15.2/2 forms a jagged loop (Fig. 3B). A full account of internucleotide interactions found in this complicated structure, many involving highly conserved nucleotides, is listed in supporting information. We point out that the available crystallographic data do not allow for explicit modeling of the bound solvent, which is expected to further complicate this structure.
Fig. 3.

Catalytic core of bacterial RNase P RNA. (A) Slab diagram representing stacking interactions in the core. Only some base pairing interactions are shown for clarity. Highly conserved helix P4 is colored in red, J3/4 in purple, J5/15 in green, J15/15.1 in yellow, J15.2/2 in cyan, J19/4 in orange, and J4/1 in blue. (B) Stereo view of an all-atom model of the core with the backbone trace shown as a round ribbon. Structural elements are colored according to A.
Nucleotide Conservation and the Catalytic Core. Of 65 nucleotides that are highly conserved (at the level of at least 80%) in the bacterial RNase P RNA, 48 are found in our model, and analysis of their spatial distribution reveals interesting trends. First, most of the absolutely (100%) and many of the highly (>80%) conserved nucleotides in this RNA are located in helix P4 and the adjacent structure formed from joining regions, as described above. Because of this evolutionary conservation and the long history of biochemical data that implicate the nucleotides in this portion of the molecule in catalytic function (22-27), we believe this region constitutes the catalytic core of this ribozyme (Fig. 4A). Patches of base conservation found throughout the rest of the structure are distributed mostly along the flat surface formed by the coaxially stacked helical subdomains and lead from the catalytic core toward helices P10 and P11, at the base of the highly conserved, so-called “specificity domain” (”S-domain”) of the RNA (28). Such spatial distribution of conserved residues probably reflects conservation of RNase P properties that are important for recognition of the substrate (below), which is expected to bind to RNase P RNA along the flat surface with the T-loop docked to the base of the S-domain and the 5′ end of the acceptor stem (site of RNase P processing) bound to the core (9, 10, 28).
Fig. 4.

Base conservation in the bacterial RNase P RNA and identification of the chemically active site. (A) Distribution of the conserved nucleotides in the bacterial RNase P RNA. Nucleotides absolutely conserved in bacteria are highlighted in red, whereas those highly conserved (≥80%) are shown in yellow. (B) Nucleotides in bacterial RNase P RNA crosslinked by a photoagent at the 5′ end of product tRNA (9). Nucleotides crosslinked in both A- and B-types of RNase P RNA are colored in red, whereas nucleotides crosslinked only in the A-type RNase P RNA are colored in yellow. (C) Proposed chemically active site (orange). Balls and sticks, nucleotides crosslinked by a photoagent at the 5′ end of product tRNA in A- and B-types of RNase P RNA. Helix P4 is colored in blue, P15 in green, and nucleotide and A256 in purple. RNA-phased anomalous difference maps from Os(III) hexamine (red mesh) and Pb(II) (yellow mesh) contoured at 4.5 σ indicate potential metal-binding sites. The metal-bindng sites discussed in the text are labeled A through E. The orientation of the model is approximately the same as in Fig. 3B.
Identification of the Chemically Active Site. The core as we define it, including helix P4 and adjacent interhelical joining regions (Fig. 3A), contains >40 nucleotides. Closer perspective on the active site of this ribozyme is provided by photoaffinity crosslinking experiments that identified a small set of nucleotides located within 9 Å from the product 5′-phosphate of tRNA in the RNase P-tRNA complex (ref. 9 and Fig. 4B). These nucleotides (homologs of A256, A332, U333, A334, and G335 in the B. stearothermophilus RNase P RNA) occur close to each other in our model and, while providing little information on the identity of the catalytically important residues, lead us to propose that the chemical site of catalysis by RNase P is located in the cleft formed by nucleotides A256, A332-G335, A390, C391 (backbone), and G275-A277, as depicted in Fig. 4C.
Metal-Binding Sites. RNase P is a metalloenzyme, dependent for catalysis on divalent metal ions, preferably magnesium (29, 30). Three major functions performed by divalent metal ions in RNase P include contribution to the correct folding of the ribozyme (31), participation in substrate binding (32), and direct involvement in hydrolysis of the substrate phosphodiester bond (31, 33, 34). Some insight into the distribution of the metal-binding sites is provided by the anomalous signal of the heavy atom derivatives, most informatively the derivative with Os(III) hexamine, which mimics a fully hydrated octahedrally coordinated Mg2+ ion, and is expected to bind RNA only by outer-sphere coordination (35, 36). A similar mode of Mg2+ binding to RNA is rare (one or two inner-sphere contacts with RNA are made most frequently; ref. 37), but many metal-binding sites in RNA can accommodate a variety of metal ions that differ in hydration state and coordination preferences (38). Of the 27 strongest Os(III) hexamine sites in the RNase P RNA structure, 23 are found in major grooves of double-stranded helices, often distorted from A-form geometry by helical junctions and noncanonical base pairing. Twenty-one of the 27 Os(III) hexamine ions appear to be coordinated by Hoogsteen faces of purines, including at least one guanine. Both of the two Pb(II)-binding sites are found in the major grooves of helices distorted from the A-form geometry. Most of these metal-binding sites overlap with significant residual density in the σA-weighted Fo-Fc maps calculated with native amplitudes. This suggests that ordered solvent, presumably hydrated magnesium ions that would be detectable at this resolution, is indeed bound at these sites in the native RNA structure.
Of particular interest are metal-binding sites in the vicinity of highly conserved helix P4. Biochemical (39-42) and physical data (43, 44) indicate the importance of magnesium-binding sites in this region for the activity of RNase P RNA. Three Os(III) hexamine (sites A, B, and C, Fig. 4C) and one Pb(II) site are bound by the major groove of P4 and the adjacent highly conserved nucleotides. The Os(III) hexamine site A, which is formed by consecutive base pairs U56-A392 and G57-C391, overlaps with one of the Pb(II) sites, which suggests low cation specificity for this site. Two more sites, D and E (Fig. 4C), deserve special attention: they are found in the cleft we identify as the chemically active center. Although the latter metal-binding sites can be the candidates for direct involvement in chemistry, sites A and B, in the major groove of P4, are remote from the proposed chemically active site and thereby most probably serve a different function. Docking of tRNA onto the RNase P RNA structure (below) positions the backbone of the acceptor stem of tRNA directly in the major groove of helix P4. Therefore, we propose that the primary role of the metal-binding sites in helix P4 is to screen electronegative repulsion between the negatively charged substrate and ribozyme RNAs.
Reconstruction of the Full-Size RNA and Its Complex with the Product. Homologs of 73 nucleotides in the present structure are found in the recently determined S-domain structure of the Bacillus subtilis RNase P RNA (PDB ID code 1NBS; ref. 45); 67 of the overlapping nucleotides are identical between the B. subtilis and B. stearothermophilus RNase P RNAs. Initial comparison of the structures revealed differences in the orientation of helix P8 and in the conformations adopted by loop L8. This loop is docked into the minor groove of helix P4 in our structure but is involved in the formation of crystal contacts in the free S-domain structure. P8 and L8, therefore, were excluded from the superimposition, and only 51 nucleotides, comprising P7, P10, P11 and parts of P9 and P10.1, were used to superimpose the two structures, with rms deviations of 1.98 Å for the phosphorous atoms in the selected nucleotides. As shown in Fig. 5A, the resulting model of the entire RNase P RNA is still remarkably flat, but in agreement with the RNA packing in the crystals (see supporting information).
Fig. 5.

Reconstructed entire B-type RNase P RNA and its complex with tRNA. (A) Reconstructed entire RNase P RNA. Nucleotides in P7, P10, P11, and parts of P9 and P10.1 of B. stearothermophilus RNA structure used in superimposition are colored in purple. The missing substructure, imposed from the B. subtilis S-domain structure, is colored in red. (B) Photoaffinity crosslinking data (9, 10) used in reconstruction of the complex with tRNA. S-domain of RNase P RNA (Left) is colored in gray. Colored spheres in tRNA (Right) represent different sites of photoagent attachment. Colored spheres in RNase P RNA represent sites of crosslinking by those photoagents within the RNase P-tRNA complex. (C) Reconstructed complex. Small gray spheres indicate nucleotide G230. Reconstruction was performed interactively with the computer program o (13); high-resolution tRNAphe structure (PDB ID code 1EH Z; ref. 53) was used in the modeling.
Photoaffinity crosslinking data (9, 10) allow reconstruction, in general detail, of the complex between RNase P RNA and product tRNA. Photoagents chemically attached at different tRNA nucleotides (Fig. 5B) form crosslinks to a limited set of nucleotides in RNase P RNA that consequently are expected to be within 9 Å from the site of the photoagent in the tRNA. The docked complex reconstructed on the basis of the crosslinking data is shown in Fig. 5C. This model of the complex satisfies available photoaffinity crosslinking data and provides a working model for the RNase P RNA-tRNA complex.
Discussion
Extensive previous phylogenetic comparative studies had identified the elements of secondary structure and defined the conserved elements of the RNA. The comparative structure was useful in the interpretation of electron density maps, which in turn agreed with structure predicted on phylogenetic grounds (see supporting information). Patterns of coaxial stacking of the helices and their overall spatial arrangement agree remarkably well between the crystal and phylogenetic-crosslinking structure models. Thus, as with the structure of the ribosome (46-48), crystallography validates the phylogenetic-comparative approach to RNA structure. Comparative and modeling studies with RNase P also indicated some tertiary structural elements, although these generally proved, in the light of the crystal structure, to be more complex than predicted. Phylogenetic comparisons perceive only base interactions and so cannot detect the wealth of backbone and other contacts that are revealed by the crystal structure.
The crystal structure also provides a framework for understanding functional aspects of previous comparative, crosslinking, and other biochemical analyses. Thus, those data were instrumental in inference of the location of the active site of the ribozyme. Our assignment of the chemically active site is further corroborated by the spatial proximity of structural elements implicated in the recognition of the substrate precursors of tRNA. The base of the highly conserved A256 is thought to pair with nucleotide N-1 of the 5′-precursor sequence (49, 50) and is positioned next to the cleft. Loop L15, shown to be involved in binding the substrate 3′-CCA (51), although partially disordered is also within consistent distance to the proposed active site (Fig. 4C).
The global structure of RNase P RNA as seen in the crystal is unusual in that it is remarkably flat and extended. At least a few differences between the crystal and solution structures are expected. One possible distortion in the crystal structure involves P15.2, which possibly is pulled from the body of the RNA by formation of crystal contacts. Biochemical data also suggest that some rearrangement of the RNase P RNA structure may occur upon tRNA binding. The homolog of nucleotide G230, at the base of the S-domain (Fig. 5C), is protected from chemical modification in B. subtilis RNase P RNA (52) but is separated by >11 Å from the nearest tRNA atom in the reconstructed complex. Other potential distortions of the crystal structure from the solution structure are possible, indicating the need for further structural study of additional examples of this universal RNA.
Supplementary Material
Acknowledgments
We thank Drs. R. T. Batey and J. Jankarik for fruitful discussions and help at different stages of this project and Howard Hughes Medical Institute for beam-line access during scanning for the heavy atom derivatives. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. This work was supported by National Institutes of Health Grant GM 34527 (to N.R.P.).
Abbreviations: tRNA, transfer RNA; A-type, ancestral type; B-type, Bacillus type; S-domain, specificity domain.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2A64).
Footnotes
Paired sequences with Watson-Crick complementarity are named P1, P2, etc., in the order of their occurrence in the RNA sequence, from the 5′ to the 3′ end. Loops capping such elements are named according to the name of the elements that they cap, e.g., L3 denotes a loop capping helix P3. Interhelical joining regions are named according to the names of the helical elements that they join, e.g., J3/4 denotes a sequence element that joins helices P3 and P4.
References
- 1.Frank, D. N. & Pace, N. R. (1998) Annu. Rev. Biochem. 67, 153-180. [DOI] [PubMed] [Google Scholar]
- 2.Altman, S. & Kirsebom, L. (1999) in The RNA World, eds. Gesteland, R. F., Cech, T. R. & Atkins, J. F. (Cold Spring Harbor Lab. Press, Plainview, NY), 2nd Ed., p. 380.
- 3.Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N. & Altman, S. (1983) Cell 35, 849-857. [DOI] [PubMed] [Google Scholar]
- 4.Harris, J. K., Haas, E. S., Williams, D., Frank, D. N. & Brown, J. W. (2001) RNA 7, 220-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massire, C., Jaeger, L. & Westhof, E. (1997) RNA 3, 553-556. [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, J. W., Nolan, J. M., Haas, E. S., Rubio, M. A. T., Major, F. & Pace, N. R. (1996) Proc. Natl. Acad. Sci. USA 93, 3001-3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanner, M. A. & Cech, T. R. (1995) RNA 1, 349-350. [PMC free article] [PubMed] [Google Scholar]
- 8.Tallsjö, A., Svard, S. G., Kufel, J. & Kirsebom, L. A. (1993) Nucleic Acids Res. 21, 3927-3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgin, A. B. & Pace, N. R. (1990) EMBO J. 9, 4111-4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, J.-L., Nolan, J. M., Harris, M. E. & Pace, N. R. (1998) EMBO J. 17, 1515-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pflugrath, J. W. (1999) Acta Crystallogr. D 55, 1718-1725. [DOI] [PubMed] [Google Scholar]
- 12.Brunger, A. T. et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 13.Jones, T. A., Zou, J. Y., Cowan, S. & Kjeldgaard, M. (1991) Acta Crystallogr. A 46, 110-119. [DOI] [PubMed] [Google Scholar]
- 14.Brown, J. W. (1999) Nucleic Acids Res. 27, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massire, C., Jaeger, L. & Westhof, E. (1998) J. Mol. Biol. 279, 773-793. [DOI] [PubMed] [Google Scholar]
- 16.Haas, E. S., Banta, A. B., Harris, J. K., Pace, N. R. & Brown, J. W. (1996) Nucleic Acids Res. 24, 4775-4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore, P. B. (1999) Annu. Rev. Biochem. 68, 287-300. [DOI] [PubMed] [Google Scholar]
- 18.Nissen, P., Ippolito, J. A., Ban, N., Moore, P. B. & Steitz, T. A. (2001) Proc. Natl. Acad. Sci. USA 98, 4899-4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura, M. & Holbrook, S. R. (2002) J. Mol. Biol. 320, 455-474. [DOI] [PubMed] [Google Scholar]
- 20.Harris, M. E., Kazantsev, A. V., Chen, J.-L. & Pace, N. R. (1997) RNA 3, 561-576. [PMC free article] [PubMed] [Google Scholar]
- 21.Klosterman, P. S., Hendrix, D. K., Tamura, M., Holbrook, S. R. & Brenner, S. E. (2004) Nucleic Acids Res. 32, 2342-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegel, R. W., Banta, A. B., Haas, E. S., Brown, J. W. & Pace, N. R. (1996) RNA 2, 452-462. [PMC free article] [PubMed] [Google Scholar]
- 23.Haas, E. S., Armbruster, D. W., Vucson, B. M., Daniels, C. J. & Brown, J. W. (1996) Nucleic Acids Res. 24, 1252-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazantsev, A. V. & Pace, N. R. (1998) RNA 4, 937-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris, M. E. & Pace, N. R. (1995) RNA 1, 210-218. [PMC free article] [PubMed] [Google Scholar]
- 26.Loria, A. & Pan, T. (1996) RNA 2, 551-563. [PMC free article] [PubMed] [Google Scholar]
- 27.Pan, T. (1995) Biochemistry 34, 902-909. [DOI] [PubMed] [Google Scholar]
- 28.Pan, T., Loria, A. & Zhong, K. (1995) Proc. Natl. Acad. Sci. USA 92, 12510-12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beebe, J. A. & Fierke, C. A. (1994) Biochemistry 33, 10294-10304. [DOI] [PubMed] [Google Scholar]
- 30.Smith, D. & Pace, N. R. (1993) Biochemistry 32, 5273-5281. [DOI] [PubMed] [Google Scholar]
- 31.Fang, X. W., Pan, T. & Sosnick, T. R. (1999) Nat. Struct. Biol. 6, 1091-1095. [DOI] [PubMed] [Google Scholar]
- 32.Beebe, J. A., Kurz, J. C. & Fierke, C. A. (1996) Biochemistry 35, 10493-10505. [DOI] [PubMed] [Google Scholar]
- 33.Warnecke, J. M., Sontheimer, E. J., Piccirilli, J. A. & Hartmann, R. K. (2000) Nucleic Acids Res. 28, 720-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warnecke, J. M., Held, R., Busch, S. & Hartmann, R. K. (1999) J. Mol. Biol. 290, 433-445. [DOI] [PubMed] [Google Scholar]
- 35.Misra, V. K. & Draper, D. E. (1999) Biopolymers 48, 113-135. [DOI] [PubMed] [Google Scholar]
- 36.Cate, J. H. & Doudna, J. A. (1996) Structure (Cambridge, U.K.) 4, 1221-1229. [DOI] [PubMed] [Google Scholar]
- 37.Klein, D. J., Moore, P. B. & Steitz, T. A. (2004) RNA 10, 1366-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Batey, R. T. & Doudna, J. A. (2002) Biochemistry 41, 11703-11710. [DOI] [PubMed] [Google Scholar]
- 39.Christian, E. L., Kaye N. M. & Harris, M. E. (2002) EMBO J. 21, 2253-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasmussen, T. A. & Nolan, J. M. (2002) Gene 294, 177-185. [DOI] [PubMed] [Google Scholar]
- 41.Christian, E. L., Kaye, N. M. & Harris, M. E. (2002) RNA 6, 511-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frank, D. N., Ellington, A. E. & Pace, N. R. (1996) RNA 2, 1179-1188. [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitz, M. (2004) Nucleic Acids Res. 32, 6358-6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitz, M. & Tinoco I., Jr. (2000) RNA 6, 1212-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krasilnikov, A. S., Yang, X., Pan, T. & Mondragon, A. (2003) Nature 421, 760-764. [DOI] [PubMed] [Google Scholar]
- 46.Ban, N., Nissen, P., Hansen, J., Moore, P. B. & Steitz, T. A. (2000) Science 289, 905-920. [DOI] [PubMed] [Google Scholar]
- 47.Wimberly, B. T., Brodersen, D. E., Clemons W. M., Jr., Morgan-Warren, R. J., Carter, A. P., Vonrhein, C., Hartsch, T. & Ramakrishnan, V. (2000) Nature 407, 327-339. [DOI] [PubMed] [Google Scholar]
- 48.Yusupov, M. M., Yusupova, G. Zh., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H. D. & Noller, H. F. (2001) Science 292, 883-896. [DOI] [PubMed] [Google Scholar]
- 49.Zahler, N. H., Sun, L., Christian, E. L. & Harris, M. E. (2005) J. Mol. Biol. 345, 969-985. [DOI] [PubMed] [Google Scholar]
- 50.Zahler, N. H., Cristian, E. L. & Harris, M. E. (2003) RNA 9, 734-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh, B. K. & Pace, N. R. (1994) Nucleic Acids Res. 22, 4087-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Odell, L., Huang, V., Jakacka, M. & Pan, T. (1998) Nucleic Acids Res. 26, 3717-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi, H. & Moore, P. B. (2000) RNA 6, 1091-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.